

Abstract
A reversible structural unlocking reaction, in which the close-packed van der Waals interactions break cooperatively, has been found for the villin headpiece subdomain (HP35) using triplet-triplet-energy transfer to monitor conformational fluctuations from equilibrium. Unlocking is associated with an unfavorable enthalpy change (ΔH0 = 35 ± 4 kJ/mol) which is nearly compensated in free energy by the entropy change (ΔS0 = 112 ± 20 J·mol-1·K-1). The unlocking reaction has a time constant of about 1 μs at 5 °C and is enthalpy-limited with an activation energy of 32 ± 1 kJ/mol and a large Arrhenius preexponential factor of A = 7.5 × 1011 s-1. In the unlocked state a fast local conformational fluctuation with a time constant of 170 ns and a low activation barrier of 17 ± 1 kJ/mol leads to unfolding of the C-terminal helix and to its undocking from the core. On a much slower time scale, global unfolding occurs from the unlocked state. These results suggest that native protein structures are locked into conformations with low amplitude motions. Large scale motions and global unfolding require an initial structural unlocking step leading to a state with properties of a dry molten globule. The experiments additionally yielded information on the dynamics of loop formation between different positions in unfolded HP35. Comparison of the results with dynamics in unstructured model peptides indicates slightly decelerated kinetics of local loop formation in the C-terminal region which points at residual, nonrandom structure. Dynamics of long-range loop formation, in contrast, are not influenced by residual structure, which argues against unfolded state properties as molecular origin for ultrafast folding of HP35.
Keywords: dry molten globule, native-state fluctuations, protein dynamics, protein folding, unfolded state dynamics
Native states of proteins are known to be close-packed but almost nothing is known about at which stage of the folding process packing occurs. Shaknovich and Finkelstein proposed in 1989 (1) that heat-induced protein unfolding begins with a slight expansion of the protein, such that at least some of the close-packing interactions are broken but water cannot yet penetrate into the interior. This dry molten globule state was postulated to be located on the unfolded side of the major folding barrier but it was argued that it should not be experimentally detectable for water soluble proteins (1). Later, GdmCl-induced unfolding experiments on ribonuclease A (2) and dihydrofolate reductase (DHFR) (3) monitored by time resolved NMR spectroscopy observed rapid formation of an intermediate located on the native side of the major unfolding barrier with properties resembling those of the predicted dry molten globule. The observation of an intact hydrogen-bonding network in the unfolding intermediate of ribonuclease A supported the dry nature of this state (4). Recently, FRET-detected unfolding kinetics of monellin showed rapid expansion of the native state prior to global unfolding (5), which is also in line with the transient formation of a dry molten globule.
We tested for the presence of intermediates on the native side of the major folding/unfolding barrier in villin headpiece subdomain (HP35) by monitoring conformational fluctuations in the native state using triplet-triplet-energy transfer (TTET). Irreversible TTET coupled to a conformational equilibrium is able to monitor equilibrium fluctuations on the time scale of ns to μs and was recently applied to analyze fast equilibrium fluctuations in α-helical peptides (6). TTET between the triplet donor xanthone (Xan) and the triplet acceptor naphthylalanine (Nal) is a fast (on the 2 ps timescale) and diffusion-controlled process that requires van der Waals contact between the reporter groups (7–11). When triplet labels are attached to a protein at positions that are not in contact in the native state (N), TTET can only occur after at least partial unfolding leads to a state (I) that is flexible enough in the region of the triplet probes to allow contact between donor and acceptor (Scheme [1]).  1
1
Contact between the labels in I leads to TTET (I*), which can be easily monitored by changes in the triplet absorbance bands (7, 8) of both Xan (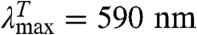 ) and Nal (
) and Nal (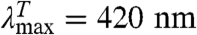 ). This coupling of an irreversible test reaction to a conformational equilibrium is analogous to hydrogen-deuterium (H/D) exchange (12, 13) and allows the determination of rate constants for local or global structural opening (kop) and closing (kcl) without applying an external perturbation to the conformational transition (6). Different kinetic regimes can be discriminated for this mechanism, depending on the relative values of kop, kcl, and kc. A detailed discussion of the different scenarios is given in SI Text. In general, analysis of the observed TTET kinetics allows the determination of all microscopic rate constants for a conformational equilibrium with dynamics on a similar timescale or slower than TTET (6). Since TTET through loop formation occurs on the 10 to 100s of ns timescale (7, 8, 14, 15), it allows us to probe dynamics of transitions that are 106 to 108 times faster than those accessible to H/D exchange. TTET is thus perfectly suited to test for fast equilibrium fluctuations on the native side of the major folding barrier indicative for the loss of close packing.
). This coupling of an irreversible test reaction to a conformational equilibrium is analogous to hydrogen-deuterium (H/D) exchange (12, 13) and allows the determination of rate constants for local or global structural opening (kop) and closing (kcl) without applying an external perturbation to the conformational transition (6). Different kinetic regimes can be discriminated for this mechanism, depending on the relative values of kop, kcl, and kc. A detailed discussion of the different scenarios is given in SI Text. In general, analysis of the observed TTET kinetics allows the determination of all microscopic rate constants for a conformational equilibrium with dynamics on a similar timescale or slower than TTET (6). Since TTET through loop formation occurs on the 10 to 100s of ns timescale (7, 8, 14, 15), it allows us to probe dynamics of transitions that are 106 to 108 times faster than those accessible to H/D exchange. TTET is thus perfectly suited to test for fast equilibrium fluctuations on the native side of the major folding barrier indicative for the loss of close packing.
