

Abstract
Firefly luciferase (FLuc), an ATP-dependent bioluminescent reporter enzyme, is broadly used in chemical biology and drug discovery assays. PTC124 (Ataluren; (3-[5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid) discovered in an FLuc-based assay targeting nonsense codon suppression, is an unusually potent FLuc-inhibitor. Paradoxically, PTC124 and related analogs increase cellular FLuc activity levels by posttranslational stabilization. In this study, we show that FLuc inhibition and stabilization is the result of an inhibitory product formed during the FLuc-catalyzed reaction between its natural substrate, ATP, and PTC124. A 2.0 Å cocrystal structure revealed the inhibitor to be the acyl-AMP mixed-anhydride adduct PTC124-AMP, which was subsequently synthesized and shown to be a high-affinity multisubstrate adduct inhibitor (MAI; KD = 120 pM) of FLuc. Biochemical assays, liquid chromatography/mass spectrometry, and near-attack conformer modeling demonstrate that formation of this novel MAI is absolutely dependent upon the precise positioning and reactivity of a key meta-carboxylate of PTC124 within the FLuc active site. We also demonstrate that the inhibitory activity of PTC124-AMP is relieved by free coenzyme A, a component present at high concentrations in luciferase detection reagents used for cell-based assays. This explains why PTC124 can appear to increase, instead of inhibit, FLuc activity in cell-based reporter gene assays. To our knowledge, this is an unusual example in which the “off-target” effect of a small molecule is mediated by an MAI mechanism.
Keywords: Ataluren, multisubstrate adduct inhibitor, protein stability, reporter gene assay, X-ray crystallography
Firefly luciferase from Photinus pyralis (FLuc) is an ATP-dependent luciferase widely used as a reporter enzyme for cell-based gene expression assays, principally due to the high sensitivity and large dynamic range bioluminescence affords (1, 2). However, as an enzyme that catalyzes the bimolecular reaction between two small molecule substrates, D-luciferin and ATP, it is prone to inhibition by a variety of low-molecular-weight heterocyclic compounds typically found in screening collections (3, 4). Expressed intracellularly as a reporter, the FLuc enzyme has a short protein half-life (t1/2 ∼ 3–4 h) relative to other reporters (5, 6). Although ideal for following dynamic responses in cell-based assays, reporter instability can be offset by ligand-induced stabilization. Stabilization of FLuc by compounds acting as inhibitors can lead to a relative increase in enzyme levels (compared to untreated basal levels) and correspondingly increased FLuc activity—a counterintuitive result for compounds characterized as FLuc inhibitors (5–8).
We have previously shown that PTC124, a compound with putative nonsense codon suppression activity (9) (Ataluren; 1, Table 1), and related 3,5-diaryl oxadiazoles, are FLuc inhibitors that stabilize the enzyme intracellularly, leading to a transcriptional- and translational-independent increase in the observed enzyme activity over basal levels (7, 8). Past efforts to profile the National Institutes of Health Molecular Libraries Small Molecule Repository for compounds that inhibit FLuc originally identified MLS000062114 (2; PubChem Chemical Identifier: 2876651, Table 1)—a molecule equivalent to the des-carboxylate analog of PTC124—as a FLuc inhibitor (3). Although 2 showed low micromolar potency in our profile study (3, 8), we observed that introduction of an m-carboxylate at R2 (i.e., PTC124; 1) resulted in a significantly more potent FLuc inhibitor (IC50 ∼ 10 nM) (8). Paradoxically, we also found that PTC124 caused an activation response in FLuc cell-based reporter gene assays, including those expressing FLuc from a wild-type luc+ gene, with typical compound incubation times (16–24 h) indicative of potent enzyme stabilization (8) by PTC124.
Table 1.
Potency and cellular activity of analogs in the enzymatic (FLuc) and cell-based (FLucUGA) assays
| R1 | R2 | FLuc IC50, μM | FLucUGA EC50, μM | FLucUGA % activation | ||
| o-F | m-COOH | (1) | 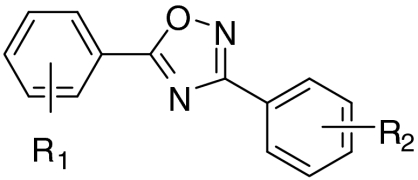 |
0.014 ± 0.008 | 0.11 ± 0.09 | 1345 ± 391 |
| o-F | H | (2) | 0.99 ± 0.59 | 11.9 ± 5.8 | 812 ± 268 | |
| o-F | m-CONH2 | (3) | 4.6 ± 1.9* | 4.3 ± 2.9 | 1019 ± 262 | |
| o-F | m-tetrazole | (4) | 2.9 ± 1.7 | 10.2 ± 5.2 | 618 ± 59 | |
| m-Cl | m-COOH | (5) | 0.023 ± 0.01 | 0.25 ± 0.23 | 1013 ± 319 | |
| o-F | m-CO(AMP) | (6) | 0.020 ± 0.01 | N/A | N/A | |
| o-F | p-COOH | (7) | 0.72 ± 0.45 | 3.1 ± 4.9 | 1288 ± 510 | |
| o-F | p-CONH2 | (8) | 4.3 ± 2.0* | 0.77 ± 0.05 | 1816 ± 982 | |
| o-F | p-tetrazole | (9) | 14.5 ± 5.0 | 16.5 ± 3.0 | 528 ± 189 | |
| o-F | o-COOH | (10) | 11.7 ± 3.2* | 23.0 ± 10.8 | 369 ± 209 | |
| o-F | o-CONH2 | (11) | 18.9 ± 7.6* | 8.2 ± 9.7 | 118 ± 92 | |
| o-F | m-CONH(sa-Ade) | (12) | 0.005 ± 0.001 | N/A | N/A | |
| o-F | p-CONH(sa-Ade) | (13) | 0.003 ± 0.001 | N/A | N/A |
N/A, not applicable; sa-Ade, acylsulfamide adenosine; error is SD from replicates performed on different days for enzyme (n = 3) and cell-based (n = 2) assays.
