

Abstract
The essential double-ring eukaryotic chaperonin TRiC/CCT (TCP1-ring complex or chaperonin containing TCP1) assists the folding of ∼5–10% of the cellular proteome. Many TRiC substrates cannot be folded by other chaperonins from prokaryotes or archaea. These unique folding properties are likely linked to TRiC’s unique heterooligomeric subunit organization, whereby each ring consists of eight different paralogous subunits in an arrangement that remains uncertain. Using single particle cryo-EM without imposing symmetry, we determined the mammalian TRiC structure at 4.7-Å resolution. This revealed the existence of a 2-fold axis between its two rings resulting in two homotypic subunit interactions across the rings. A subsequent 2-fold symmetrized map yielded a 4.0-Å resolution structure that evinces the densities of a large fraction of side chains, loops, and insertions. These features permitted unambiguous identification of all eight individual subunits, despite their sequence similarity. Independent biochemical near-neighbor analysis supports our cryo-EM derived TRiC subunit arrangement. We obtained a Cα backbone model for each subunit from an initial homology model refined against the cryo-EM density. A subsequently optimized atomic model for a subunit showed ∼95% of the main chain dihedral angles in the allowable regions of the Ramachandran plot. The determination of the TRiC subunit arrangement opens the way to understand its unique function and mechanism. In particular, an unevenly distributed positively charged wall lining the closed folding chamber of TRiC differs strikingly from that of prokaryotic and archaeal chaperonins. These interior surface chemical properties likely play an important role in TRiC’s cellular substrate specificity.
Keywords: asymmetric reconstruction, atomic model, subunit structure
Defective protein folding is emerging as the molecular basis underlying a growing number of human diseases, ranging from cancer and heart disease (1) to aggregation-linked neurodegenerative diseases such as Alzheimer’s, Huntington, and mad cow disease (2, 3). The eukaryotic group II chaperonin TRiC (also known as CCT) is a central mediator of cytosolic protein folding and assembly (4, 5). TRiC also appears important for the prevention of protein aggregation and toxicity (6–8). TRiC is essential for cell viability, as it assists the folding of many essential proteins, including actin, tubulin, and many cell cycle regulators and signaling molecules (9, 10). Notably, a number of TRiC substrates cannot be folded by other chaperonins, suggesting that TRiC possesses unique structural and mechanistic properties that distinguish it functionally from other chaperonins (11, 12).
All chaperonins share a double-ring architecture, where each ring contains a central cavity that binds and folds substrate proteins. TRiC, a 1 MDa group II chaperonin, facilitates folding through the ATPase driven closure of a built-in lid that encloses the substrate in the central chamber (13–16). Each ring of TRiC consists of eight distinct but related subunits sharing 27–39% sequence identity (Fig. S1) (13, 17). In contrast, bacterial (18) and archael chaperonins (19) only have 1–3 different types of subunits, and for those archaea with three types of subunits, it is unclear whether in the natural organism they form homo- or heterooligomeric chaperonins (20). The divergence of TRiC subunits occurred early in the evolution of eukaryotes, because all eukaryotes sequenced to date carry genes for all eight subunits; orthologs of the various subunits across species are more similar than paralogs within a single species (17). Having eight distinct subunits may have allowed the diversification of substrate binding sites and activities within the ring of TRiC. Actin, tubulin (21, 22), Huntingtin (8), and the von Hippel–Lindau tumor suppressor (23) are all recognized by distinct subsets of TRiC subunits through specific motifs; this directed binding may provide specificity for TRiC in the folding process. Accordingly, elucidating the subunit arrangement within the complex is essential to determine how TRiC affects the conformation and folding pathway of its substrates.
Understanding the molecular basis of the specific folding capacity of TRiC has been hindered by the paucity of structural information on this complex. An intraring subunit arrangement proposed from analysis of spontaneously dissociated TRiC complexes remains untested (24). Previous cryo-EM studies of TRiC achieved only up to ∼15-Å resolution with imposed 8-fold symmetry (13, 25, 26), inadequate to resolve the asymmetry between the eight structurally similar subunits. Here we present a high-resolution cryo-EM structure of mammalian TRiC, derived without imposing any symmetry among the eight subunits. Our analysis reveals (i) a 2-fold axis of symmetry between the two rings of TRiC, and (ii) its intraring subunit arrangement. The structure-derived subunit arrangement of TRiC is supported by cross-linking analysis. Our study provides a structural baseline to elucidate the complicated mechanisms of substrate recognition, folding and cooperativity in TRiC.
Results
4.7-Å Resolution Map with No Symmetry Enforced.