The three helix bundle protein HP35 was chosen as a model system to test for native-state fluctuations since it is one of the smallest known protein units that folds cooperatively (16) and its structure and folding behavior have been extensively studied both experimentally and by simulations. High-resolution NMR (17) and X-ray (18–20) structures show a defined fold with a close-packed hydrophobic core. T-jump experiments (18, 19, 21–23) and NMR line-shape analysis (24) revealed that folding occurs on the timescale of μs. T-jump experiments showed the transient population of an unfolding intermediate (19, 21, 22). Molecular dynamics (MD) simulations have addressed the properties of different states of HP35 (25–28), and complete folding to the native state was achieved using replica exchange MD (27). To investigate the dynamics of different regions in the native state of HP35, we measured TTET kinetics for several HP35 variants with the triplet donor Xan and the acceptor Nal placed at different positions in the protein. The experiments additionally yielded the dynamics of loop formation in the unfolded state, which allowed us to test whether experimentally observed residual structure in the unfolded state of HP35 (29, 30) influences unfolded state dynamics and accelerates conformational search during folding. Thus, a single set of TTET experiments gives information on dynamics on either side of the major folding/unfolding barrier in HP35.
Results
Introduction of Triplet Labels into HP35.
Four variants of HP35 with donor-acceptor pairs at the positions Xan0/Nal23, Xan0/Nal35, Xan7/Nal23, and Nal23/Xan35 were chemically synthesized to probe dynamics in different regions of the protein. All variants have the labels solvent-exposed at the protein surface (Fig. 1). In addition, donor-only reference variants for all Xan positions were synthesized to determine the intrinsic donor triplet lifetimes in the absence of an acceptor. In all variants Met12 was replaced by norleucine (Nle) to avoid quenching of the Xan triplet state (14). In three of the four variants Trp23, which would also interfere with TTET, was replaced by the triplet acceptor Nal, which is a commonly used Trp analogue. In the Xan0/Nal35 variant, Trp23 was replaced by Phe and Phe35 was replaced by Nal. Triplet lifetimes in all donor-only reference peptides were around 10 μs independent of the donor position and of the GdmCl concentration indicating the absence of intramolecular triplet quenching processes (Fig. 2 A, D, and G). This lifetime sets the upper limit for the timescale of fluctuations that can be observed by TTET.
Fig. 1.

Positions of the donor (Xan; blue) and acceptor (Nal; red) groups for TTET in the different HP35 variants. The models are based on the X-ray structure (18) (PDB 1YRF) and were prepared using MolMol (44).
Fig. 2.

GdmCl-dependence of TTET in the indicated HP35 variants at 5.0 °C. (A, D, and G) Xan triplet decay curves measured by the absorbance change at 590 nm. For comparison, the intrinsic xanthone triplet lifetimes in the native state measured in the respective donor-only variants are shown in gray. Solid lines represent fits with the rate constants shown in B, E, and H and their normalized amplitudes shown in C, F, and I (see also Table S1). The fractions of unfolded molecules determined by CD-detected equilibrium unfolding transitions are shown for comparison in C, F, and I (open circle). The Xan0/Nal35 variant exhibits an additional fast unresolved phase (D). The amplitude of this reaction is included in F (solid blue circle).
Structure and Stability of the HP35 Variants.
All labeled HP35 variants show α-helical far-UV CD spectra and one-dimensional NMR spectra comparable to those of the wild-type protein (31) indicating a folded structure (Figs. S1 and S2). GdmCl-induced equilibrium unfolding transitions at 5.0 °C monitored by the change in ellipticity at 222 nm show cooperative transitions for all variants (Fig. 2 C, F, and I). Table S1 summarizes the stability data obtained from fitting the transitions with the two-state model. The ΔG0-values range from -6 to -9 kJ/mol, indicating a 3–6 kJ/mol reduction in stability compared to wild-type HP35 [ΔG0 ≈ -12 kJ/mol; (20)]. A similar decrease in stability was observed for a Met12Leu variant (32) and for the variants used in T-jump experiments (23), which facilitates the comparison of results obtained by TTET and T-jump. This comparison suggests that the Met12Nle replacement is the major source for decreased protein stability. A hydrophobic contact between Trp23 and Pro21 was shown to contribute to HP35 stability. The Trp to Nal substitution at position 23 should not have major destabilizing effects on this interaction since Nal has similar properties as Trp. The meq-values (meq = ∂ΔG0/∂[GdmCl] for HP35 unfolding are around 3 (kJ/mol)/M, which is identical to reported values for the wild-type protein (16) and in agreement with the expected value based on the change in solvent accessible surface area (SASA) (33), which demonstrates that the compactness of the native state of HP35 is not affected by the labels.
Fluctuations in the N-Terminal Part of HP35.
