

Abstract
Based on studies in animal models, viral infections, in particular by enteroviruses, can accelerate or halt type 1 diabetes (T1D) development. Among factors that determine the outcome are the degree of viral replication in the target organ (viral titres), the tropism of the virus for β cells, and the precise time-point of infection in relation to the diabetogenic process. Mechanisms underlying these phenomena have been assessed in mouse studies and should now be verified for human T1D. For enhancement of diabetes development, up-regulation of interferon pathways, expression of class-I major histocompatibility complexes and Toll-like receptor-dependent immunity appear important. In contrast, prevention of T1D involves pathways that the immune system usually invokes to shut down anti-viral responses to limit immunopathology, and which can ‘clean out’ autoreactive memory effector T cells as a bystander phenomenon: up-regulation of inhibitory molecules and invigoration of regulatory T cell (Treg) function. Importantly, these immunoregulatory processes also appear to foster and sustain persistent viral infections. Induction of immunoregulatory mechanisms, and in particular the phenotype and function of Tregs, is of interest therapeutically and will be discussed.
Keywords: regulatory T cells, suppressor cytokines, type 1 diabetes, viral infection, viral persistence
Introduction
The immune response mounted against viral infections has the potential to cause severe immunopathology and is therefore highly regulated. A number of immunoregulatory mechanisms are in place to ensure that the cells participating in anti-viral immunity are eliminated rapidly or kept under control following their activation. Among those is expression of inhibitory molecules [e.g. programmed death (PD)-1, cytotoxic T lymphocyte antigen (CTLA)-4] on activated T cells, activation or induction of regulatory T cells (Tregs), and/or production of suppressor cytokines [e.g. interleukin (IL)-10, transforming growth factor (TGF)-β]. Of importance is that these different factors all constitute major regulators of not only anti-viral but also autoimmune processes, and in particular type 1 diabetes (T1D). With similar parameters controlling anti-viral immunity and autoimmunity, an intriguing question is whether and how viral infections might affect the outcome of autoimmune diseases such as T1D. The aetiology of T1D is complex, because many genes that protect from the disease as well as enhance the pathogenesis act in concert to confer susceptibility, and in addition to genetic predisposition environmental factors such as viral infections can modulate the disease process [1,2]. In some cases viruses can cause presentation of β cell antigens to autoreactive T cells and promote T1D, while in others trigger immunoregulatory mechanisms that prevent disease development (supporting the ‘hygiene hypothesis’[3–5]. While immunoregulation is beneficial in the face of autoimmune diseases, it can hinder the immune response mounted against viruses, thereby enabling them to establish persistence. In sum, fine-tuning of the balance between anti-viral immunity and immunoregulation is a crucial aspect ensuring that neither autoimmune disease nor viral persistence occurs.
How viruses enhance autoimmunity in T1D
Viruses, notably human enteroviruses, infect various organs causing a range of disorders, among which is damage to β cells. These viruses have been proposed to be associated with T1D development [6]. Enterovirus infections are more frequent in siblings developing T1D compared to non-diabetic siblings, and enterovirus antibodies are elevated in pregnant mothers whose children develop T1D later [7]. Cocksackie virus B4 (CVB4) is the most common enteroviral strain found in prediabetic and diabetic individuals. CVB RNA has been detected in blood from patients at the onset or during the course of T1D [8,9]. In addition, immune responses to CVB antigens were found to be enhanced in T1D patients after the onset of the disease [10]. Recently, Dotta et al. were also able to detect CVB4 in pancreatic tissue specimens from three of six T1D patients [11,12]. Elshebani et al. also found that enterovirus isolates obtained from newly diagnosed T1D patients could infect and induce destruction of human islet cells in vitro[13]. Also recently, Oikarinen et al. have isolated enteroviruses from intestinal biopsy samples in 75% of T1D cases versus 10% of control patients, possibly reflecting persistent enteroviral infection of gut mucosa in T1D patients [14]. In sum, isolation of enteroviral antigens from diabetic individuals, particularly after recent onset, is becoming a fairly reproducible finding, supporting a role for these viruses in disease development.
Enteroviruses such as CVB4 might not commonly cause T1D in those individuals with low genetic risk, but rather predispose β cells to autoimmune attack as a consequence of their tropism for these cells (Fig. 1). Most, if not all, viral infections trigger interferon production by the host immune system. Interferons primarily limit viral replication to prevent damage to the infected cell, but in the case of β cells, interferons and other inflammatory factors such as IL-1β and tumour necrosis factor (TNF)-α have harmful effects [15]. Interferons also increase the visibility of virally infected cells to the immune system, by enhancing their expression of surface recognition molecules called major histocompatibility complexes (MHCs). In the case of β cells, expression of MHC class-I (MHC-I) molecules in response to interferon enables their recognition and attack by autoreactive T cells [16]. Following viral infection, MHC-I expression on β cells is increased 100-fold. Unmasking of β cells to the immune system – triggered by viral infection and mediated by interferons – could be an essential step in the development of T1D [17].