We used cryo-EM to determine the structure of bovine TRiC in the presence of ATP-AlFx. This biochemical preparation generated a homogeneous both-ring-closed conformation in all the particles (Fig. 1A). Using ∼101,000 particle images, we calculated a density map without any imposed symmetry employing EMAN, a single particle reconstruction package (27, 28). At 4.7-Å resolution of this asymmetric map (Fig. 1B and Fig. S2A), it was possible to unambiguously delineate the 16 individual subunits in the complex. Moreover, the α-helix pitch, β-strand separation, and some of the bulky side-chain densities and loop regions were resolved in all subunits (Fig. S2B). Of note, this level of detail is sufficient to observe relatively small structural differences, i.e., insertions and differences in amino acid composition, among the subunits. To avoid any initial arrangement bias in our analysis, we have arbitrarily named the rings a, encompassing subunits ai–aviii, and b, with subunits bi–bviii (Fig. 1B), labeled clockwise looking down on either end of the map.
Fig. 1.

TRiC 4.7 Å asymmetric cryo-EM density map and the identification of the 2-fold axis between its two rings. (A) A typical 300 kV image of ice embedded TRiC in the both-ring-closed conformation. Representative particles are highlighted by white boxes. (B) End-on view of a ring and side view of both rings of TRiC with different subunits in different colors. The identified 2-fold axis is indicated by black ellipsoid in the side view. (C) Rotational correlation coefficient analysis between a ring and b ring. At 0° the two rings have the best correlation score, indicating the location of the 2-fold axis as illustrated in B. Due to the periodicity of the TRiC map, 0° peak of the curve is split, with the other half of the peak at ∼359°. (D) Cartoon diagram depicts the relative orientation of the two rings of TRiC.
One outstanding question about the organization of TRiC is whether both rings have the same subunit arrangement and, if so, what is the relative orientation of the rings. Because the structure was determined with no symmetry imposed, we were able to compare the two rings as well as use unique structural features in each subunit to answer these questions unambiguously.
An exhaustive correlation analysis, carried out by pairwise comparison between each computationally extracted subunit in one ring and each of the eight subunits in the other ring, reveals that the ordering of the subunits in the two rings is identical. A subsequent rotational correlation analysis between the two rings shows one pronounced peak at 0° (Fig. 1C), pinpointing the location of the 2-fold axis between the rings (Fig. 1B). These analyses identify the relative orientation of the rings and localize the 2-fold symmetry axis between them. In this arrangement, two opposing subunits (ai and av) are directly aligned with the same subunits in the opposing ring (bi and bv). As a result, two subunits (aiii and avii) are related 180° azimuthally from their equivalent subunits in b ring and the remaining four subunits (aii, aiv, avi, and aviii) are related 90° azimuthally to their equivalents in the b ring (Fig. 1D). This two-ring arrangement pattern is different from the previous report hypothesized from low-resolution cryo-EM maps (26).
Identification of Intraring Subunit Arrangement.
Exploiting the existence and position of the 2-fold symmetry axis, a new TRiC structure was computed enforcing the 2-fold symmetry. This produced an improved map with ∼4.0-Å resolution (Fig. S2), in which a large proportion of the side-chain densities was visible in all the subunits (Fig. 2). These identifiable structural features were used to unambiguously localize subunit-specific sequences and thus assign which subunits corresponded to the densities within the ring.
Fig. 2.

The match of the side-chain densities in an apical domain region with the corresponding optimized model for each of the eight subunits. (A) Location of this stretch (Sky Blue, including the protruding helix H8 and the connected loop) in a complete TRiC subunit (Gray). The three domains are labeled. (B) Sequence alignment of bovine TRiC eight subunits in that apical domain region as shown in A. Unique sequence stretches of each subunit are highlighted by red characters. Either one or a combination of several such characteristic stretches can serve as a fingerprint for each TRiC subunit. (C) Subunit ai map (Blue meshes) with the optimized model of the best matching CCT8(θ) (Red). The residues with the clearly observable side-chain densities are labeled in black or red. Here the red labels correspond to the residues in the unique stretches of CCT8(θ) as in B. (D–J) Similar rendering style as in C for each of the subunit maps and the corresponding optimized models in the equivalent region.