*Equals partial inhibition of FLuc observed, e.g., % inhibition (x) was 40% < x ≤ 80%. See SI Text for additional details.
Ligand-receptor affinity arises from alignment of shape and electrostatic features between receptor and ligand (10). On rare occasions, the unique arrangement of these elements can lead to enzymatically assisted formation of an even higher affinity multisubstrate adduct inhibitor (MAI). By combining components of both substrates into a single molecule, the resulting conjugate gains a large entropic advantage, and its affinity can often be estimated by the product of the substrate KDs (11). The ligation of two substrates with micromolar affinity can thus result in an MAI with potency in the subnanomolar range (12). In recent years, this principle has been used in the domain of fragment-based ligand design (13). Although the majority of MAIs described in the literature have been prepared synthetically (14), precedence exists for MAI generation by the target enzyme itself (15, 16). A FLuc-generated MAI—the mixed-anhydride dehydroluciferyl-AMP (L-AMP)—is naturally formed by the oxidation of luciferyl-AMP (LH2-AMP), an intermediate in the conversion of D-luciferin to oxyluciferin (17, 18) (Fig. 1A and Fig. S1A). The L-AMP MAI acts as a potent inhibitor of FLuc, with an IC50 = 6 nM (KD = 500 pM) (17). The enzyme-catalyzed production of an MAI from PTC124 and ATP could explain the exceptional affinity of PTC124 against FLuc in both enzyme and cell-based assays.
Fig. 1.

Chemical and cocrystal structures. (A) Comparison of the structure of the mixed anhydrides of AMP (Left) with PTC124, D- luciferin (LH2), dehydroluciferin (L), and DLSA. (B) Fo-Fc omit map of the PTC124-AMP mixed anhydride contoured at 3σ. (C) Hydrogen-bonded interactions between PTC124-AMP MAI (6, ball and stick) and luciferase residue (sticks) are indicated as dashed lines. (D) Cocrystal X-ray structure of FLuc and PTC124-AMP MAI.
With this study, we describe the mechanism of formation, high-affinity binding, and stabilization of FLuc by PTC124. The X-ray cocrystal structure of FLuc bound to PTC124 and the substrate ATP revealed the presence of an enzyme-bound PTC124-AMP mixed-anhydride (6) analogous to the L-AMP intermediate in the FLuc catalytic reaction. PTC124 binds to the luciferin pocket allowing reaction of the m-carboxylate of PTC124 with ATP yielding the mixed-anhydride PTC124-AMP—an MAI. Using a series of 3,5-diaryl oxadiazole analogs containing nonreactive carboxyl biosteres, we demonstrate that formation of the MAI critically depends on the aryl m-carboxylate moiety of PTC124. The MAI mechanism for PTC124 was not anticipated because classical MAIs are based on substrate analogs (19) and the 3,5-diaryl oxadiazole core is not a luciferin analog. This represents a unique example in which a synthetic compound mediates an off-target effect by an MAI mechanism. Further, and seemingly contradictory, is the ability of the apparent MAI-bound FLuc reporter activity to be fully measured in bioluminescent endpoint assays. This is possible because the MAI bound to FLuc is sensitive to FLuc-mediated thiolysis by free coenzyme A (CoASH), a component of most FLuc detection reagents. The findings presented here provide essential guidance for the use of luciferase assays in chemical biology and drug discovery efforts, while more specifically identifying the mechanism of action of PTC124 in FLuc cell-based assays.
Results
Crystal Structures.
The structure of an apo form of luciferase [Luc:apo; Protein Data Bank (PDB): 3IEP] was determined to 2.1-Å resolution by molecular replacement (see Methods, Table S1, and Fig. S2). Apo crystals were soaked in the presence of PTC124 (1, Table 1) and ATP and the resulting cocrystal structure (PDB: 3IES) electron density maps were examined for ligand binding. Surprisingly, clear density was observed for an adduct of AMP with PTC124 (PTC124-AMP; 6, Table 1 and Fig. 1A and B), apparently resulting from nucleophilic displacement of pyrophosphate from ATP by the m-carboxylate of PTC124 (Fig. S1B). The AMP portion of 6 forms hydrogen bonds with luciferase residues (Fig. 1C) and the diaryl oxadiazole group is positioned in a hydrophobic pocket near Ala 348 and Ile 351 and shows aromatic stacking with Phe 247 (Fig. 2A and B). Therefore, PTC124-AMP fully occupies the active site of FLuc (Fig. 1D). The PTC124-AMP structure was found to make critical H bonds with several invariant residues including Asp 422, which is within H-bonding distance to the 2′-OH of the ribose ring (Fig. 2A and B) (20).
Fig. 2.

Substrate binding pocket of FLuc with PTC124-AMP bound. (A, B) Two pocket orientations. (C) Structure of FLuc (Gray Ribbon) liganded with PTC124-AMP (Turquoise) overlaid on the structure of DLSA (Gold) bound to LcrLuc (PDB: 2D1S) showing the close similarity of ligand binding modes. Leu286 in FLuc corresponds to Ile288 in LcrLuc. These residues are located at the end of the respective ligand binding pockets, but whereas Ile288 moves upon ligand binding, Leu286 does not. (D) Overlay of the structures of apo-FLuc (Magenta) and FLuc liganded with the PTC124-AMP MAI adduct (6, Turquoise) showing movement of a loop between residues Ser314 and Leu319 upon binding of ligand (ball and stick).