In the Xan0/Nal23 variant donor and acceptor are separated by helix 1 and helix 2 which places them far apart from each other in the native state (Fig. 1). TTET between these positions can only occur after large-scale unfolding in the N-terminal region including disruption of the hydrophobic core. Fig. 2A shows TTET kinetics for the Xan0/Nal23 variant at various GdmCl concentrations measured by the decrease in the Xan triplet absorbance at 590 nm. All triplet decay curves can be described by the sum of two exponentials with time constants of 8 μs (84% amplitude) and 630 ns (16% amplitude) at 1 M GdmCl (Fig. 2B). The relative amplitudes of the fast and slow phases correspond to the fraction of unfolded and folded molecules, respectively, measured in the CD-detected equilibrium transition (Fig. 2C) which demonstrates that the slow triplet decay occurs in native molecules and the fast triplet decay represents loop formation in unfolded molecules. The time constant of the slow reaction is virtually identical to the Xan lifetime in the donor-only variant (Fig. 2A) indicating that no TTET occurs in N, which is confirmed by the absence of an increase in Nal triplet absorbance at 420 nm (Fig. S3). This result demonstrates the absence of large-scale unfolding reactions in the N-terminal part of HP35 on the ns to μs timescale and suggests that disruption of the interface involving helices 1 and 2 only occurs during global unfolding. Since no TTET is observed in N, global unfolding is slower than spontaneous Xan triplet decay, in agreement with results from T-jump experiments which yielded time constants for global unfolding of 400 μs in the absence of GdmCl and of 30 μs in 4 M GdmCl at 20 °C (21, 34).
The fast triplet decay observed in the Xan0/Nal23 variant represents TTET by loop formation in the unfolded state (kc), which is confirmed by a concomitant increase in the Nal triplet absorbance at 420 nm (Fig. S3). ln kc depends linearly on the denaturant concentration [D] (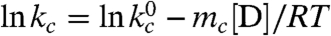 ) with similar mc-values (Fig. 2B) as in unstructured model peptides (8, 35). Extrapolating kc to zero GdmCl gives
) with similar mc-values (Fig. 2B) as in unstructured model peptides (8, 35). Extrapolating kc to zero GdmCl gives 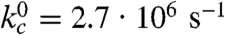 (τ = 370 ns; Table S1).
(τ = 370 ns; Table S1).
A small fraction (∼10%) of the slow triplet decay is still observed at high denaturant concentrations when HP35 is > 99% unfolded. This reaction may result from molecules with photo-inactive acceptor groups or from molecules which cannot form contact e.g. due to formation of small oligomers. This phenomenon is observed in all HP35 variants and has also been reported for model peptides (8).
Fluctuations in the Central Region of HP35.
Moving Xan to the side chain of Lys7 (Xan7/Nal23) gives information on large-scale dynamics around helix 2. This variant shows similar TTET kinetics as the Xan0/Nal23 protein with the amplitudes of a slow and a fast phase reflecting the populations of native and unfolded molecules, respectively (Fig. S4). The triplet decay in the native state corresponds to the Xan lifetime, indicating the absence of opening reactions around helix 2 on the ns to μs timescale. TTET in the unfolded state has a rate constant, 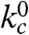 , of 4·106 s-1 (Table S1) which is faster than for the Xan0/Nal23 variant in accordance with a shorter loop between the labels (8, 15).
, of 4·106 s-1 (Table S1) which is faster than for the Xan0/Nal23 variant in accordance with a shorter loop between the labels (8, 15).
Dynamics Between the N- and C-Terminal Regions.
N- and C-terminus are in close vicinity in native HP35 (Fig. 1). TTET in the unfolded state of the Xan0/Nal35 variant thus yields the maximally possible rate constant for formation of this native interaction, whereas TTET in the native state is expected to be rapid. In the absence of GdmCl, TTET in the Xan0/Nal35 variant shows a fast unresolved amplitude change of about 60% within the 12 ns dead time of the experiment (Fig. 2D) indicating a large fraction of very rapid triplet transfer with k > 3·108 s-1. In the remaining molecules, the Xan triplet decay is double exponential (Fig. 2E) with rate constants of 3·107 s-1 (30% amplitude) and 3·106 s-1 (10% amplitude). Both processes become slower with increasing GdmCl concentration. The amplitude of the slowest reaction increases with increasing GdmCl concentration and corresponds to the fraction of unfolded molecules measured by CD, indicating that it monitors loop formation in U, which occurs with a time constant of 340 ns in the absence of GdmCl. The amplitude of the faster resolved process decreases with increasing GdmCl concentration indicating that this reaction represents a native state fluctuation. This observation points at conformational heterogeneity in the terminal regions of native HP35. The major native conformation (70% of the native molecules) has the ends in close contact leading to the observed unresolved very fast triplet transfer after excitation. In the remaining native molecules the ends meet with a time constant around 30 ns. This reaction becomes slightly slower with increasing GdmCl concentration [m = 0.7 ± 0.1 (kJ/mol)/M].
Fluctuations in the C-Terminal Region of HP35.
The Nal23/Xan35 variant gives information on local dynamics in the C-terminal region consisting of helix 3 and the conserved terminal amino acids Gly-Leu-Phe (GLF-motif) at residues 33–35 (Fig. 1). NMR and H/D exchange data had indicated that the GLF-region is flexible in the folded state of HP35, whereas helix 3 is well-defined (16, 20, 36). According to the X-ray structures, a stable C-terminal helix should prevent contact formation between positions 23 and 35 (Fig. 1). TTET kinetics in this variant are complex (Fig. 2 H and I). Two kinetic phases with time constants of 170 ns (20% amplitude) and 1 μs (80% amplitude; Fig. 2 G, H) are observed at zero denaturant. Under these conditions the population of the native state is about 96% (Fig. 2I) indicating that the two TTET processes occur in folded molecules. Both reactions are faster than the Xan triplet lifetime, pointing at native-state dynamics as the origin of both processes. The rate constant of the slower process decreases slightly with increasing GdmCl concentration indicating that the rate-limiting step of this reaction is associated with only small changes in SASA. The faster reaction becomes strongly accelerated with increasing GdmCl concentrations with a large m-value of -1.8 ± 0.2 (kJ/mol)/M, which shows that this process represents a partial unfolding reaction with a rate-limiting step that exposes about 50% of the SASA compared to the change between N and U (Table S1). Above 2.5 M GdmCl this reaction becomes independent of the GdmCl concentration with a time constant of 30 ns indicating a change in the rate-limiting step. Loop formation in a partially folded state may become rate-limiting at high denaturant concentrations. Alternatively, the changing slope may indicate a change from EX2 to EX1 mechanism for TTET at high GdmCl concentrations, which would indicate a time constant of 30 ns for the unfolding reaction (see SI Text).