To start the identification process, we first constructed an initial homology model for each subunit of bovine TRiC based on the homologous group II chaperonins (13): the thermosome [1A6E, (19)] and KS-1 [1Q3Q, (29)]. Herein we denote the TRiC subunits by both their CCT number and a Greek letter, i.e., CCT1(α), CCT2(β), CCT3(γ), CCT4(δ), CCT5(ϵ), CCT6(ζ), CCT7(η), and CCT8(θ). Because all eight TRiC subunits share a high level of sequence identity (Fig. S1A), we would anticipate that the subunit densities are quite similar. However, sequence alignment of the eight bovine TRiC subunits (Fig. S1A) reveals substantial variations in the apical domain, particularly helix H8 and the adjacent connecting long loop (Fig. 2A and B). In addition, unique insertions are found in several subunits, including CCT1(α) (equatorial domain, 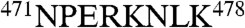 ), and CCT4(δ) (apical domain,
), and CCT4(δ) (apical domain, 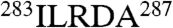 ) (Fig. S1A). These insertions are predicted to form loops based on our homology models and provide characteristic features that, together with the diagnostic apical domain regions, allowed us to identify the individual subunits in the maps.
) (Fig. S1A). These insertions are predicted to form loops based on our homology models and provide characteristic features that, together with the diagnostic apical domain regions, allowed us to identify the individual subunits in the maps.
Next, each subunit density was evaluated against each of the eight possible homology models, with a special focus on the most divergent sequence regions in the apical domain (Fig. 2A and B). For instance, the side-chain densities of the subunit ai map gives the best match to the model CCT8(θ) as shown in Fig. 2C. The unique sequence stretches of CCT8(θ) located in the helix H8 and connecting long loop including 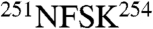 ,
, 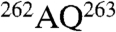 , and
, and 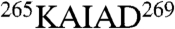 (red, highlighted in Fig. 2B) matched the larger side-chain densities unambiguously visualized in the ai map (red characters in Fig. 2C). The side-chain densities in these apical regions were similarly matched to unique sequences in the other seven subunits (Fig. 2D–J).
(red, highlighted in Fig. 2B) matched the larger side-chain densities unambiguously visualized in the ai map (red characters in Fig. 2C). The side-chain densities in these apical regions were similarly matched to unique sequences in the other seven subunits (Fig. 2D–J).
Analyses of other unique sequence insertions (Fig. S3) and characteristic sequence stretches (Fig. S4) provided independent evidence for the identification of the eight subunits. For instance, CCT6(ζ) has a unique insertion loop (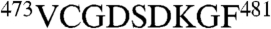 ) right after helix H8 (Fig. S1A). Fig. S3C demonstrates that the subunit aiv map most closely matches the CCT6(ζ) model; indeed the aiv map clearly depicts the extra density in the corresponding insertion loop region, along with the unique stretch
) right after helix H8 (Fig. S1A). Fig. S3C demonstrates that the subunit aiv map most closely matches the CCT6(ζ) model; indeed the aiv map clearly depicts the extra density in the corresponding insertion loop region, along with the unique stretch 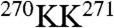 of CCT6(ζ) and some of the bulky side-chain densities (i.e., D278, K279, and F281). In contrast, fitting any of the other seven models did not produce a match, leaving this insertion loop density unoccupied (e.g., Fig. S3D shows that CCT2(β) did not fit to the aiv subunit map). Furthermore, the insertion loops of several other subunits, including CCT3(γ) (Fig. S3A), CCT1(α) (Fig. S3E), and CCT4(δ) (Fig. S3G), were also identified as the best match to the corresponding densities. The above two sets of independent evidence (Fig. 2 and Figs. S3 and S4) provide a consistent intraring subunit arrangement of TRiC.
of CCT6(ζ) and some of the bulky side-chain densities (i.e., D278, K279, and F281). In contrast, fitting any of the other seven models did not produce a match, leaving this insertion loop density unoccupied (e.g., Fig. S3D shows that CCT2(β) did not fit to the aiv subunit map). Furthermore, the insertion loops of several other subunits, including CCT3(γ) (Fig. S3A), CCT1(α) (Fig. S3E), and CCT4(δ) (Fig. S3G), were also identified as the best match to the corresponding densities. The above two sets of independent evidence (Fig. 2 and Figs. S3 and S4) provide a consistent intraring subunit arrangement of TRiC.
We next optimized the Cα model for each subunit from the initial homology model with the constraint of the corresponding cryo-EM subunit map, concurrently improving the peptide geometry. The optimized Cα model of the TRiC complex is shown in Fig. 3A. Viewed from the top of the map (Fig. 3A and B), the subunits follow the following clockwise arrangement: CCT8(θ), CCT3(γ), CCT2(β), CCT6(ζ), CCT1(α), CCT7(η), CCT5(ϵ), and CCT4(δ). The arrangement is identical in the trans ring, thus creating two pairs of homotypic interring interactions: CCT1(α)–CCT1(α) and CCT8(θ)–CCT8(θ) (Fig. 3B). Our structure-based subunit ordering differs substantially from the previous proposal based on the analysis of spontaneously dissociated dimers in extracts (24).