The binding mode of this inhibitor is similar to that observed for ligand-bound Japanese firefly (Luciola cruciata) luciferase (LcrLuc) (21). Alignment of our FLuc cocrystal structure and the structure of LcrLuc cocrystallized with the L-AMP analog 5′-O-[N-(dehydroluciferyl)-sulfamoyl] adenosine (DLSA) (PDB: 2D1S) (21, 22) shows that PTC124-AMP binds with the 3,5-diaryl oxadiazole within the luciferin pocket with the m-carboxylate specifically located to form the mixed anhydride with AMP (Fig. 3A and B) in a manner that is isostructural with DLSA (Fig. 2C and Fig. S3A). The planar nature of the 3,5-diaryl oxadiazole provides for an orientation in the active site similar to the natural substrate D luciferin and positions the carboxylate to fulfill a rare case of enzyme inhibition wherein the enzyme catalyzes the formation of its own inhibitor. Remarkably, PTC124-AMP represents a structural mimicry of the luciferyl-AMP intermediate (LH2-AMP; Fig. 1A and Fig. S1A) that gives rise to luminescence via dioxetanone formation, and to formation of the naturally occurring potent inhibitor of the enzyme, L-AMP via a side reaction (18) (Fig. 1A and Fig. S1A).
Fig. 3.

NAC analysis of carboxylate PTC124 regioisomers. (A) Schematic of bond formation as the m-carboxylate of PTC124 displaces the pyrophosphate from ATP. R = 5-(2-fluorophenyl)-1,2,4-oxadiazole. (B) NAC modeling of PTC124/analogs within the FLuc active site. The m-carboxylate of PTC124 (1) provides an optimal NAC as the 3,5-diaryl oxadiazole is optimally positioned within the luciferin binding pocket. However, the p-carboxylate analog (7), must rotate by ∼20° within the constraints of a productive NAC model to be able to form a NAC and react with ATP. For the o-carboxylate analog (10) to maintain the NAC, a classic biaryl steric clash between the o-carboxylate and the adjacent oxadiazole occurs necessitating out of plane rotation which effectively pushes the analog out of the luciferin binding pocket, preventing MAI formation. (C, D) Graphs show formation of adenylate adducts by FLuc as analyzed by LC-MS. Total ion current (TIC) data for regioisomers of PTC124 mass (C), or the corresponding AMP adduct mass (D), following incubation of 20 μM PTC124 in the presence of 2 mM ATP for the regioisomers (indicated in D). Percentages represent the percent remaining of the parent ion peak, calculated from the ratio of the peak areas for the compound incubated with ATP in the presence and absence of FLuc.
The structure of FLuc:PTC124-AMP is quite similar overall to the apo structure with an overall rmsd of 0.60 Å between Cα atoms (Fig. S3B). The largest structural difference is present in the loop between Ser 314 and Leu 319, which moves to accommodate ligand binding (Fig. 2D). This loop shows well-defined electron density in the ligand-bound form and has a similar conformation to the homologous loop of LcrLuc bound to DLSA.
Solution Formation of the MAI by FLuc.
Liquid chromatography/mass spectrometry (LC-MS) was used to confirm the FLuc-dependent synthesis of PTC124-AMP in solution. Incubation of PTC124 and ATP alone did not yield any mass corresponding to PTC124-AMP, with only the parent compound peak of PTC124 observed (Fig. S4A). Addition of FLuc to the same sample resulted in loss of the parent compound peak and appearance of the PTC124-AMP mass (Fig. S4A). Additionally, the yield of PTC124-AMP obtained could be titrated by varying the FLuc concentration ( Fig. S4B). The lack of reaction in the absence of FLuc is expected due to the unfavorable energetics required for the negatively charged carboxylate group to come into van der Waals contact with the triphosphate moiety of ATP, as well as the necessary desolvation of the carboxylate water shell. However, in the active site of the enzyme, the correct “near-attack conformers” (NACs) of the reactants are brought within van der Waals distance allowing bond formation, presumably via SN2 displacement (Fig. 3A) (23).
Meta-Carboxylate Provides the Optimal NAC.
To further understand the reactivity of the m-carboxylate, we modeled the ortho-, meta-, and para-carboxylate regioisomers of PTC124 (i.e., compounds 10, 1, and 7, respectively) into the crystal structure (Fig. 3B). This showed that reaction with ATP requires the p-isomer (7) to rotate approximately 20° in-plane away from the position in which m-isomer (1) binds (Fig. 3B). This is somewhat unfavorable energetically, as indicated by the decreased potency of p-isomer (7) as an inhibitor (Table 1). However, formation of the MAI from o-PTC124 (10) would require complete rotation out of the luciferin binding pocket, suggesting that this NAC could not be achieved in the FLuc active site (Fig. 3B, 10, and Fig. S3C). Consistent with these models, using equivalent incubation times (∼10 min) and high FLuc concentrations (20 μM), we were able to detect the presence of p-PTC124-AMP at an intermediate level upon incubation of 7 with FLuc and ATP (Fig. 3C and D). However, when we incubated 10 with FLuc and ATP under the same conditions, we did not observe any conversion and no product corresponding to o-PTC124-AMP was observed (Fig. 3C and D).
Potency Depends on the Aryl m-Carboxylate.