Additional information on the nature of the two native-state TTET processes in the Nal23/Xan35 variant was obtained from the temperature dependence of TTET kinetics (Fig. 3A) which yields activation energies (Ea) of 17 ± 1 kJ/mol and 32 ± 1 kJ/mol for the faster and slower process, respectively (Fig. 3A), with respective preexponential factors (A) of 1.3·1010 s-1 and 7.5·1011 s-1. The equilibrium is shifted towards the minor conformation with increasing temperature with ΔH0 = 35 ± 4 kJ/mol and ΔS0 = 112 ± 20 J·mol-1·K-1 (Fig. 3B). A similar shift in equilibrium is observed with increasing denaturant concentration (Fig. 2I).
Fig. 3.

(A) Teperature dependence of the fast (open circles) and the slow (solid circles) native-state TTET process in the Nal23/Xan35 variant at zero denaturant. A fit to the Arrhenius equation (see SI Text) gives A = 1.3·1010 s-1 and Ea = 17 ± 1 kJ/mol for the faster reaction and A = 7.5·1011 s-1 and Ea = 32 ± 1 kJ/mol are the slower process. (B) Van’t Hoff plot for the equilibrium between N and N′. The equilibrium constant (K = [N′]/[N]) was determined by the ratio of the amplitudes of the fast and the slow native-state TTET process. A fit to the van’t Hoff equation (see SI Text) gives ΔH0 = 35 ± 4 kJ/mol and ΔS0 = 112 ± 20 J·mol-1·K-1.
At higher denaturant concentrations a third kinetic phase is observed with an intermediate time constant of 300 ns extrapolated to zero denaturant. The amplitude of this reaction corresponds to the fraction of unfolded molecules indicating that it reflects TTET in the unfolded state (Fig. 2I).
Discussion
Native-state Heterogeneity in HP35.
Native-state TTET in the Xan0/Nal35 and Nal23/Xan35 variants reveals two alternative native conformations with different dynamic properties. Based on the X-ray and NMR structures the Xan0/Nal35 variant should have the labels close together in the folded state, which is observed for about 70% of native molecules that undergo TTET between the ends in a subnanosecond process. In the alternative, minor conformation N- and C-terminus are further apart and TTET occurs with a time constant of about 30 ns. This reaction becomes slower with increasing GdmCl concentrations indicative of a partial folding reaction. An equilibrium between two native conformations is also observed in native-state TTET of the Nal23/Xan35 variant which occurs in two processes with time constants of 1 μs (80% amplitude) and 170 ns (20% amplitude). The similar amplitudes of the two native-state TTET processes in the Xan0/Nal35 and Nal23/Xan35 variants suggest that they monitor the same conformational heterogeneity, which is supported by a similar increase in the fraction of the minor conformation with increasing GdmCl concentration in the two variants (Fig. 2F, I). Comparison of the results from TTET experiments in the two variants reveals the structural and dynamic properties of the two native conformations. In the major conformation, C- and N-terminus are in close vicinity and only slow contact formation between the ends of helix 3 is observed without major unfolding in the rate-limiting step as indicated by the low m-value of this reaction. However, the large activation energy for TTET between Xan35 and Nal23 in this conformation shows that large-scale fluctuations from this state encounter major enthalpic barriers indicating breaking or weakening of interactions. In the minor conformation, C- and N-terminus are not in direct contact and positions 23 and 35 can meet fast without encountering large barriers although major structural opening occurs in the rate-limiting step of this process leading to exposure of about 50% of the SASA relative to the complete unfolding reaction. Undocking of helix 3 from helix 1 and helix 2 would lead to a relative change in SASA of only 25%, whereas undocking and unfolding of helix 3 would expose the experimentally observed 50% change in SASA. Native-state fluctuations in proteins including unfolding of complete α-helices have also been observed in H/D exchange experiments (37, 38). Since these experiments were carried out in the EX2 regime (see SI Text), no information on the dynamics of this process could be obtained.
Structural Unlocking.