Fig. 3.

The optimized Cα model of the entire TRiC complex with its 16 subunits in the spatial arrangement determined by our cryo-EM structure. (A) End-on and side views of the TRiC complex. Same color scheme as in Fig. 1. (B) Cartoon diagram illustrating the arrangement of the 16 subunits in the two rings. We denote the TRiC subunits only by their CCT number. (C) Summary of near neighbors TRiC subunits identified by chemical cross-linking. Neighboring subunit pairs consistent with the indicated models are marked with a “+,” and those inconsistent with the indicated models are marked with a “−.”
Validation of Subunit Arrangement by Biochemical Near-Neighbor Analysis.
An independent assessment of the TRiC subunits arrangement was provided by chemical cross-linking of adjacent subunits in the intact chaperonin (Fig. S5, Table S1, and SI Methods). Formaldehyde, which has one of the shortest cross-linking spans (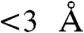 ) was used to yield specific covalent adducts between neighboring chaperonin subunits both within a single ring and across the rings (30, 31). Given the similarity in molecular weight and isoelectric point of the TRiC subunits, and hence of their covalently linked dimers, we separated the cross-linked subunits by 2D-PAGE (Fig. S5B). Well-separated spots corresponding in size to a dimer of TRiC subunit were excised, and MS was used to identify the subunits present in each spot. Covalently linked near neighbors were inferred from these spots that had multiple peptides corresponding to only two TRiC subunits. This approach identified several unambiguous neighboring subunit pairs (Fig. 3C, Fig. S5C, and Table S1), all of which were consistent with our cryo-EM-derived subunit arrangement (Fig. 3B). Of note, several of these determined cross-links were inconsistent with previously proposed subunit arrangement of TRiC (24, 26). For instance, the prominent CCT2(β)-CCT5(ϵ) cross-link (Fig. 3C and Fig. S5C) is fully inconsistent with previous models (24, 26), but agrees with an interring contact proposed by our cryo-EM-derived structure (Fig. 3B). In case of the CCT8(θ)–CCT8(θ) dimer, obtained by cross-linking (Fig. 3C and Fig. S5C), this covalent adduct can only arise from the homotypic interring contact of two CCT8(θ) subunits, as shown by our cryo-EM-derived structure (Fig. 3B). This biochemical analysis thus supports our cryo-EM-derived subunit arrangement and model.
) was used to yield specific covalent adducts between neighboring chaperonin subunits both within a single ring and across the rings (30, 31). Given the similarity in molecular weight and isoelectric point of the TRiC subunits, and hence of their covalently linked dimers, we separated the cross-linked subunits by 2D-PAGE (Fig. S5B). Well-separated spots corresponding in size to a dimer of TRiC subunit were excised, and MS was used to identify the subunits present in each spot. Covalently linked near neighbors were inferred from these spots that had multiple peptides corresponding to only two TRiC subunits. This approach identified several unambiguous neighboring subunit pairs (Fig. 3C, Fig. S5C, and Table S1), all of which were consistent with our cryo-EM-derived subunit arrangement (Fig. 3B). Of note, several of these determined cross-links were inconsistent with previously proposed subunit arrangement of TRiC (24, 26). For instance, the prominent CCT2(β)-CCT5(ϵ) cross-link (Fig. 3C and Fig. S5C) is fully inconsistent with previous models (24, 26), but agrees with an interring contact proposed by our cryo-EM-derived structure (Fig. 3B). In case of the CCT8(θ)–CCT8(θ) dimer, obtained by cross-linking (Fig. 3C and Fig. S5C), this covalent adduct can only arise from the homotypic interring contact of two CCT8(θ) subunits, as shown by our cryo-EM-derived structure (Fig. 3B). This biochemical analysis thus supports our cryo-EM-derived subunit arrangement and model.
Optimization and Validation of the TRiC Model.
Our cryo-EM structure clearly resolves many side chains within the subunits. In X-ray crystallography, density maps determined at ∼4.0-Å resolution range are often considered marginal for determining the atomistic structures (32). However, recent studies have shown that it is indeed possible to reliably build a de novo Cα model directly from cryo-EM density map in this resolution range (28, 33). It should also be noted that our density map (Fig. 1B) was directly computed from the raw images (Fig. 1A) and not biased by any atomic model.