Fig. 1.
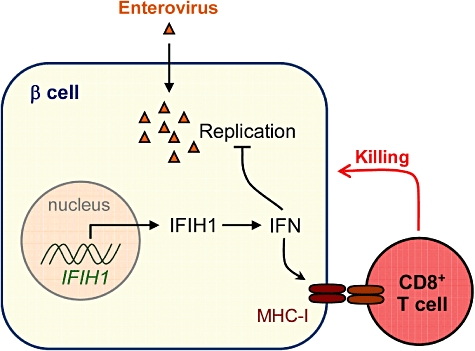
How infection by pancreatotropic viruses might promote type 1 diabetes (T1D). On infection with viruses such as human enteroviruses, interferon response genes, including IFIH1, are activated in insulin-producing pancreatic β cells, leading to increased levels of interferon. These immune mediators not only inhibit viral replication, but also enhance the expression of surface major histocompatibility complex class-I (MHC-I) molecules. Cytotoxic CD8+ T cells recognize infected β cells through the MHC-I molecules on their surface, damaging and eventually killing them. Thus, viral infection can contribute to the development of T1D. In support of this proposal, Nejentsev et al.[18] show that rare variations in IFIH1 protect against T1D.
Different mechanisms can lead to production of interferons and other inflammatory cytokines in response to viral infection. Interferon-induced helicase C domain-containing protein 1 (IFIH1) is a viral sensing helicase enzyme (also known as MDA5) which triggers interferon secretion. In a recent study, Nejentsev et al. identified four genetic variations independently lowering the risk of T1D by about 50%, all of which are located in the IFIH1 gene which controls the expression and structure of IFIH1 [18]. In other words, genetic disabling of IFIH1 appears to lower the risk of T1D. Another recent study by Liu et al. also supports the notion that IFIH1 expression in peripheral blood cells is associated with T1D [19]. Another means through which viral infections cause the release of interferons is through Toll-like receptor (TLR) signalling. TLRs are usually referred to as ‘danger-sensing’ molecules expressed by professional antigen-presenting cells (APCs) that play a central part in triggering inflammation and immunity in response to infection [20]. Different TLRs each recognizing specific molecular patterns have been identified in humans and mice. TLR signalling might promote the development of autoimmunity. For instance, both TLR-3 [21] and TLR-9 [22] signalling can cause T1D when triggered in the presence of β cell antigens. Similarly, TLR-2 was shown to cause APC activation and TNF-α production upon binding to byproducts of late apoptotic β cells, and thereby contribute to the initiation of autoimmune responses in the non-obese diabetic (NOD) mouse model for T1D [23].
In the case of humans, accumulating evidence supports a role for viral infections in the development of T1D. Indeed, all the main ingredients seem to be present. First, CD8+ T cells are the most common cell type present in human pancreatic islets [24], and clones of these cells that can kill human β cells have been isolated [25]. Secondly, and more importantly, increased expression of MHC-I molecules (and in some cases interferons) was observed in pancreatic islets of patients with T1D but not those of controls [12]. As many of these patient islets were not yet targeted by the immune system, an underlying viral infection is a plausible explanation for these observations. Indeed, some studies have documented the presence of human enterovirus proteins in islets of T1D patients but not in those of healthy individuals [26], further thickening the plot for a role of these viruses in T1D. It is now possible to use well-preserved, freshly frozen human organ samples, such as those provided by nPOD (network for pancreatic organ donors with diabetes) (http://www.jdrfnpod.org), to examine islets for the presence of viral footprints such as interferons and increased levels of MHC-I. If viral infection is proved to be a contributing factor, we could aim for lowering the risk of T1D through designing suitable vaccines against human enteroviruses.
How viruses could protect from T1D: virally induced immunoregulation
Just as in humans, an association exists between T1D in mouse models and infection with the enterovirus CVB [27]. CVB4 was shown to be associated tightly with the initiation of T1D in the NOD model, although the influence of the virus appears to be contingent upon the precise point in time at which infection occurs [28,29]. Conversely, certain strains of the CVB3 serotype mediate significant protection against T1D development in NOD mice, regardless of the time of infection [30]. Protective and causative CVB strains appear to differ regarding tropism for beta cells, and the extent of damage they inflict to these cells. Interestingly, other viruses that are also thought to be associated with the pathogenesis of T1D in animals have been shown to mediate protective effects in some instances. For instance, lymphocytic choriomeningitis virus (LCMV) is used as a means to induce T1D experimentally in NOD (and other) mice that express LCMV antigens transgenically in their β cells [31–33], but this virus otherwise prevents T1D efficiently in these mice [34,35]. In sum, while the use of animals has established clearly that particular viruses are capable of inducing autoimmune diabetes, it has also shown that these viruses can play a preventive role in the development of the disease.