The different properties of the conformational fluctuations in the alternative native states of HP35 lead to the minimal kinetic model shown in Fig. 4. The two native states, N and N′, are in equilibrium and have all three helices folded but show different conformations in the C-terminal region. Large-scale fluctuations cannot occur in the major conformation, N, whereas in N′ undocking and unfolding of helix 3 is fast (1/kop = 170 ns) leading to an intermediate state (I). TTET from N first requires formation of N′ with a time constant of 900 ns (1/k-l) at 5 °C. This unlocking of the structure upon formation of N′ is the rate-limiting step for TTET from N, which explains the low m-value for the slow native-state TTET process (Fig. 2H), although major unfolding in the C-terminal region has to occur to allow the labels to meet. Since N and N′ are populated to similar amounts, the locking and unlocking reactions have similar microscopic time constants around 900 ns. The increasing population of N′ with increasing GdmCl concentration shows that 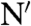 is slightly more solvent-exposed than N indicating a slight protein expansion. The observed ΔH0 of 35 kJ/mol and ΔS0 of 112 J·mol-1·K-1 for the equilibrium between N and N′ (Fig. 3B) indicate weakened interactions and increased structural flexibility in N′. Partial loss of interaction energy and increased flexibility is already observed in the transition state for the N - > N′ reaction as indicated by a high activation energy of 32 kJ/mol and a large preexponential factor of 7.5·1011 s-1 pointing at favorable entropic contributions to the transition state for unlocking. The large-scale fluctuations occurring from N′ seem to be mainly limited to the C-terminal helix of HP35 since no native-state TTET is observed in the variants with the labels in the N-terminal region indicating that the partially unfolded I has helices 1 and 2 folded and helix 3 unfolded (Fig. 4). This finding is in contrast to results from MD simulations that suggested an I with helices 2 and 3 forming native interactions and helix 1 undocked (27). However, studies on peptide fragments reported native-like interactions between helices 1 and 2 in the absence of helix 3 (29, 30). Recent results from NMR experiments on HP35 also revealed native-state fluctuations on the μs timescale which were interpreted by a native-like alternative conformation on the native side of the unfolding barrier (39). Larger amplitudes of the fluctuations in helix 3 compared to helices 1 and 2 observed by NMR suggested increased flexibility in the C-terminal part of the alternative conformation, as observed for the unlocked state in TTET experiments. Native-state heterogeneity was not observed in any of the reported X-ray structures (18–20).
is slightly more solvent-exposed than N indicating a slight protein expansion. The observed ΔH0 of 35 kJ/mol and ΔS0 of 112 J·mol-1·K-1 for the equilibrium between N and N′ (Fig. 3B) indicate weakened interactions and increased structural flexibility in N′. Partial loss of interaction energy and increased flexibility is already observed in the transition state for the N - > N′ reaction as indicated by a high activation energy of 32 kJ/mol and a large preexponential factor of 7.5·1011 s-1 pointing at favorable entropic contributions to the transition state for unlocking. The large-scale fluctuations occurring from N′ seem to be mainly limited to the C-terminal helix of HP35 since no native-state TTET is observed in the variants with the labels in the N-terminal region indicating that the partially unfolded I has helices 1 and 2 folded and helix 3 unfolded (Fig. 4). This finding is in contrast to results from MD simulations that suggested an I with helices 2 and 3 forming native interactions and helix 1 undocked (27). However, studies on peptide fragments reported native-like interactions between helices 1 and 2 in the absence of helix 3 (29, 30). Recent results from NMR experiments on HP35 also revealed native-state fluctuations on the μs timescale which were interpreted by a native-like alternative conformation on the native side of the unfolding barrier (39). Larger amplitudes of the fluctuations in helix 3 compared to helices 1 and 2 observed by NMR suggested increased flexibility in the C-terminal part of the alternative conformation, as observed for the unlocked state in TTET experiments. Native-state heterogeneity was not observed in any of the reported X-ray structures (18–20).
Fig. 4.

Schematic free energy diagram for folding of HP35. The native state (N) and the unlocked state (N′) are in equilibrium of the native side of the major unfolding barrier. The diagram indicates that the reaction from N′ to N may become rate-limiting for folding of very fast folding variants even if the step from N′ to U represents the mayor unfolding barrier. A high-energy intermediate (I) with helix 3 unfolded is accessible from N′ and was detected by native-state TTET. Comparison with results from theoretical work (23) suggests that I is located on the folding/unfolding pathway.
N′ and I are both located on the folded side of the major unfolding barrier (Fig. 4), since global unfolding occurs on a much slower timescale of several hundred μs (23) and is thus too slow to be observed in TTET experiments (Fig. 2). Our experiments further reveal that I does not become populated in equilibrium at any temperature investigated and thus represents a high-energy intermediate. The experiments cannot discriminate, whether I is located on the unfolding/folding pathway or whether it is formed by a conformational fluctuation parallel to global unfolding. The detected high-energy I in HP35 is, however, in agreement with the observation that high-energy on-pathway intermediates are frequently located after the rate-limiting step for folding of apparent two-state folders (40).
Dry Molten Globule.
The results from TTET experiments in the different HP35 variants show that N and N′ both have native secondary structure but N′ has weakened side-chain interactions, increased flexibility, and slightly increased SASA compared to N. These results suggest that the unlocked state corresponds to the dry molten globule state previously observed during unfolding of the larger proteins ribonuclease A (2, 4) (124 amino acids), DHFR (3) (159 amino acids), and monellin (5) (94 amino acids). As for HP35, the dry molten globule state of ribonuclease A is in equilibrium with the native state and becomes favored with increasing GdmCl concentration (2). Dry molten globules may be a common feature of folding and unfolding pathways indicating that structural unlocking is required before local and global unfolding events can occur. Up-to-date MD simulations on HP35 have not reported an unlocked state with native-like secondary and tertiary structure. Because N′ has native secondary structure and native-like topology, it may be difficult to distinguish from N in MD simulations.
Folding Free Energy Landscape of HP35.