In order to assess the ultimate quality of our map and model, we optimized the full atomic model from the initial Cα chain trace by matching it to the map and the visible side-chain densities for three randomly chosen subunits: CCT2(β), CCT1(α), and CCT7(η). Fig. 4A shows the atomic model of the CCT2(β) subunit. The Ramachandran plot of this optimized model shows that over 95% of the main chain dihedral angles fall within allowable regions (Fig. 4B), demonstrating the quality of the final model to follow the protein stereochemistry. Optimized atomic models of the other two subunits show similar quality.
Fig. 4.

The atomic model of CCT2(β) subunit and the match between the aiii density and the model. (A) Optimized atomic model of CCT2(β) with the N terminus in blue and C terminus in red. Zoom in views show the match between three stretches of densities highlighted in different color frames and the corresponding model. (B) The Ramachandran plot of the CCT2(β) model calculated by MolProbity (48) shows that over 95% of the dihedral angles fall within allowable regions.
To further validate our model, we compare the only available TRiC domain crystal structure [i.e., the apical domain of mouse CCT3(γ) (34)] with our corresponding model. Because the crystal structure was not used as a template to build our model, 1GML can serve as a quality check. The good match between our model and 1GML (Fig. S6) validates the reliability of our model and thus, the quality of our map.
Discussion
Unlike most chaperonins, which have one to three distinct subunits, TCP1-ring complex or chaperonin containing TCP1 (TRiC/CCT) has eight distinct, but similar, subunits. The subunits are sufficiently similar that determining individual particle orientation with sufficient precision would require very high resolution. Considering also the roughly spherical shape of the both-ring-closed conformation (Fig. 1A and B), the determination of particle orientation of TRiC presented even more challenges than other asymmetric structures studied so far by cryo-EM. Approximately 35% of the ∼160,000 particles lacked sufficient contrast even at this resolution to unambiguously assign their orientation. Identifying and eliminating these particles using custom software (35) and our recently developed 2D fast rotation matching method (FRM2D) (36, 37) for the image alignment was critical in achieving the final resolution. This approach provided unprecedented resolution for an asymmetric structure by cryo-EM and allowed the unambiguous identification of the TRiC subunit arrangement.
Our TRiC structure has a number of important biological and mechanistic implications. First, it shows that each type of subunit has a fixed position within the ring, as suggested previously based on biochemical and genetic data (38). It also identifies the location of a 2-fold axis between the rings and reveals a unique interring arrangement that produces three pairs of heterotypic interring contacts, [CCT3(γ)–CCT4(δ), CCT2(β)–CCT5(ϵ), and CCT6(ζ)–CCT7(η)], and two sets of homotypic interring contacts, namely, CCT8(θ)–CCT8(θ) and CCT1(α)–CCT1(α). This unexpected subunit arrangement has important implications for the assembly mechanism and allosteric regulation of TRiC. Like other chaperonins, TRiC exhibits positive intraring cooperativity and strong negative interring cooperativity (15, 39). Interestingly, close analysis of the interring contacts for the various subunit pairs reveals different types of interfaces, with some pairs exhibiting a more extensive set of contacts (Fig. 3A). These data may open the way for understanding the molecular basis of the negative cooperativity, likely achieved through interring contacts (15). Furthermore, it has been proposed that the positive intraring cooperativity follows a sequential KNF model, whereby the conformational change is initiated at a specific subunit (39). Because this would be determined by the chemical and enzymatic properties of the individual subunits within the ring, as well as the substrate, identifying the subunit arrangement is critical to test this mechanism.
Our model also provides insights into the unique ability of TRiC to fold certain substrates. The eight different subunits appear to bind different motifs within nonnative substrates and thus may position their folding intermediates in a set of defined orientations. Lid closure confines the substrate to the inner chamber, where folding of stringent substrates takes place (14). In the group I bacterial chaperonin GroEL-ES, the inner chamber of the closed cis-ring is polar, and the pattern of charges in the chamber is important for the folding ability of GroEL (40). We thus computed the electrostatic surface property of TRiC from our model and compared it with those of the thermosome, an archaeal group II chaperonin, and GroEL-ES (Fig. 5). TRiC, like the thermosome (Fig. 5E and Fig. S7B) and GroEL-ES (Fig. 5F and Fig. S7C), has a large proportion of hydrophilic residues lining the inner surface of the closed chamber (Fig. 5D and Fig. S7A). Notably, both TRiC (Fig. 5D) and thermosome (Fig. 5E) have a lower percentage of hydrophobic residues lining the chamber than GroEL-ES (Fig. 5F). Although the charge distribution in both thermosome (Fig. 5B) and GroEL-ES (Fig. 5C) is symmetrical, we observe varying surface properties among the different subunits of TRiC (Fig. 5A); e.g., CCT6(ζ) is more positively charged, whereas CCT2(β) is more negatively charged. Notably, the wall of the TRiC chamber is dominated by positively charged patches (Fig. 5A), particularly on the side containing subunits CCT1(α), CCT6(ζ), CCT2(β), and CCT3(γ). The thermosome (1A6E) closed cavity is also dominated by positive charges, but unlike TRiC, the charge distribution is symmetric (Fig. 5B). In contrast, the inner chamber of GroEL-ES is dominated by negatively charged patches (41).