Animal studies have shown that human enteroviruses that replicate fast in vivo cause T1D [36], whereas those that grow more slowly actually protect from this disease [30,37]. In addition, pancreatic/islet tropism and replication rate in β cells might be crucial factors determining whether autoimmunity is promoted or prevented. In fact, viruses more commonly spare β cells and abrogate autoimmunity. We have reported recently that the ability of two different viruses to trigger immunoregulatory mechanisms (and no β cell damage) accounts for the capacity of these viruses to protect from T1D [38] (Fig. 2). Infection of prediabetic NOD mice with CVB3 or LCMV delayed diabetes onset and reduced disease incidence. Delayed T1D onset was due to transient up-regulation of programmed death-1 ligand 1 (PD-L1) on lymphoid cells, which was caused partly by interferon and curbed the accumulation of diabetogenic CD8+ T cells expressing PD-1. Reduced T1D incidence was due to expansion and invigoration of CD4+CD25+ Tregs which maintained long-term tolerance. Full protection from T1D resulted from synergy between PD-L1 and CD4+CD25+ Tregs. These results provided evidence that viruses can activate regulatory pathways that participate in controlling not only anti-viral immune responses but also autoimmunity. While these immunoregulatory pathways usually have only a minimal impact on viral elimination, with the goal to limit T cell-mediated immunopathology [39], they appear able to control autoimmune responses efficiently when acting in synergy. These findings provided useful insight for the development of novel and safe therapies to treat T1D [40].
Fig. 2.
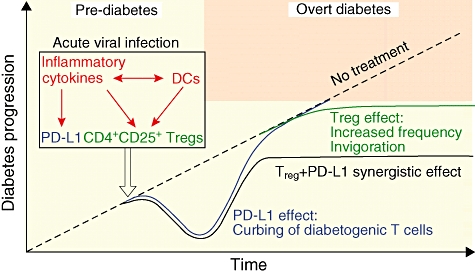
Proposed model for prevention of type 1 diabetes (T1D) by acute viral infection. On infection by β cell non-lytic viruses during the prediabetic phase, release of inflammatory cytokines causes a ‘wave’ of programmed death-1 ligand 1 (PD-L1) up-regulation in lymphoid organs, which curbs diabetogenic CD8+ T cells and delays the onset of overt diabetes. In parallel, natural regulatory T cells (Tregs) stimulated (by, e.g. dendritic cells) under inflammatory conditions expand and acquire enhanced suppressor function, notably via transforming growth factor-β production. Such invigorated Tregs take additional advantage of PD-L1/PD-1-mediated demise of autoreactive effectors to control the remaining diabetogenic T cells efficiently, and thus prevent T1D with high efficacy.
In addition, our findings further supported the notion that viral tropism/replication rate in β cells, which causes their subsequent damage, is a crucial determinant of the diabetogenic property of viruses. In order to expand autoreactive T cells and cause diabetes, viral infections have to either lyse β cells directly or inflict damage to these cells via antigenic recognition of β cell proteins within the proinflammatory milieu. Conversely, our results suggested that proinflammatory signals, when β cells are spared, can invoke important immunoregulatory mechanisms that protect from autoimmunity. In sum, bystander inflammation and anti-viral immunity systemically or in the vicinity of the pancreatic islets may not be detrimental but, rather, beneficial in T1D when occurring in the absence of direct β cell damage. Accordingly, CVB and LCMV cause T1D in NOD mice in contexts where they mediate β cell injury [CVB4 [28,29] and rat insulin promoter (RIP)–LCMV mice [31–33], respectively], but prevent T1D when infection does not directly harm β cells (CVB3 [30] and wild-type mice [34,35], respectively). This suggests that induction versus prevention of T1D by viruses depends upon the extent to which anti-viral inflammation and immunity alter the balance between β cell injury and immunoregulation.