The existence of an unlocked dry molten globule state in equilibrium with N explains the complex unfolding kinetics of HP35 observed in T-jump experiments that showed an initial fast process on the 100 ns timescale at 25 °C (19, 21, 22) which was interpreted as formation of an obligatory intermediate before the rate-limiting step for global unfolding (23). Theoretical work suggested that this intermediate has helices 1 and 2 formed and helix 3 unfolded (23). Our results from TTET indicate that the I observed in T-jump experiments corresponds to N′ which becomes increasingly populated with increasing temperature (Fig. 3B). The kinetics of TTET starting from N are exclusively determined by the unlocking reaction (k-l) whereas the fast reaction in T-jump reflects the sum of k-l and kl, which are both around 400 ns at 25 °C (Fig. 3A) and would result in a relaxation time of about 200 ns in T-jump experiments, similar to the observed value. Our results further indicate that the kinetic intermediate postulated from theoretical work with helix 3 unfolded and helices 1 and 2 folded is rather a high-energy intermediate located between N′ and the major unfolding barrier (Fig. 4).
The time constant of several hundred ns for relocking is similar to the folding time constant of the fastest folding HP35 variants which show values as low as 700 ns (19). This comparison suggests that relocking may be rate-limiting for folding of HP35 under certain conditions or for very fast folding variants, although the unlocked state is on the native side of the major unfolding barrier. In this case the free energy difference between the unfolded state and the major transition state for unfolding would be lower than the barrier between N′ and N (Fig. 4), which is most likely restricted to folding of very small and ultrafast folding proteins. This model is in accordance with results from recent freeze-quenching experiments on HP35 analyzed by solid-state NMR, which detected an intermediate with native-like secondary structure accumulating before the rate-limiting step of folding (41).
Loop Formation in the Unfolded State.
The TTET experiments performed on the different HP35 variants yield rate constants for loop formation in the unfolded state between the various donor/acceptor positions (kc). Loop formation of all investigated variants is described by a single exponential function, which points to a rapid equilibration of chain conformations in the denatured state ensemble (7, 42). All variants exhibit a linear GdmCl-dependence of ln kc both at 5 °C (Figs. 2 B, E, and H) and at 22.5 °C (Fig. S5). Except for the Nal23/Xan35 variant, the mc-values are similar to the values found for unfolded model polypeptide chains (35) indicating the absence of major structural transition in the unfolded state that would affect the kinetics of loop formation (Table S1). Loop formation in the Nal23/Xan35 variant shows a slightly smaller mc-value, which may indicate nonrandom structure in the C-terminal region. Fig. 5 compares the rate constants for loop formation in the unfolded state of the HP35 variants with the dynamics of end-to-end loop formation in poly(Gly-Ser) and in poly(Ser) model peptides of different length (8, 15). All variants except for Xan0/Nal35 show slower kinetics for loop formation compared to polyserine chains. In the variants Xan0/Nal23, Xan7/Nal23, and Nal23/Xan35, however, either one or both labels are located in the interior of the chain, which slows down loop formation up to a factor of 2.5 (15). Taking this effect into account only the Nal23/Xan35 loop is formed slower than expected (Fig. 5), which indicates the presence of nonrandom structure in the unfolded state in the region of helix 3 and agrees with the weaker GdmCl-dependence of loop formation in this variant. The region of helix 1 and helix 2 was reported to retain helical structure and native side-chain clustering in a peptide fragment that lacks helix 3 (29, 30). Formation of stable helices in this region should strongly influence the dynamics of loop formation as observed in TTET on helical peptides (6). Our result may indicate a low population of the residual structure that does not affect the dynamics for loop formation. Alternatively, the peptide fragment comprising helices 1 and 2 may not represent the unfolded state but form a folded structure that is similar to the intermediate (I) with an unfolded C-terminal helix observed in native-state fluctuations of the Nal23/Xan35 variant (Fig. 4). The dynamics in the unfolded state are not influenced by the ionic strength of the GdmCl solutions. Also the urea mc-value measured in the Xan0/Nal23 variant (Fig. S6A) corresponds to the value expected from studies on polyserine chains (35) and the addition of salt in the presence of high urea concentrations has no effect on kc (Fig. S6B).
Fig. 5.

Comparison of kc at 25 °C, 0 M GdmCl with the values determined for end-to-end contact formation in unstructured poly(Gly-Ser) and poly(Ser) chains of different length (8). Additional tails were shown to slow down kc up to maximally 2.5-fold as indicated by the dashed line (15).
Contact formation in the GdmCl-unfolded state of HP35 was previously measured by quenching of the Trp23 triplet state by a cysteine at the N-terminus (34) and yielded about 20-fold slower rate constants for contact formation compared to TTET between the same positions. This discrepancy can be attributed to slow triplet quenching by cysteine, which is not diffusion controlled (43).
In summary, the global dynamic properties of the denatured state ensemble of HP35 under native conditions do not differ from those of unstructured model chains. Formation of the native contact between N- and C-terminus is not accelerated in unfolded HP35 compared to poly(Ser) chains arguing against special properties of unfolded HP35 as molecular origin for ultrafast folding.
Materials and Methods
Preparation and Labelling of HP35.
All HP35 variants were synthesized using standard 9-fluorenylmethyloxycarbonyl chemistry on an Applied Biosystems 433A synthesizer. The HP35 variants with canonical sequence
 |
were labeled with xanthonic acid (Xan) and naphthylalanine (Nal) at the position indicated in the text. Details on chemical synthesis, purification, and labeling of HP35 and on the spectroscopic and thermodynamic characterization are given in SI Text.
Experimental Conditions.
All measurements were performed in degassed solutions of 10 mM potassium phosphate buffer at pH 7.0. For experimental details on CD and NMR experiments, see SI Text.
TTET Experiments.
Xanthone triplet states were produced by a 4 ns laserflash at 354.6 nm (∼50 mJ). Transient absorption traces were recorded at 590 nm to monitor decay of the xanthone triplet state and at 420 nm to monitor formation of the naphthyl triplet state. Peptide concentrations were 50 μM.