Fig. 5.

Surface property of the central cavity of TRiC, thermosome (1A6E) and the cis-ring of GroEL-GroES (1AON). (A–C) The inner cavity electrostatic potential of the TRiC/thermosome/GroEL-GroES are shown in cutaway views of the apical and intermediate domains, including GroES for C. Blue represents positively charged patches, red negatively charged patches, and white neutral patches. The smaller panel illustrates the viewing angle. Of note, the surfaces are approximate and variances due to model/map resolution may affect the fine details of surface potential. (D–F) Inner wall surface property of TRiC/thermosome/GroEL-GroES with side-chain properties is shown: hydrophilic (Sky Blue), hydrophobic (Yellow), and main chain (White).
The differences between TRiC and other simpler chaperonins, particularly the diversified surface properties of the inner chamber of TRiC, might be related to TRiC’s differential ability to fold some substrates that cannot be folded by other chaperones. Our structure will open the way to understand the mechanism and physical properties that underlie this selectivity.
Materials and Methods
TRiC sample purification (42) and cryoEM sample preparation follow our established procedures (14). Data were collected on a JEM3200FSC electron microscope with an in-column energy filter (energy slit of 15 eV) under the following condtions: 300 kV, ∼50,000x magnification, ∼20 electrons/Å2 dose, and 101 K specimen temperature. Images were recorded on Kodak SO163 film and digitized on a Nikon 9000 ED scanner with a 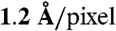 sampling. The majority of the defocus ranges from 1.2–2.7 μm.
sampling. The majority of the defocus ranges from 1.2–2.7 μm.
Approximately 160,000 particles were selected from 1,500 micrographs with the EMAN2 tool e2boxer. Contrast transfer function parameters were determined using ctfit of EMAN (27, 28, 43). A recently developed FRM2D algorithm for image alignment (36, 37, 44), available in EMAN 1.8+ (frm2d option in refine program), was adopted in the refinement steps. We used a previously determined 15-Å resolution 8-fold symmetrized map of closed TRiC (13) as the initial model of the reconstruction. Other than that, in the asymmetric reconstruction and refinement process, no symmetry was imposed. The final map was computed from ∼101,000 particle images, after eliminating particles that were not consistently classified in the same orientation between iterations. The map resolution was based on the 0.5 Fourier shell correlation (FSC) criterion (45). The final map was filtered and scaled to optimized map resolvability (46, 47).
Detailed procedures about map similarity analysis, homology model building and model optimization, and cross-linking and nearest-neighbor analysis are provided in SI Materials and Methods.
Supplementary Material
Acknowledgments.
We would like to thank Dr. Michael F. Schmid for very helpful discussions and suggestions. This research is supported by grants from National Institutes of Health (PN1EY016525, P41RR02250, and GMR01-074074) and National Science Foundation (IIS-0705644), and Department of Energy Contract DE-AC02-05CH11231.
Footnotes
Conflict of interest statement: The Sponsor is an investigator of the Nanomedicince Development Center with the support of National Institutes of Health Grant (5PN2EY016525), which is directed by the corresponding author.
This article contains supporting information online at www.pnas.org/cgi/content/full/0913774107/DCSupplemental.
Data deposition: The density maps and models have been deposited in the Electron Microscopy Data Bank (accession numbers EMD-5145 and EMD-5148) and Protein Data Bank, www.pdb.org (PDB ID codes 3KTT and 3IYG).