In this regard, while TLR-mediated activation of innate immunity and inflammation in the context of β cell damage might promote T1D, activation of TLR signalling is not systematically causative for T1D. Treatment with agonists of TLR-2 [41], TLR-3 [42], TLR-4 [43] or TLR-9 [44] protect from T1D when given in the absence of β cell antigens. Interestingly, previous work has shown that CD4+CD25+ Tregs, which express TLR-2, TLR-4, TLR-5, TLR-7 and TLR-8 [45–47], become activated and control T cell-mediated wasting disease following exposure to lipopolysaccharide (LPS), a TLR-4 agonist. In addition, while binding to TLR-2 by endogenous antigens causes APC activation and promotes T1D [23], it was also reported to enhance the function of CD4+CD25+ Tregs[41,48]. In fact, activation of TLR-2 signalling in CD4+CD25+ Tregs may cause a transient loss of their function but in parallel trigger their expansion [46,47]. Of note, a study in the NOD model showed that TLR-2 and myeloid differentiation primary response gene 88 (MyD88) were dispensable for the development of T1D [49], thereby contrasting with previous work involving these molecules in the initiation of autoimmune responses directed against β cells [23]. Our recent findings indicate that activation of the TLR-2 signalling pathway, in a naive context or on viral infection, can confer protection from T1D, in part by promoting the expansion of invigorated CD4+CD25+ Tregs (Filippi et al., manuscript in preparation). As discussed above, TLR-2 was shown previously to promote rather than hinder T1D, notably by inducing TNF-α production by APCs [23]. Such opposing roles of TLR-2, and other TLRs, in T1D development might reflect the importance of not only timing [2], but also of β cell antigen release concomitant to TLR signalling for autoimmunity to develop. It thus appears that similar phenomena account for both T1D development and prevention. This suggests that the recurring occurrence of infectious events during early life might promote autoimmunity but will also drive the immune system to build increased immunoregulatory force.
Do immunoregulatory pathways play a role in chronic viral infections?
It is established that acute viral infections trigger immunoregulatory pathways that control anti-viral responses with the goal of limiting immunopathology. For instance, herpes simplex virus (HSV) and influenza infections are associated with expansion of CD4+CD25+ Tregs and IL-10-producing T cells, respectively [39,50]. However, while this notion suggests that viruses could thereby control autoimmunity [38], it also raises the possibility that these regulatory pathways could constitute a means for certain viruses to establish persistence by inhibiting crucial components of the anti-viral immune response (Beutler, this issue). Interestingly, in persistent infections with hepatitis C virus (HCV), human immunodeficiency virus (HIV) or Epstein–Barr virus (EBV), an increase in systemic IL-10 production can be observed. IL-10 is an immunomodulatory cytokine that can be produced by different cell types in humans and mice, including monocytes, macrophages, B cells, CD4+ and CD8+ T cells. Importantly, while APCs and T cells are the main producers of IL-10, they are also important targets of this cytokine, which plays its immunosuppressive role through interplay between these two cell types. IL-10 can inhibit cytokine production directly by T cells in vitro[51], but also prevents the maturation of DCs, rendering them ineffective in activating T and other immune cells. IL-10 plays a role in blocking proinflammatory cytokine production, co-stimulation, MHC class II expression and chemokine secretion. By acting on APCs, IL-10 regulates the proliferation and differentiation of Th1 cells, which are helper T cells that control not only host defence, but also many crucial effector immune responses in vivo such as, for example, anti-tumour immunity. In the case of HIV, IL-10-mediated hampering of APC maturation reduces the efficacy of antigen presentation by these cells and induces T cell-dependent suppression of anti-viral responses [52,53].
It is intriguing to hypothesize that IL-10, along with other factors such as PD-1 [54], may play a role in maintaining chronicity and pathogenicity in chronic infections (Fig. 3). However, absolute proof of concept in humans has not yet been achieved, and solid evidence is not currently available for certain chronic situations such as HIV infection. Importantly, it was shown that the significant amounts of IL-10 produced during HCV infection can be attributed to the generation of a subset of suppressor cells thought to hamper viral resolution by T helper type 1/cytotoxic T (Th1/Tc1) cells [55]. In addition, administration of IL-10 to HCV patients with the intention of dampening chronic immunopathology was found to actually increase viral loads [56]. Furthermore, single nucleotide polymorphisms in the IL-10 region have been associated with natural clearance of HCV in some populations [57]. Thus, the overall concept emerging from clinical studies examining chronic HCV infection supports the hypothesis that influencing IL-10 signalling may have an impact on this chronic viral infection and its deleterious secondary consequences, such as liver cancer, immunodeficiency and lymphomas. In a previous study using the mouse model of protracted viral infection induced by LCMV Clone 13, we observed that a significant amount of IL-10 was produced systemically which, interestingly, coincided with the loss of CTL responses directed against the virus [58]. We used systemic administration of a blocking monoclonal antibody (mAb) against the IL-10 receptor (IL-10R) and observed rapid resolution of the persistent infection. Similar results were obtained simultaneously in an independent study by Michael Oldstone's group [59]. In our study, successful anti-IL-10R therapy occurred in the absence of systemic side effects or immunopathology, and showed efficacy in mice in which the persistent infection was already established.