Supplementary Material
ACKNOWLEDGMENTS.
We thank Robert Baldwin, Beat Fierz, Annett Bachmann, and Claus Seidel for discussion and comments on the manuscript, Judith Habazettl for help with NMR measurements, Josef Wey for the synthesis of 9oxoxanthene2carboxylic acid, and Jianmin Gao and Jeff Kelly for the data of the NMR spectrum of wild-type HP35 (see Fig. S2). This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 749) and the Munich Center for Integrated Protein Science.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0910001107/DCSupplemental.
References
- 1.Shaknovich EI, Finkelstein AV. Theory of cooperative transitions in protein folding. II. Phase diagram for a protein molecule in solution. Biopolymers. 1989;28:1681–1694. doi: 10.1002/bip.360281004. [DOI] [PubMed] [Google Scholar]
- 2.Kiefhaber T, Labhardt AM, Baldwin RL. Direct NMR evidence for an intermediate preceding the rate-limiting step in the unfolding of ribonuclease A. Nature. 1995;375:513–515. doi: 10.1038/375513a0. [DOI] [PubMed] [Google Scholar]
- 3.Hoeltzli SD, Frieden C. Stopped-flow NMR spectroscopy: Real-time unfolding studies of 6-19F-tryptophan-labeled Escherichia coli dihydrofolate reductase. Proc Natl Acad Sci USA. 1995;92:9318–9322. doi: 10.1073/pnas.92.20.9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kiefhaber T, Baldwin RL. Kinetics of hydrogen bond breakage in the process of unfolding of ribonuclease A measured by pulsed hydrogen exchange. Proc Natl Acad Sci USA. 1995;92:2657–2661. doi: 10.1073/pnas.92.7.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jha SK, Udgaonkar JB. Direct evidence for a dry molten globule intermediate during the unfolding of a small protein. Proc Natl Acad Sci USA. 2009;106:12289–12294. doi: 10.1073/pnas.0905744106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fierz B, Reiner A, Kiefhaber T. Local conformational fluctuations in α-helices measured by fast triplet transfer. Proc Natl Acad Sci USA. 2009;106:1057–1062. doi: 10.1073/pnas.0808581106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bieri O, et al. The speed limit for protein folding measured by triplet-triplet energy transfer. Proc Natl Acad Sci USA. 1999;96:9597–9601. doi: 10.1073/pnas.96.17.9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krieger F, Fierz B, Bieri O, Drewello M, Kiefhaber T. Dynamics of unfolded polypeptide chains as model for the earliest steps in protein folding. J Mol Biol. 2003;332:265–274. doi: 10.1016/s0022-2836(03)00892-1. [DOI] [PubMed] [Google Scholar]
- 9.Heinz B, et al. On the unusual fluorescence properties of xanthone in water. Phys Chem Chem Phys. 2006;8:3432–3439. doi: 10.1039/b603560d. [DOI] [PubMed] [Google Scholar]
- 10.Satzger H, et al. Ultrafast quenching of the xanthone triplet by energy transfer: New insight into the intersystem crossing kinetics. J Phys Chem A. 2004;108:10072–10079. [Google Scholar]
- 11.Möglich A, Joder K, Kiefhaber T. End-to-end distance distributions and intrachain diffusion constants in unfolded polypeptide chains indicate intramolecular hydrogen bond formation. Proc Natl Acad Sci USA. 2006;103:12394–12399. doi: 10.1073/pnas.0604748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hvidt A, Nielsen SO. Hydrogen exchange in proteins. Adv Protein Chem. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- 13.Linderstrøm-Lang K. Deuterium exchange between peptides and water. Chem Soc Spec Publ. 1955;2:1–20. [Google Scholar]
- 14.Krieger F, et al. Intrachain diffusion in a protein loop fragment from carp parvalbumin. Chem Phys. 2004;307:209–215. [Google Scholar]
- 15.Fierz B, Kiefhaber T. End-to-end vs interior loop formation kinetics in unfolded polypeptide chains. J Am Chem Soc. 2007;129:672–679. doi: 10.1021/ja0666396. [DOI] [PubMed] [Google Scholar]
- 16.McKnight CJ, Doering DS, Matsudaira PT, Kim PS. A thermostable 35-residue subdomain within villin headpiece. J Mol Biol. 1996;260:126–134. doi: 10.1006/jmbi.1996.0387. [DOI] [PubMed] [Google Scholar]
- 17.McKnight CJ, Doering DS, Kim PS. NMR structure of the 35-residue villin headpiece subdomain. Nat Struct Biol. 1997;4:180–184. doi: 10.1038/nsb0397-180. [DOI] [PubMed] [Google Scholar]
- 18.Chiu TK, et al. High-resolution x-ray crystal structures of the villin headpiece subdomain, an ultrafast folding protein. Proc Natl Acad Sci USA. 2005;102:7517–7522. doi: 10.1073/pnas.0502495102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kubelka J, Chiu TK, Davies DR, Eaton WA, Hofrichter J. Sub-microsecond protein folding. J Mol Biol. 2006;359:546–553. doi: 10.1016/j.jmb.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 20.Bi Y, et al. Rational design, structural and thermodynamic characterization of a hyperstable variant of the villin headpiece helical subdomain. Biochemistry. 2007;46:7497–7505. doi: 10.1021/bi6026314. [DOI] [PubMed] [Google Scholar]
- 21.Kubelka J, Eaton WA, Hofrichter J. Experimental tests of villin subdomain folding simulations. J Mol Biol. 2003;329:625–630. doi: 10.1016/s0022-2836(03)00519-9. [DOI] [PubMed] [Google Scholar]