To further investigate the effect of the m-carboxylate on potency, we synthesized several carboxylate R2 regioisomers and nonreactive analogs (Table 1) and determined their IC50s against purified FLuc using [S] ∼ KM (e.g., both ATP and D-luciferin were at concentrations near their KMs so that the observed IC50 approximated the true inhibitor affinity, KI). Varying the meta substituent to either an amide (3) or the tetrazole (4) carboxylate biostere resulted in > 100-fold loss in potency against the enzyme (Table 1). The des-carboxylate analog of PTC124 (2) demonstrated 80-fold lower potency, suggesting that meta substitution can add little toward the binding energy in the absence of adduct formation. Significantly, the potency of the synthetic PTC124-AMP (6) was approximately the same as that determined with PTC124 in the presence of ATP and FLuc, consistent with solution formation of adduct. Additionally, we measured the competition of synthetically prepared LH2-AMP from FLuc by PTC124-AMP and PTC124 (in the absence of ATP). From these data, we were able to estimate a KD = 120 ± 30 pM for PTC124-AMP and 1.4 ± 0.4 μM for PTC124 using the method described by Rhodes and McElroy (Fig. S5) (17). Therefore, the KD of PTC124 for FLuc improves by ∼10,000-fold upon acyl-phosphoryl anhydride MAI formation.
We previously noted a decrease in potency for p-carboxylate analogs (8), which is confirmed here. In agreement with this series being less competent to form the PTC124-AMP MAI, we observed smaller shifts in potency between p-carboxylate and nonreactive derivatives. Changing the p-carboxylate substituent to either an amide (8) or tetrazole (9) resulted in potency shifts of ∼10-fold (Table 1), significantly less than those observed between the m-carboxylate analogs (1, 3, and 4). Again in line with our previously determined pharmacological correlation between potency in the FLuc enzyme assay and the cell-based FLuc nonsense codon suppression assay (8), both PTC124 (1) and the m-chloro analog (5) demonstrated potent (< 100 nM) activity in either assay, while the p-carboxylate analog of PTC124 (7) demonstrated weak (μM) potency in these assays. As well, we found that ortho-carboxylate analogs (10 and 11) demonstrated the greatest reduction in potency in both assays (Table 1).
In an effort to further investigate the potency differences between the meta- and para-carboxylate analogs, we prepared stable analogs of the meta- and para-adducts (12 and 13; Table 1). These were prepared as acylsulfamide derivatives, so as to limit any possibility of hydrolysis influencing the potency measurement. We found that both stable adducts (12 and 13) showed potent inhibition similar to anhydride 6 (Table 1). Further, when we preincubated the p-carboxylate analog (7) or PTC124 with ATP and FLuc (10 nM) in the enzyme assay, we observed no time-dependent increase in inhibition by either compound (IC50 = 4 ± 3 nM without preincubation and 4 ± 0.6 nM with a 24 h preincubation for PTC124; for 7, IC50 = 1.1 ± 0.3 μM and 3.8 ± 0.3 μM for the respective conditions). Therefore, under these enzymological conditions (i.e., low FLuc concentrations), formation of p-PTC124-AMP is kinetically unfavorable. Thus, given the near equal inhibition potencies for the premade adduct analogs (6, 12, and 13) we conclude that the much weaker inhibition potency observed for 7 is due to a kinetic effect. This is further supported by the NAC models, with the potency governed by the rates of formation and breakdown of the adduct.
FLuc Thermal Stabilization Parallels Inhibitor Potency.
To further probe the interaction of FLuc with PTC124 and its analogs, we used a fluorescence-based thermal denaturation assay designed to assess the stabilization of proteins by small molecules (24). The melting curves for FLuc in the presence of varying concentrations of PTC124 showed a maximum ΔTm = 8 °C (at 200 μM concentration), whereas inclusion of 2 mM ATP in the buffer significantly enhanced this by 4.6 °C (Fig. S6). Overall, the ΔTm shifts of FLuc in the presence of compound correlated well with the potency of the compound against the enzyme. The most dramatic enhancement in FLuc stabilization occurred in the presence of either PTC124 or an analog in which the m-carboxylate was preserved (ΔTm = 12–13 °C in the presence of ATP; Fig. 4A; 1 and 5). This was followed closely by the p-carboxylate analog of PTC124 (7). On the other hand, the m-substituted analogs containing moieties incapable of adduct formation, such as the m-amide (3) or m-tetrazole (4) displayed much lower ΔTm enhancements in the presence of ATP. Not surprisingly, the lowest ΔTm values were obtained for the o-carboxylate (10) and o-amide (11) analogs. Significantly, we also found a very large enhancement in thermal stability of FLuc in the presence of synthetically prepared PTC124-AMP (6), which showed a ΔTm = 28 °C (Fig. 4B and Fig. S6C and D). The large ΔTm observed for synthetically prepared PTC124-AMP lends further support to the MAI stabilization hypothesis.
Fig. 4.

Correlation between stabilization and inhibitor potency. Compounds are identified in Table 1. (A) The stabilizing effect of PTC124 analogs, as demonstrated by ΔTm, on 3.6 μM Fluc in the presence of 2 mM ATP correlates with their IC50s for FLuc inhibition in vitro. The average maximum ΔTm is plotted and error bars represent the SD from two determinations. Compounds showing decreasing ΔTm also show lower inhibitor potency against the enzyme. (B) The ΔTm values for PTC124 (1), PTC 124 + 2 mM ATP, and the synthetically prepared PTC124-AMP adduct (6) are shown in either PBS (i) or Tris-acetate buffer (ii). The higher ΔTm value obtained for the synthetic adduct is likely due to the fact that this adduct is measured in the absence of ATP (2 mM). Adding an equivalent concentration of ATP to 6 yielded the same ΔTm value (12.6 °C) as was observed with PTC124 and ATP. (C) A correlation plot of the EC50 values in the cell-based FLuc nonsense codon suppression assay plotted against their IC50 values in the purified FLuc enzymatic assay for PTC124, a regioisomer, and related analogs (values from Table 1). Inhibition of purified FLuc in an enzymatic assay follows the same trend as apparent activation in a cell-based FLuc reporter gene assay. (D) Fold-shift in IC50 values for PTC124 analogs relative to PTC124 (1) in the FLuc enzyme assay (i) and the nonsense codon suppression cell-based assay (ii) indicate that the potency trends remain largely similar in both types of assay.