Fig. 3.
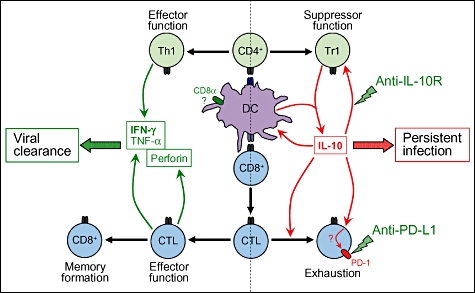
Establishment of viral persistence through induction of interleukin (IL)-10 production. The left panel illustrates how an acute viral infection usually triggers the activation of virus-specific CD8+ T cells by dendritic cells (DCs) driving their subsequent expansion and differentiation into cytotoxic T lymphocytes (CTLs). These cells exert their anti-viral properties by producing inflammatory and cytolytic cytokines such as interferon (IFN)-γ, tumour necrosis factor (TNF)-α and perforin. This phenomenon is generally accompanied by the activation of CD4+ T cells and their polarization into T helper type 1 (Th1) cells capable of producing IFN-γ and TNF-α. The consequence of the anti-viral T cell response is the rapid elimination of the invading virus, followed by the formation of T cell memory. The right-hand panel illustrates how under certain circumstances the initially active CTLs can become exhausted and lose their anti-viral properties. Current evidence indicates that exhaustion may be the consequence of programmed death-1 (PD-1) expression by virus-specific CD8+ T cells and systemic IL-10 production. IL-10 may be produced directly by virally infected DCs, or/and by CD4+ T cells (Tr1) as a consequence of their activation by DCs bearing the intrinsic ability to drive IL-10 responses (e.g. CD8α- DCs [58]). The outcome is the establishment of a feedback loop in which IL-10 maintains its own production, thereby acting in conjunction with PD-1 to keep anti-viral CD8+ T cells in an inactive state.
The observation by us and others that persistent viral infection can be resolved by neutralizing the effect of IL-10 in vivo suggests that successful treatment of other chronic infections also associated with high IL-10 production may be achieved using the same approach. Interestingly, induction of IL-10 production appears to be a common strategy for different viruses to achieve immunosuppression and escape anti-viral immunity. Importantly, however, current therapies such as interferon or anti-viral drugs have proved efficient in lowering viral titres and thus should not be abandoned. These strategies could be revised to incorporate an additional component with the goal to modulate the class of the anti-viral response. Disruption of IL-10 responses may be an essential ingredient for a complete cure. In particular, it may enable successful treatment of patients who do not respond to conventional therapy. Neutralizing IL-10 can restore immune responses mounted by T cells isolated from patients harbouring chronic infection. For example, defective T cell immunity in asymptomatic HIV-infected patients can be enhanced in vitro by mAbs neutralizing IL-10 [60].
Although it is possible to restore immunity against persistent viruses in vitro and other pathogens in vivo by blocking the IL-10 signalling pathway, will this strategy function in HIV- or HCV-infected patients? Recent important advances should enable us to address this issue in preclinical studies soon. Although a mouse model for HCV infection is not yet available, the recent identification of human occludin as a necessary factor for entry of HCV into mouse cells constitutes a major step in the right direction [61]. Whether or not neutralization of IL-10 represents a promising therapeutic approach to treat HCV infection should be addressed in mouse models when they become available. With regard to HIV, a number of models using humanized mice are currently available, but the issue of IL-10 involvement/blockade in viral persistence might be trickier than in HCV or other persistent infections. HIV replicates well in CD4+ T cells, and therefore any strategy that will trigger T cell expansion might well promote viral spread rather than containment. In other words, IL-10 might have dual roles in HIV infection; interfering with immunity and clearance, but also suppressing T cell expansion and prohibiting viral propagation. In support of this hypothesis are data showing that mutations in the IL-10 promoter favouring increased IL-10 production diminish susceptibility to HIV infection in human populations [62]. Therefore, one could argue that after HIV infection has been established, blockade of the IL-10 signalling pathway could have adjuvant properties; however, before infection, using a vaccine that induces anti-viral Tregs (e.g. producing IL-10) instead of effector cells might prevent infection. Of important note, this might be the case for HIV but not hold true for HCV or other non-T cell tropic infections.
Disclosure
The authors declare no conflict of interest.