References
- 1.Scott MD, Frydman J. Aberrant protein folding as the molecular basis of cancer. Method Mol Biol. 2003;232:67–76. doi: 10.1385/1-59259-394-1:67. [DOI] [PubMed] [Google Scholar]
- 2.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dobson CM. Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol. 2004;15:3–16. doi: 10.1016/j.semcdb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 4.Frydman J. Folding of newly translated proteins in vivo: The role of molecular chaperones. Annu Rev Biochem. 2001;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- 5.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 6.Behrends C, et al. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol Cell. 2006;23:887–897. doi: 10.1016/j.molcel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 7.Kitamura A, et al. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat Cell Biol. 2006;8:1163–1170. doi: 10.1038/ncb1478. [DOI] [PubMed] [Google Scholar]
- 8.Tam S, Geller R, Spiess C, Frydman J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat Cell Biol. 2006;8:1155–1162. doi: 10.1038/ncb1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunn AY, Melville MW, Frydman J. Review: Cellular substrates of the eukaryotic chaperonin TRiC/CCT. J Struct Biol. 2001;135:176–184. doi: 10.1006/jsbi.2001.4380. [DOI] [PubMed] [Google Scholar]
- 10.Yam AY, et al. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat Struct Mol Biol. 2008;15:1255–1262. doi: 10.1038/nsmb.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leroux MR, Hartl FU. Protein folding: Versatility of the cytosolic chaperonin TRiC/CCT. Curr Biol. 2000;10:R260–264. doi: 10.1016/s0960-9822(00)00432-2. [DOI] [PubMed] [Google Scholar]
- 12.Spiess C, Meyer AS, Reissmann S, Frydman J. Mechanism of the eukaryotic chaperonin: protein folding in the chamber of secrets. Trends Cell Biol. 2004;14:598–604. doi: 10.1016/j.tcb.2004.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Booth CR, et al. Mechanism of lid closure in the eukaryotic chaperonin TRiC/CCT. Nat Struct Mol Biol. 2008;15:746–753. doi: 10.1038/nsmb.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer AS, et al. Closing the folding chamber of the eukaryotic chaperonin requires the transition state of ATP hydrolysis. Cell. 2003;113:369–381. doi: 10.1016/s0092-8674(03)00307-6. [DOI] [PubMed] [Google Scholar]
- 15.Reissmann S, Parnot C, Booth CR, Chiu W, Frydman J. Essential function of the built-in lid in the allosteric regulation of eukaryotic and archaeal chaperonins. Nat Struct Mol Biol. 2007;14:432–440. doi: 10.1038/nsmb1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutsche I, Essen LO, Baumeister W. Group II chaperonins: New TRiC(k)s and turns of a protein folding machine. J Mol Biol. 1999;293:295–312. doi: 10.1006/jmbi.1999.3008. [DOI] [PubMed] [Google Scholar]
- 17.Archibald JM, Blouin C, Doolittle WF. Gene duplication and the evolution of group II chaperonins: Implications for structure and function. J Struct Biol. 2001;135:157–169. doi: 10.1006/jsbi.2001.4353. [DOI] [PubMed] [Google Scholar]
- 18.Xu Z, Horwich AL, Sigler PB. The crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperonin complex. Nature. 1997;388:741–750. doi: 10.1038/41944. [DOI] [PubMed] [Google Scholar]
- 19.Ditzel L, et al. Crystal structure of the thermosome, the archaeal chaperonin and homolog of CCT. Cell. 1998;93:125–138. doi: 10.1016/s0092-8674(00)81152-6. [DOI] [PubMed] [Google Scholar]
- 20.Kapatai G, et al. All three chaperonin genes in the archaeon Haloferax volcanii are individually dispensable. Mol Microbiol. 2006;61:1583–1597. doi: 10.1111/j.1365-2958.2006.05324.x. [DOI] [PubMed] [Google Scholar]
- 21.Valpuesta JM, Martin-Benito J, Gomez-Puertas P, Carrascosa JL, Willison KR. Structure and function of a protein folding machine: The eukaryotic cytosolic chaperonin CCT. FEBS Lett. 2002;529:11–16. doi: 10.1016/s0014-5793(02)03180-0. [DOI] [PubMed] [Google Scholar]
- 22.Etchells SA, et al. The cotranslational contacts between ribosome-bound nascent polypeptides and the subunits of the hetero-oligomeric chaperonin TRiC probed by photocross-linking. J Biol Chem. 2005;280:28118–28126. doi: 10.1074/jbc.M504110200. [DOI] [PubMed] [Google Scholar]