MAI Formation Explains FLuc Cell-Based Activity.
To examine if PTC124 and analogs increased luciferase enzyme activity in cells in a manner consistent with protein stabilization by MAI formation, compounds were tested in a cell-based assay similar to that used to discover PTC124 (referred to here as the FLucUGA assay) (9). This assay employs cells expressing FLuc from a cDNA with a nonsense mutation at codon 190 of the FLuc gene (pFLuc190UGA) (8). Although a nonsense mutation in the coding region is present in the FLuc mRNA produced in these cells, preventing efficient translation of full-length protein, there is still an extremely low, but detectable, basal level of FLuc activity due to sporadic translational readthrough of the nonsense mutation (25). When cells expressing pFLuc190UGA were treated with compound, we again found that PTC124 (1) showed the most potent activation, followed by the p- and o-carboxylate analogs (7 and 10), respectively, which follows the same trend established by thermal denaturation experiments and enzyme inhibition data (Fig. 4C and Fig. S7A). Efficacy in the cell-based assay was similar for both the m- and p-carboxylate analogs (1 and 7; FLucUGA % activation in Table 1; and Fig. S7A) suggesting that intracellulary, a stabilized enzyme-inhibitor complex is also formed with these compounds, although the concentration at which this is achieved varies. However, the o-carboxylate (10), which is not predicted to form the MAI, demonstrates significantly reduced activation in the cell-based assay (Table 1) with only partial concentration-response curves (CRCs) (Fig. S7A). Shifts in cell-based activation for regioisomers and analogs correlated well with enzymatic inhibitory potency (Fig. 4D), as an example, both the p-carboxylate (7) and the m-tetrazole (4) analogs displayed reduced potency in both assays (∼100-fold less potent). Further, as expected from the NAC analysis, the potency of the o-carboxylate (10) and o-amide (11) were relatively weak in both the enzyme and cell-based assays. The only inconsistency seen between the cell-based and biochemical assay was for the des-carboxylate analog (2), which, though still supporting the critical nature of the m-carboxylate, displayed weaker activity in the cell-based assay (EC50 ∼ 10 μM) than in the purified FLuc enzyme assay (IC50 ∼ 1 μM; Table 1). This might reflect poor cell-permeability of the compound due to serum binding or poor solubility in the cell media, ultimately resulting in decreased potency for this compound in the cell-based assay. Overall, however, potency in the cell-based assay followed the order of reactivity of the compound in forming the MAI, that is, the carboxylate reactivity of meta > para≫ortho, as predicted by the FLuc cocrystal structure and corresponding NAC models for the active site of FLuc.
CoASH Effects on PTC124 Potency.
CoASH has been shown to significantly reduce FLuc inhibition by the L-AMP MAI via an enzymatically catalyzed thiolytic reaction that converts L-AMP to L-CoA (see Fig. S1) (18). CoASH is also a common constituent of detection reagents used for firefly luciferase assays, because it increases the bioluminescent signal through relief of L-AMP inhibition and thus provides more efficient use of the substrate D-luciferin (18). As PTC124 forms a potent PTC124-AMP MAI, we sought to determine if addition of CoASH to the FLuc enzyme reaction would also relieve PTC124-AMP MAI inhibition, potentially via the same mechanism demonstrated for L-AMP—thiolytic conversion of PTC124-AMP to PTC124-CoA (Fig. S1). We found this to be the case with addition of 500 μM CoASH (a concentration likely equal to what is typically used in commercial detection reagents for cell-based assays) to purified FLuc enzyme assays reducing the potency of PTC124 (1) by approximately 35-fold, on average (Fig. S7B–E and Table S2). The potency of the p-carboxylate analog (7) was reduced to a lesser degree (∼10-fold), consistent with the reduced propensity for this analog to form the MAI. Consistent with thiolysis as the reason for reduced potency (versus a potential allosteric role for CoASH binding), we did not observe any shift in potency for the stable MAI analogs, 12 and 13, in the presence of CoASH (IC50 remaining at 3 ± 1 nM in either condition; Fig. S7D and E and Table S2). Of note, addition of a higher concentration of cysteine (1 mM) did not affect the potency of PTC124 against FLuc (Fig. S7B and C), indicating that the reaction with CoASH is specific, and cannot be generalized to simple reducing reagents or nucleophiles (18).
Discussion
This study demonstrates that the molecular basis for high-affinity binding of PTC124 to FLuc arises from the enzyme-catalyzed formation of an MAI through a key m-carboxylate group. The cocrystal structure is similar to that reported for LcrLuc bound to DLSA, a stable sulfamoyl analog of oxidized luciferin and AMP (21). The differential activity of 3,5-diaryl oxadiazole carboxylate regioisomers in FLuc cell-based assays can be explained by the crystal structure-supported NAC models. These models indicate that placement of the PTC124 carboxylate at the ortho or para position either prevents or impairs, respectively, alignment of the necessary functionalities needed for MAI formation. Experimentally, it was also found that FLuc-dependent formation of ortho- and para-carboxylate adducts was either nonexistent or proceeded with dramatically reduced efficiency. Further, the PTC124-AMP adduct formed either enzymatically, or by direct chemical synthesis, results in an inhibitor with nanomolar potency and high-affinity binding to FLuc (KD of ∼100 pM).