References
- 1.Redondo MJ, Jeffrey J, Fain PR, Eisenbarth GS, Orban T. Concordance for islet autoimmunity among monozygotic twins. N Engl J Med. 2008;359:2849–50. doi: 10.1056/NEJMc0805398. [DOI] [PubMed] [Google Scholar]
- 2.Filippi CM, von Herrath MG. Viral trigger for type 1 diabetes: pros and cons. Diabetes. 2008;57:2863–71. doi: 10.2337/db07-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okada H, Kuhn C, Feillet H, Bach J-F. 99th Dahlem Conference on Infection, Inflammation and Chronic Inflammatory Disorders: The ‘hygiene hypothesis’ for autoimmune and allergic diseases: an update. Clin Exp Immunol. 2010;160:1–9. doi: 10.1111/j.1365-2249.2010.04139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rook GAW. 99th Dahlem Conference on Infection, Inflammation and Chronic Inflammatory Disorders: Darwinian medicine and the ‘hygiene’ or ‘old friends’ hypothesis. Clin Exp Immunol. 2010;160:70–9. doi: 10.1111/j.1365-2249.2010.04133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.von Mutius 99th Dahlem Conference on Infection, Inflammation and Chronic Inflammatory Disorders: Farm lifestyles and the hygiene hypothesis. Clin Exp Immunol. 2010;160:130–5. doi: 10.1111/j.1365-2249.2010.04138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Getts MR, Miller SD. 99th Dahlem Conference on Infection, Inflammation and Chronic Inflammatory Disorders: Triggering of autoimmune diseases by infections. Clin Exp Immunol. 2010;160:15–21. doi: 10.1111/j.1365-2249.2010.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hyoty H, Hiltunen M, Knip M, et al. A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Childhood Diabetes in Finland (DiMe) Study Group. Diabetes. 1995;44:652–7. doi: 10.2337/diab.44.6.652. [DOI] [PubMed] [Google Scholar]
- 8.Clements GB, Galbraith DN, Taylor KW. Coxsackie B virus infection and onset of childhood diabetes. Lancet. 1995;346:221–3. doi: 10.1016/s0140-6736(95)91270-3. [DOI] [PubMed] [Google Scholar]
- 9.Andreoletti L, Hober D, Hober-Vandenberghe C, et al. Detection of coxsackie B virus RNA sequences in whole blood samples from adult patients at the onset of type I diabetes mellitus. J Med Virol. 1997;52:121–7. doi: 10.1002/(sici)1096-9071(199706)52:2<121::aid-jmv1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 10.Juhela S, Hyoty H, Roivainen M, et al. T-cell responses to enterovirus antigens in children with type 1 diabetes. Diabetes. 2000;49:1308–13. doi: 10.2337/diabetes.49.8.1308. [DOI] [PubMed] [Google Scholar]
- 11.Roep BO, Kleijwegt FS, van Halteren AGS, et al. Islet inflammation and CXCL10 in recent-onset type 1 diabetes. Clin Exp Immunol. 2010;159:338–43. doi: 10.1111/j.1365-2249.2009.04087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dotta F, Censini S, van Halteren AG, et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci USA. 2007;104:5115–20. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elshebani A, Olsson A, Westman J, Tuvemo T, Korsgren O, Frisk G. Effects on isolated human pancreatic islet cells after infection with strains of enterovirus isolated at clinical presentation of type 1 diabetes. Virus Res. 2007;124:193–203. doi: 10.1016/j.virusres.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Oikarinen M, Tauriainen S, Honkanen T, et al. Detection of enteroviruses in the intestine of type 1 diabetic patients. Clin Exp Immunol. 2008;151:71–5. doi: 10.1111/j.1365-2249.2007.03529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–26. doi: 10.1038/nrendo.2009.21. [DOI] [PubMed] [Google Scholar]
- 16.Seewaldt S, Thomas HE, Ejrnaes M, et al. Virus-induced autoimmune diabetes: most beta-cells die through inflammatory cytokines and not perforin from autoreactive (anti-viral) cytotoxic T-lymphocytes. Diabetes. 2000;49:1801–9. doi: 10.2337/diabetes.49.11.1801. [DOI] [PubMed] [Google Scholar]
- 17.von Herrath MG, Fujinami RS, Whitton JL. Microorganisms and autoimmunity: making the barren field fertile? Nat Rev Microbiol. 2003;1:151–7. doi: 10.1038/nrmicro754. [DOI] [PubMed] [Google Scholar]
- 18.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–9. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu S, Wang H, Jin Y, et al. IFIH1 polymorphisms are significantly associated with type 1 diabetes and IFIH1 gene expression in peripheral blood mononuclear cells. Hum Mol Genet. 2009;18:358–65. doi: 10.1093/hmg/ddn342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beutler B, Jiang Z, Georgel P, et al. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu Rev Immunol. 2006;24:353–89. doi: 10.1146/annurev.immunol.24.021605.090552. [DOI] [PubMed] [Google Scholar]