- 23.Spiess C, Miller EJ, McClellan AJ, Frydman J. Identification of the TRiC/CCT substrate binding sites uncovers the function of subunit diversity in eukaryotic chaperonins. Mol Cell. 2006;24:25–37. doi: 10.1016/j.molcel.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liou AK, Willison KR. Elucidation of the subunit orientation in CCT (chaperonin containing TCP1) from the subunit composition of CCT micro-complexes. EMBO J. 1997;16:4311–4316. doi: 10.1093/emboj/16.14.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Llorca O, et al. The “sequential allosteric ring” mechanism in the eukaryotic chaperonin-assisted folding of actin and tubulin. EMBO J. 2001;20:4065–4075. doi: 10.1093/emboj/20.15.4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin-Benito J, et al. The inter-ring arrangement of the cytosolic chaperonin CCT. EMBO Rep. 2007;8:252–257. doi: 10.1038/sj.embor.7400894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ludtke SJ, Baldwin PR, Chiu W. EMAN: Semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 28.Ludtke SJ, et al. De novo backbone trace of GroEL from single particle electron cryomicroscopy. Structure. 2008;16:441–448. doi: 10.1016/j.str.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 29.Iizuka R, et al. Role of the helical protrusion in the conformational change and molecular chaperone activity of the archaeal group II chaperonin. J Biol Chem. 2004;279:18834–18839. doi: 10.1074/jbc.M400839200. [DOI] [PubMed] [Google Scholar]
- 30.Nadeau OW, Carlson GM. Protein interactions captured by chemical cross-linking. In: Golemis EA, Adams PD, editors. Protein–Protein Interactions: A Molecular Cloning Manual. 2nd Ed. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2005. pp. 105–128. [Google Scholar]
- 31.Rice NA, Nadeau OW, Yang Q, Carlson GM. The calmodulin-binding domain of the catalytic γ subunit of phosphorylase kinase interacts with its inhibitory alpha subunit: Evidence for a Ca2+ sensitive network of quaternary interactions. J Biol Chem. 2002;277:14681–14687. doi: 10.1074/jbc.M201229200. [DOI] [PubMed] [Google Scholar]
- 32.Blow D. Outline of Crystallography for Biologists. New York: Oxford University Press; 2002. pp. 191–204. [Google Scholar]
- 33.DiMaio F, Tyka MD, Baker ML, Chiu W, Baker D. Refinement of protein structures into low-resolution density maps using rosetta. J Mol Biol. 2009;392:181–190. doi: 10.1016/j.jmb.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pappenberger G, et al. Crystal structure of the CCTgamma apical domain: Implications for substrate binding to the eukaryotic cytosolic chaperonin. J Mol Biol. 2002;318:1367–1379. doi: 10.1016/s0022-2836(02)00190-0. [DOI] [PubMed] [Google Scholar]
- 35.Tang G, et al. EMAN2: An extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 36.Cong Y, Kovacs JA, Wriggers W. 2D fast rotational matching for image processing of biophysical data. J Struct Biol. 2003;144:51–60. doi: 10.1016/j.jsb.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 37.Cong Y, et al. Fast rotational matching of single-particle images. J Struct Biol. 2005;152:104–112. doi: 10.1016/j.jsb.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 38.Miller EJ, Meyer AS, Frydman J. Modeling of possible subunit arrangements in the eukaryotic chaperonin TRiC. Protein Sci. 2006;15:1522–1526. doi: 10.1110/ps.052001606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rivenzon-Segal D, Wolf SG, Shimon L, Willison KR, Horovitz A. Sequential ATP-induced allosteric transitions of the cytoplasmic chaperonin containing TCP-1 revealed by EM analysis. Nat Struct Mol Biol. 2005;12:233–237. doi: 10.1038/nsmb901. [DOI] [PubMed] [Google Scholar]
- 40.Tang YC, et al. Structural features of the GroEL-GroES nano-cage required for rapid folding of encapsulated protein. Cell. 2006;125:903–914. doi: 10.1016/j.cell.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 41.Horwich AL, Fenton WA, Chapman E, Farr GW. Two families of chaperonin: Physiology and mechanism. Annu Rev Cell Dev Biol. 2007;23:115–145. doi: 10.1146/annurev.cellbio.23.090506.123555. [DOI] [PubMed] [Google Scholar]
- 42.Ferreyra RG, Frydman J. Purification of the cytosolic chaperonin TRiC from bovine testis. Method Mol Biol. 2000;140:153–160. doi: 10.1385/1-59259-061-6:153. [DOI] [PubMed] [Google Scholar]
- 43.Ludtke SJ, Jakana J, Song JL, Chuang DT, Chiu W. A 11.5 A single particle reconstruction of GroEL using EMAN. J Mol Biol. 2001;314:253–262. doi: 10.1006/jmbi.2001.5133. [DOI] [PubMed] [Google Scholar]
- 44.Cong Y, et al. Structural mechanism of SDS-induced enzyme activity of scorpion hemocyanin revealed by electron cryomicroscopy. Structure. 2009;17:749–758. doi: 10.1016/j.str.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harauz G, van Heel M. Exact filters for general geometry three dimensional reconstruction. Optik. 1986;73:146–156. [Google Scholar]
- 46.Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 47.Fernandez JJ, Luque D, Caston JR, Carrascosa JL. Sharpening high resolution information in single particle electron cryomicroscopy. J Struct Biol. 2008;164:170–175. doi: 10.1016/j.jsb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 48.Davis IW, et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.