Enhancement of protein stability through ligand-induced conformational changes or modulation of folding pathway equilibria is well documented (26, 27) and can be reflected by increases in protein thermal stability. Thermal denaturation assays of FLuc in the presence of PTC124-AMP show a large ΔTm of nearly 30 °C, significantly greater than the sum of the ΔTm values for PTC124 and ATP at the same concentration, suggesting that the formation of the adduct is essential for the observed increase in protein stability. Analogs containing nonreactive acid isosteres at the meta position (3-5), and carboxylate-containing regioisomers at the ortho (10) or para (7) positions, also have significantly reduced thermal shifts, further indicating that FLuc protein stability is dependent on PTC124-AMP formation.
The apparent activity (or inactivity) of PTC124 in the cell-based assay depends on the assay protocol and the commercial detection reagent used (28, 29). As mammalian cells lack D-luciferin, intracellular binding of PTC124 to FLuc occurs without competing endogenous ligands. Cellular ATP saturates the FLuc nucleotide binding site, thus facilitating PTC124-AMP formation, high-affinity binding, and both inhibition and stabilization of the enzyme. Detection of the associated increase in relative FLuc activity in cell-based assays can be readily achieved by washout of the inhibitor prior to detection, and/or addition of appropriate detection reagents containing excess substrates and cofactors (8, 9, 28, 29).
Catalysis of the ATP-dependent oxidation of D-luciferin to oxyluciferin, AMP, CO2, and light involves an LH2-AMP intermediate that can be oxidized to form an L-AMP by-product, a potent FLuc inhibitor (17, 18) (Fig. S1). Commercially available FLuc detection reagents (e.g., BrightGlo, Promega) contain high levels of CoASH that relieve FLuc inhibition by L-AMP via thiolysis, thereby optimizing FLuc luminescence (30). We show that addition of high micromolar concentrations of CoASH to the FLuc enzymatic assay significantly decreases the potency of PTC124 and m-carboxylate analogs. Conceivably, CoASH in detection reagents reacts with PTC124-AMP to create a PTC124-CoA adduct. The resulting shift in potency parallels that observed for the conversion of L-AMP (6 nM) to L-CoA (5 μM) (18). The application of commercial detection mixes (which have relatively high concentrations of D-luciferin, ATP, and CoASH) will promote thiolysis and dissociation of the PTC124-AMP components from the enzyme, facilitating an interpretation of increased enzyme activity as apparent activation in the reporter gene assay.
It is not uncommon for a compound to demonstrate a higher potency when assayed against its isolated molecular target compared to that obtained for the target in a cellular context, as is seen for PTC124 (1), in which there is an ∼10-fold decrease in potency for the cell-based assay (Table 1, compare IC50 and EC50). One possible explanation for this could be due to competition by intracellular substrates or cofactors. Intracellular ATP, for example, could compete directly with PTC124-AMP association. Additionally, intracellular CoASH may thiolytically react with FLuc-bound PTC124-AMP to form the less potent PTC124-CoA, which is expected to stabilize the enzyme only at higher compound concentrations. However, it is likely that in an intracellular environment lacking D-luciferin, and in which PTC124 and saturating concentrations of ATP are present, enzymatic reformation of the high-affinity MAI is favored. Therefore, the reduced potency observed in the FLucUGA cell-based assay (∼nM) relative to the measured KD of the PTC124-AMP (∼pM) against FLuc, and the fact that the cell-based structure-activity relationships parallel the catalytic requirements of FLuc for MAI formation, support the notion that enzymatic rounds of PTC124-AMP hydrolysis/reformation may occur in cells, the extent of which is dependent on the cellular quantity and availability of CoASH.
The prevalence of compounds that cause the paradoxical inhibitor-based reporter stabilization in cell-based assays can be significant, reaching 20–60% of the actives selected from cell-based FLuc assays aimed at activation (7). The counterintuitive nature of this phenomenon defies post hoc conventional wisdom and requires counterscreens designed to recognize compound-mediated reporter stabilization. One such example is a FLuc reporter system with attenuated expression (e.g., SV40 promoter) (31). Our work highlights the importance of determining the potency of compounds active in cell-based FLuc assays against purified FLuc in the presence of defined concentrations of substrates ([S] ∼ Km) as opposed to using formulated detection reagents whose components can mask or alter inhibitory activity (7, 8, 28, 29). Finally, reporters with mechanistically distinct modes of bioluminescence such as RLuc can be used in orthogonal assays to confirm that compound activity is not biased by choice of reporter (8).
The original identification of PTC124 arose from a FLuc cell-based assay of nonsense codon suppression, suggesting an agent of nanomolar potency (9, 28). Divergence of the structure-activity relationships associated with biochemical inhibition of FLuc and the cellular activation phenotype in FLuc-based nonsense codon suppression assays (FLucUGA) would argue for dual mechanisms of action for PTC124, supporting the existence of a cellular target mediating readthrough of premature termination codons. However, the dependence on the m-carboxylate functional group of PTC124 in the cell-based FLucUGA assay was mirrored in the FLuc biochemical inhibition assay and thermal denaturation assay. The parallel nature of these responses is due to a rare and remarkable mechanism of enzyme inhibition—the formation of an MAI—and supports our previous observations that PTC124 activity is reporter dependent and nonsense codon independent, increasing the activity of both FLucUGA and wild-type FLuc, in cell-based assays while demonstrating inactivity in a corresponding RLucUGA assay (8). Taken together, these results question the biological plausibility for a molecular target other than FLuc as the reason for increased cellular luciferase activity.