- 21.Moriyama H, Wen L, Abiru N, et al. Induction and acceleration of insulitis/diabetes in mice with a viral mimic (polyinosinic–polycytidylic acid) and an insulin self-peptide. Proc Natl Acad Sci USA. 2002;99:5539–44. doi: 10.1073/pnas.082120099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lang KS, Recher M, Junt T, et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med. 2005;11:138–45. doi: 10.1038/nm1176. [DOI] [PubMed] [Google Scholar]
- 23.Kim HS, Han MS, Chung KW, et al. Toll-like receptor 2 senses beta-cell death and contributes to the initiation of autoimmune diabetes. Immunity. 2007;27:321–33. doi: 10.1016/j.immuni.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 24.Pipeleers D, In't Veld P, Pipeleers-Marichal M, Gorus F. The beta cell population in type 1 diabetes. Novartis Found Symp. 2008;292:19–24. doi: 10.1002/9780470697405.ch3. discussion 31, 122–9, 202–3. [DOI] [PubMed] [Google Scholar]
- 25.Skowera A, Ellis RJ, Varela-Calvino R, et al. CTLs are targeted to kill beta cells in patients with type 1 diabetes through recognition of a glucose-regulated preproinsulin epitope. J Clin Invest. 2008;118:3390–402. doi: 10.1172/JCI35449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia. 2009 doi: 10.1007/s00125-009-1276-0. [DOI] [PubMed] [Google Scholar]
- 27.Coleman TJ, Gamble DR, Taylor KW. Diabetes in mice after Coxsackie B 4 virus infection. BMJ. 1973;3:25–7. doi: 10.1136/bmj.3.5870.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med. 1998;4:781–5. doi: 10.1038/nm0798-781. [DOI] [PubMed] [Google Scholar]
- 29.Serreze DV, Ottendorfer EW, Ellis TM, Gauntt CJ, Atkinson MA. Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T-cells in pancreatic islets. Diabetes. 2000;49:708–11. doi: 10.2337/diabetes.49.5.708. [DOI] [PubMed] [Google Scholar]
- 30.Tracy S, Drescher KM, Chapman NM, et al. Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: inoculating nonobese diabetic mice with CVB markedly lowers diabetes incidence. J Virol. 2002;76:12097–111. doi: 10.1128/JVI.76.23.12097-12111.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinic MM, Juedes AE, Bresson D, et al. Minimal impact of a de novo-expressed beta-cell autoantigen on spontaneous diabetes development in NOD mice. Diabetes. 2007;56:1059–68. doi: 10.2337/db05-0062. [DOI] [PubMed] [Google Scholar]
- 32.Oldstone MB, Nerenberg M, Southern P, Price J, Lewicki H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: role of anti-self (virus) immune response. Cell. 1991;65:319–31. doi: 10.1016/0092-8674(91)90165-u. [DOI] [PubMed] [Google Scholar]
- 33.Ohashi PS, Oehen S, Buerki K, et al. Ablation of ‘tolerance’ and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–17. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- 34.Christen U, Benke D, Wolfe T, et al. Cure of prediabetic mice by viral infections involves lymphocyte recruitment along an IP-10 gradient. J Clin Invest. 2004;113:74–84. doi: 10.1172/JCI200417005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oldstone MB. Prevention of type I diabetes in nonobese diabetic mice by virus infection. Science. 1988;239:500–2. doi: 10.1126/science.3277269. [DOI] [PubMed] [Google Scholar]
- 36.Kanno T, Kim K, Kono K, Drescher KM, Chapman NM, Tracy S. Group B coxsackievirus diabetogenic phenotype correlates with replication efficiency. J Virol. 2006;80:5637–43. doi: 10.1128/JVI.02361-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tracy S, Hofling K, Pirruccello S, Lane PH, Reyna SM, Gauntt CJ. Group B coxsackievirus myocarditis and pancreatitis: connection between viral virulence phenotypes in mice. J Med Virol. 2000;62:70–81. doi: 10.1002/1096-9071(200009)62:1<70::aid-jmv11>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 38.Filippi CM, Estes EA, Oldham JE, von Herrath MG. Immunoregulatory mechanisms triggered by viral infections protect from type 1 diabetes in mice. J Clin Invest. 2009;119:1515–23. doi: 10.1172/JCI38503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suvas S, Azkur AK, Kim BS, Kumaraguru U, Rouse BT. CD4+CD25+ regulatory T cells control the severity of viral immunoinflammatory lesions. J Immunol. 2004;172:4123–32. doi: 10.4049/jimmunol.172.7.4123. [DOI] [PubMed] [Google Scholar]