- 22.Brewer SH, et al. Effect of modulating unfolded state structure on the folding kinetics of the villin headpiece subdomain. Proc Natl Acad Sci USA. 2005;102:16662–16667. doi: 10.1073/pnas.0505432102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kubelka J, Henry ER, Cellmer T, Hofrichter J, Eaton WA. Chemical, physical, and theoretical kinetics of an ultrafast folding protein. Proc Natl Acad Sci USA. 2008;105:18655–18662. doi: 10.1073/pnas.0808600105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang M, et al. Dynamic NMR line-shape analysis demonstrates that the villin headpiece subdomain folds on the microsecond time scale. J Am Chem Soc. 2003;125:6032–6033. doi: 10.1021/ja028752b. [DOI] [PubMed] [Google Scholar]
- 25.Duan Y, Kollman PA. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 1998;282:740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 26.Zagrovic B, Snow CD, Khaliq S, Shirts MR, Pande VS. Native-like mean structure in the unfolded ensemble of small proteins. J Mol Biol. 2002;323:153–164. doi: 10.1016/s0022-2836(02)00888-4. [DOI] [PubMed] [Google Scholar]
- 27.Lei H, Wu C, Liu H, Duan Y. Folding free-energy landscape of villin headpiece subdomain from molecular dynamics simulations. Proc Natl Acad Sci USA. 2007;104:4925–4930. doi: 10.1073/pnas.0608432104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wickstrom L, Bi Y, Hornak V, Raleigh DP, Simmerling C. Reconciling the solution and X-ray structures of the villin headpiece helical subdomain: Molecular dynamics simulations and double mutant cycles reveal a stabilizing cation-pi interaction. Biochemistry. 2007;46:3624–3634. doi: 10.1021/bi061785+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang Y, Goger MJ, Raleigh DP. NMR characterization of a peptide model provides evidence for significant structure in the unfolded state of the villin headpiece helical subdomain. Biochemistry. 2006;45:6940–6946. doi: 10.1021/bi052484n. [DOI] [PubMed] [Google Scholar]
- 30.Meng W, Shan B, Tang Y, Raleigh DP. Native like structure in the unfolded state of the villin headpiece helical subdomain, an ultrafast folding protein. Protein Sci. 2009;18:1692–1701. doi: 10.1002/pro.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao J, Kelly JW. Toward quantification of protein backbone hydrogen bonding energies: An energetic analysis of an amide-to-ester mutation in an α-helix within a protein. Protein Sci. 2008;17:1096–1101. doi: 10.1110/ps.083439708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frank BS, Vardar D, Buckley DA, McKnight CJ. The role of aromatic residues in the hydrophobic core of the villin headpiece subdomain. Protein Sci. 2002;11:680–687. doi: 10.1110/ps.22202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buscaglia M, Kubelka J, Eaton WA, Hofrichter J. Determination of ultrafast protein folding rates from loop formation dynamics. J Mol Biol. 2005;347:657–664. doi: 10.1016/j.jmb.2005.01.057. [DOI] [PubMed] [Google Scholar]
- 35.Möglich A, Krieger F, Kiefhaber T. Molecular basis for the effect of urea and guanidinium chloride on the dynamics of unfolded polypeptide chains. J Mol Biol. 2005;345:153–162. doi: 10.1016/j.jmb.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 36.Vugmeyster L, Trott O, McKnight CJ, Raleigh DP, Palmer AG., 3rd Temperature-dependent dynamics of the villin headpiece helical subdomain, an unusually small thermostable protein. J Mol Biol. 2002;320:841–854. doi: 10.1016/S0022-2836(02)00537-5. [DOI] [PubMed] [Google Scholar]
- 37.Mayo SL, Baldwin RL. Guanidinium chloride induction of partial unfolding in amide proton exchange in ribonuclease A. Science. 1993;262:873–876. doi: 10.1126/science.8235609. [DOI] [PubMed] [Google Scholar]
- 38.Bai Y, Sosnick TR, Mayne L, Englander SW. Protein folding intermediates: Native-state hydrogen exchange. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vugmeyster L, McKnight CJ. Slow motions in chicken villin headpiece subdomain probed by cross-correlated NMR relaxation of amide NH bonds in successive residues. Biophys J. 2008;95:5941–5950. doi: 10.1529/biophysj.108.134320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sánchez IE, Kiefhaber T. Evidence for sequential barriers and obligatory intermediates in apparent two-state protein folding. J Mol Biol. 2003;325:367–376. doi: 10.1016/s0022-2836(02)01230-5. [DOI] [PubMed] [Google Scholar]
- 41.Hu K-N, Yau W-M, Tycko R. Detection of a transient intermediate in a rapid protein folding process by solid-state nuclear magnetic resonance. J Am Chem Soc. 2010;132:24–25. doi: 10.1021/ja908471n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szabo A, Schulten K, Schulten Z. First passage time approach to diffusion controlled reactions. J Chem Phys. 1980;72:4350–4357. [Google Scholar]
- 43.Yeh IC, Hummer G. Peptide loop-closure kinetics from microsecond molecular dynamics simulations in explicit solvent. J Am Chem Soc. 2002;124:6563–6568. doi: 10.1021/ja025789n. [DOI] [PubMed] [Google Scholar]
- 44.Koradi R, Billeter M, Wüthrich K. MOLMOL: A program for display and analysis of macromolecular structures. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.