Methods
Crystallization of FLuc, X-Ray Data Collection, and Processing.
All X-ray diffraction data were collected at the Advanced Photon Source, Industrial Macromolecular Crystallography Association Collaborative Access Team beamline 17BM using an Area Detector Systems Corporation Quantum 210 CCD detector. Details of the crystallization are provided in SI Methods and Table S1.
LC-MS.
Compound (20 μM) and ATP (2 mM) in PBS were premixed and further incubated with Fluc (20 μM or varied), or PBS as no-enzyme control, for ∼10 min at room temperature. Selective ion monitoring of the PTC124/analogs parent ion, the corresponding adduct, and the total ion current, was achieved on an Agilent 6130 Quadrupole MS detector. Additional information is in SI Methods.
Molecular Modeling.
Fast Rigid Exhaustive Docking (FRED) (OpenEye Scientific Software) was performed on the para- and ortho-PTC124-AMP adducts using an X-ray crystal structure of luciferase with PTC124-AMP bound. Full details are given in SI Methods.
Thermal Shift Experiments.
Using conditions described in SI Methods, data were recorded on a iQ5 Real Time PCR Detection system (Bio-Rad) using a temperature range from 20 to 95 °C in increments of 1 °C and a ramping rate of 0.1 °C/s. For more detail see SI Methods.
FLuc enzymatic assays to determine compound potency.
Compound potency was determined using a defined buffer with 10 μM of each substrate (ATP and D-luciferin) as previously described (8) using 10 nM P. pyralis luciferase (EC 1.13.12.7). Coenzyme A (C3019; Sigma) and DL-cysteine (861677; Sigma) were added to the substrate reagent for specified experiments for final concentrations of 500 μM and 1 mM, respectively. Luminescence was detected using ViewLux (PerkinElmer). For more details, see SI Methods.
Cell-Based FLuc Nonsense Codon Suppression Assays.
Grip-Tite 293 cells (Invitrogen) were transfected according to standard procedure (SI Methods) with a construct containing the reporter FLuc with a nonsense mutation at codon 190 (construct pFLuc190UGA; prepared by GenScript). In 1,536-well format, cells were seeded at 4,000 cells/well and compound was added 1 h after seeding. After 48 h, cells were washed three times with 1x PBS before detection mix was added to lyse cells and quantitate FLuc activity. Additional detail is provided in SI Methods.
Chemical Synthesis Procedures.
PTC124 (1) and analogs were prepared by methods similar to those previously described (8), and synthetic schemes and supporting analytical data are provided in SI Methods.
Supplementary Material
Acknowledgments.
This research was supported by the Molecular Libraries Initiative of the National Institutes of Health (NIH) Roadmap for Medical Research, and an NIH Center of Biomedical Research Excellence Award (5P20 RR17708) to the University of Kansas (KU). We thank Nadya Galeva from the KU Mass Spectrometry Laboratory for her assistance with the Matrix Assisted Laser Desorption-Ionization-Mass Spectrometry analysis, and Bill Leister of the NIH Chemical Genomics Center for analytical chemistry support.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. B.R. is a guest editor invited by the Editorial Board.
Data deposition: The structures have been deposited in the Protein Data Bank, www.pdb.org (Luc:PTC124-AMP, PDB: 3IES; Luc:Apo, PDB: 3IEP; Luc:Apo2, PDB: 3IER).
This article contains supporting information online at www.pnas.org/cgi/content/full/0909141107/DCSupplemental.
References
- 1.Fan F, Wood KV. Bioluminescent assays for high-throughput screening. Assay Drug Dev Technol. 2007;5(1):127–136. doi: 10.1089/adt.2006.053. [DOI] [PubMed] [Google Scholar]
- 2.Roda A, Guardigli M, Michelini E, Mirasoli M. Bioluminescence in analytical chemistry and in vivo imaging. TRAC-Trend Anal Chem. 2009;28(3):307–322. [Google Scholar]
- 3.Auld DS, et al. Characterization of chemical libraries for luciferase inhibitory activity. J Med Chem. 2008;51(8):2372–2386. doi: 10.1021/jm701302v. [DOI] [PubMed] [Google Scholar]
- 4.Heitman LH, et al. False positives in a reporter gene assay: Identification and synthesis of substituted N-pyridin-2-ylbenzamides as competitive inhibitors of firefly luciferase. J Med Chem. 2008;51(15):4724–4729. doi: 10.1021/jm8004509. [DOI] [PubMed] [Google Scholar]
- 5.Thompson JF, et al. Mutation of a protease-sensitive region in firefly luciferase alters light emission properties. J Biol Chem. 1997;272(30):18766–18771. doi: 10.1074/jbc.272.30.18766. [DOI] [PubMed] [Google Scholar]
- 6.Thompson JF, Hayes LS, Lloyd DB. Modulation of firefly luciferase stability and impact on studies of gene regulation. Gene. 1991;103(2):171–177. doi: 10.1016/0378-1119(91)90270-l. [DOI] [PubMed] [Google Scholar]