- 40.Chatenoud L, You S, Okada H, Kuhn C, Michaud B, Bach J-F. 99th Dahlem Conference on Infection, Inflammation and Chronic Inflammatory Disorders: Immune therapies of type 1 diabetes: new opportunities based on the hygiene hypothesis. Clin Exp Immunol. 2010;160:106–12. doi: 10.1111/j.1365-2249.2010.04125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karumuthil-Melethil S, Perez N, Li R, Vasu C. Induction of innate immune response through TLR2 and dectin 1 prevents type 1 diabetes. J Immunol. 2008;181:8323–34. doi: 10.4049/jimmunol.181.12.8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Serreze DV, Hamaguchi K, Leiter EH. Immunostimulation circumvents diabetes in NOD/Lt mice. J Autoimmun. 1989;2:759–76. doi: 10.1016/0896-8411(89)90003-6. [DOI] [PubMed] [Google Scholar]
- 43.Iguchi M, Inagawa H, Nishizawa T, et al. Homeostasis as regulated by activated macrophage. V. Suppression of diabetes mellitus in non-obese diabetic mice by LPSw (a lipopolysaccharide from wheat flour) Chem Pharm Bull (Tokyo) 1992;40:1004–6. doi: 10.1248/cpb.40.1004. [DOI] [PubMed] [Google Scholar]
- 44.Quintana FJ, Rotem A, Carmi P, Cohen IR. Vaccination with empty plasmid DNA or CpG oligonucleotide inhibits diabetes in nonobese diabetic mice: modulation of spontaneous 60-kDa heat shock protein autoimmunity. J Immunol. 2000;165:6148–55. doi: 10.4049/jimmunol.165.11.6148. [DOI] [PubMed] [Google Scholar]
- 45.Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197:403–11. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sutmuller RP, den Brok MH, Kramer M, et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485–94. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc Natl Acad Sci USA. 2006;103:7048–53. doi: 10.1073/pnas.0601554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zanin-Zhorov A, Cahalon L, Tal G, Margalit R, Lider O, Cohen IR. Heat shock protein 60 enhances CD4+ CD25+ regulatory T cell function via innate TLR2 signaling. J Clin Invest. 2006;116:2022–32. doi: 10.1172/JCI28423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Wen L, Ley RE, Volchkov PY, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–13. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun J, Madan R, Karp CL, Braciale TJ. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med. 2009;15:277–84. doi: 10.1038/nm.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Waal Malefyt R, Yssel H, de Vries JE. Direct effects of IL-10 on subsets of human CD4+ T cell clones and resting T cells. Specific inhibition of IL-2 production and proliferation. J Immunol. 1993;150:4754–65. [PubMed] [Google Scholar]
- 52.de Waal Malefyt R, Haanen J, Spits H, et al. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J Exp Med. 1991;174:915–24. doi: 10.1084/jem.174.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Granelli-Piperno A, Golebiowska A, Trumpfheller C, Siegal FP, Steinman RM. HIV-1-infected monocyte-derived dendritic cells do not undergo maturation but can elicit IL-10 production and T cell regulation. Proc Natl Acad Sci USA. 2004;101:7669–74. doi: 10.1073/pnas.0402431101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 55.Accapezzato D, Francavilla V, Paroli M, et al. Hepatic expansion of a virus-specific regulatory CD8(+) T cell population in chronic hepatitis C virus infection. J Clin Invest. 2004;113:963–72. doi: 10.1172/JCI20515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nelson DR, Tu Z, Soldevila-Pico C, et al. Long-term interleukin 10 therapy in chronic hepatitis C patients has a proviral and anti-inflammatory effect. Hepatology. 2003;38:859–68. doi: 10.1053/jhep.2003.50427. [DOI] [PubMed] [Google Scholar]
- 57.Oleksyk TK, Thio CL, Truelove AL, et al. Single nucleotide polymorphisms and haplotypes in the IL10 region associated with HCV clearance. Genes Immun. 2005;6:347–57. doi: 10.1038/sj.gene.6364188. [DOI] [PubMed] [Google Scholar]
- 58.Ejrnaes M, Filippi CM, Martinic MM, et al. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med. 2006;203:2461–72. doi: 10.1084/jem.20061462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12:1301–9. doi: 10.1038/nm1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clerici M, Wynn TA, Berzofsky JA, et al. Role of interleukin-10 in T helper cell dysfunction in asymptomatic individuals infected with the human immunodeficiency virus. J Clin Invest. 1994;93:768–75. doi: 10.1172/JCI117031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ploss A, Evans MJ, Gaysinskaya VA, et al. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature. 2009;457:882–6. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Naicker DD, Werner L, Kormuth E, et al. Interleukin-10 promoter polymorphisms influence HIV-1 susceptibility and primary HIV-1 pathogenesis. J Infect Dis. 2009;200:448–52. doi: 10.1086/600072. [DOI] [PMC free article] [PubMed] [Google Scholar]