- 7.Auld DS, Thorne N, Nguyen DT, Inglese J. A specific mechanism for nonspecific activation in reporter-gene assays. ACS Chem Biol. 2008;3(8):463–470. doi: 10.1021/cb8000793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Auld DS, Thorne N, Maguire WF, Inglese J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc Natl Acad Sci USA. 2009;106(9):3585–3590. doi: 10.1073/pnas.0813345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welch EM, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447(7140):87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 10.Fersht AR, et al. Hydrogen bonding and biological specificity analysed by protein engineering. Nature. 1985;314(6008):235–238. doi: 10.1038/314235a0. [DOI] [PubMed] [Google Scholar]
- 11.Jencks WP. Binding energy, specificity, and enzymic catalysis: The Circe effect. Adv Enzymol Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 12.Inglese J, Blatchly RA, Benkovic SJ. A multisubstrate adduct inhibitor of a purine biosynthetic enzyme with a picomolar dissociation constant. J Med Chem. 1989;32(5):937–940. doi: 10.1021/jm00125a002. [DOI] [PubMed] [Google Scholar]
- 13.Congreve M, Chessari G, Tisi D, Woodhead AJ. Recent developments in fragment-based drug discovery. J Med Chem. 2008;51(13):3661–3680. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- 14.Korolev S, et al. The crystal structure of spermidine synthase with a multisubstrate adduct inhibitor. Nat Struct Biol. 2002;9(1):27–31. doi: 10.1038/nsb737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inglese J, Benkovic SJ. Multisubstrate adduct inhibitors of glycinamide ribonucleotide transformylase: Synthetic and enzyme-assembled. Tetrahedron. 1991;47(14–15):2351–2364. [Google Scholar]
- 16.Greasley SE, et al. Unexpected formation of an epoxide-derived multisubstrate adduct inhibitor on the active site of GAR transformylase. Biochemistry. 2001;40(45):13538–13547. doi: 10.1021/bi011482+. [DOI] [PubMed] [Google Scholar]
- 17.Rhodes WC, McElroy WD. The synthesis and function of luciferyl-adenylate and oxyluciferyl-adenylate. J Biol Chem. 1958;233(6):1528–1537. [PubMed] [Google Scholar]
- 18.Fraga H, Fernandes D, Fontes R, Esteves da Silva JC. Coenzyme A affects firefly luciferase luminescence because it acts as a substrate and not as an allosteric effector. FEBS J. 2005;272(20):5206–5216. doi: 10.1111/j.1742-4658.2005.04895.x. [DOI] [PubMed] [Google Scholar]
- 19.Broom AD. Rational design of enzyme inhibitors: Multisubstrate analogue inhibitors. J Med Chem. 1989;32(1):2–7. doi: 10.1021/jm00121a001. [DOI] [PubMed] [Google Scholar]
- 20.Conti E, Franks NP, Brick P. Crystal structure of firefly luciferase throws light on a superfamily of adenylate-forming enzymes. Structure. 1996;4(3):287–298. doi: 10.1016/s0969-2126(96)00033-0. [DOI] [PubMed] [Google Scholar]
- 21.Nakatsu T, et al. Structural basis for the spectral difference in luciferase bioluminescence. Nature. 2006;440(7082):372–376. doi: 10.1038/nature04542. [DOI] [PubMed] [Google Scholar]
- 22.Branchini BR, Murtiashaw MH, Carmody JN, Mygatt EE, Southworth TL. Synthesis of an N-acyl sulfamate analog of luciferyl-AMP: A stable and potent inhibitor of firefly luciferase. Bioorg Med Chem Lett. 2005;15(17):3860–3864. doi: 10.1016/j.bmcl.2005.05.115. [DOI] [PubMed] [Google Scholar]
-
23.Hur S, Kahn K, Bruice TC. Comparison of formation of reactive conformers for the SN2 displacements by
 in water and by
in water and by  in a haloalkane dehalogenase. Proc Natl Acad Sci USA. 2003;100(5):2215–2219. doi: 10.1073/pnas.242721799. [DOI] [PMC free article] [PubMed] [Google Scholar]
in a haloalkane dehalogenase. Proc Natl Acad Sci USA. 2003;100(5):2215–2219. doi: 10.1073/pnas.242721799. [DOI] [PMC free article] [PubMed] [Google Scholar] - 24.Ericsson UB, Hallberg BM, Detitta GT, Dekker N, Nordlund P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem. 2006;357(2):289–298. doi: 10.1016/j.ab.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 25.Parker J. Errors and alternatives in reading the universal genetic code. Microbiol Rev. 1989;53(3):273–298. doi: 10.1128/mr.53.3.273-298.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pace CN, Shirley BA, Thomson JA. Measuring Conformational Stability of a Protein in Protein Structure. New York: IRL; 1989. [Google Scholar]
- 27.Thompson PA, et al. Identification of ligand binding by protein stabilization: Comparison of ATLAS with biophysical and enzymatic methods. Assay Drug Dev Techn. 2008;6(1):69–81. doi: 10.1089/adt.2007.100. [DOI] [PubMed] [Google Scholar]
- 28.Peltz SW, et al. Nonsense suppression activity of PTC124 (Ataluren) Proc Natl Acad Sci USA. 2009;106(25):E64. doi: 10.1073/pnas.0901936106. lett. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inglese J, Thorne N, Auld DS. Reply to Peltz et al: Post-translational stabilization of the firefly luciferase reporter by PTC124 (Ataluren) Proc Natl Acad Sci USA. 2009;106:E65. lett. [Google Scholar]
- 30.Wood KV. The chemical mechanism and evolutionary development of beetle bioluminescence. Photochem Photobiol. 1995;62(4):662–673. [Google Scholar]
- 31.Lyssiotis CA, et al. Reprogramming of murine fibroblasts to induced pluripotent stem cells with chemical complementation of Klf4. Proc Natl Acad Sci USA. 2009;106(22):8912–8917. doi: 10.1073/pnas.0903860106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.