

Abstract
We used differential display analysis to identify mRNAs that accumulate to enhanced levels in human cytomegalovirus-infected cells as compared with mock-infected cells. RNAs were compared at 8 hr after infection of primary human fibroblasts. Fifty-seven partial cDNA clones were isolated, representing about 26 differentially expressed mRNAs. Eleven of the mRNAs were virus-coded, and 15 were of cellular origin. Six of the partial cDNA sequences have not been reported previously. All of the cellular mRNAs identified in the screen are induced by interferon α. The induction in virus-infected cells, however, does not involve the action of interferon or other small signaling molecules. Neutralizing antibodies that block virus infection also block the induction. These RNAs accumulate after infection with virus that has been inactivated by treatment with UV light, indicating that the inducer is present in virions. We conclude that human cytomegalovirus induces interferon-responsive mRNAs.
Human cytomegalovirus (HCMV) alters gene expression through multiple pathways. For example, the virion gB and gH glycoproteins induce cellular transcription factors when they interact with their cell surface targets (1). Virion proteins, such as pp71 (2–4), activate transcription (5); and viral proteins synthesized after infection, such as IE1 and IE2, regulate expression from a variety of promoters (6–10). Further, HCMV infection has been shown to perturb cell cycle progression (11–14), which leads to changes in gene expression.
Viral factors, induced cellular factors, and changes in cell cycle progression have the potential to exert profound effects on host cell gene expression, but relatively few cellular genes have been identified whose activity changes after HCMV infection (15). A more global understanding of HCMV-induced changes in cellular gene expression should help us to better understand how the virus interacts with its host cell during the replication process and might direct us to new targets for therapeutic intervention in HCMV disease.
We used differential display analysis (16, 17) to catalog changes in cellular RNA levels that occur after HCMV infection, and we identified 15 cellular RNAs that accumulate to higher levels in infected cells than in uninfected cells. We sequenced the cDNA segments, and six of 15 sequences have not been reported previously. All 15 RNAs accumulate in response to interferon α, and their induction is mediated by the virus particle.
MATERIALS AND METHOD
Cells and Viruses.
Primary human foreskin (HF) cells were cultured in medium containing 10% fetal calf serum. Cells were held at confluence for 3–4 days before experimentation. To avoid cell stimulation by fresh serum, treated confluent cultures were returned to the medium in which they were previously maintained. HF cells were treated with 500 units/ml of interferon α or β (Sigma) for 4 hr, and 100 μg/ml of cycloheximide was used to block protein synthesis.
HF cells were infected with HCMV strain AD169 (18), Towne (19), or Toledo (20). Wild-type adenovirus, dl309 (21), and herpes simplex virus type 1 (HSV-1) also were used. Infections with HCMV or HSV-1 were at a multiplicity of five plaque-forming units/cell, and adenovirus was used at 30 plaque-forming units/cell. For UV inactivation, 5 ml of HCMV stock was placed in a 15 cm-diameter dish and irradiated at 2J/m2 per sec for 10 min with mixing every 2 min. UV-treated stocks failed to produce detectable IE1 and IE2 protein at 8 or 36 hr after infection. For neutralization, 50 μl of HCMV stock was incubated with 20 μl of human neutralizing antibody (pooled sera from HCMV antibody positive donors; gift from Jay Nelson, University of Oregon) for 1 hr at room temperature. Neutralization was confirmed by plaque assay. HCMV particles were concentrated and purified as described (22). HCMV membrane and tegument/capsid proteins were separated by detergent stripping (23).
Differential Display Assay.
For differential display analysis (16–17), HF cells were mock-infected or infected with AD169 or UV-inactivated AD169. Total RNA was isolated 8 hr later by using the TRIZOL Reagent (Life Technologies, Grand Island, NY). First-strand cDNAs were synthesized by using oligo(dT) as primer, amplified in parallel PCRs in the presence of [33P]dCTP by using 135 combinations of 19 primers (CLONTECH), and products were separated by electrophoresis on 5% polyacrylamide gels containing 8 M urea. Differentially expressed bands were cut out of the gel, reamplified, cloned into the pT7Blue T-Vector (Novagen), and sequenced, and the results were analyzed by blast search (National Center for Biotechnology Information).
Assays for RNAs and Proteins.
For Northern blot assays 5 μg of RNA was probed with random hexanucleotide-primed 32P-labeled cDNA clones. The probes for mxA, isg15K, and interferon β were the partial cDNA sequences from I.M.A.G.E. Consortium clones (Genome Systems, St. Louis). For Western blot assays, three mouse mAbs that recognize HCMV proteins, anti-IE1/IE2 (mAb810, Chemicon), anti-pp65 (2), and anti-glycoprotein B (Goodwin Institute, Plantation, FL), were used as the primary antibodies. mAb810 and anti-pp65 also were used for immunofluorescent staining.
RESULTS
Analysis of Cytomegalovirus-Induced RNAs.
HCMV could alter host cell gene expression through the action of virion proteins or by the synthesis of new viral proteins after infection. To distinguish between these possibilities, we compared competent virus (HCMV) to UV-inactivated virus (UV HCMV). To test the effect of UV treatment, the delivery of the pp65 virion protein to the cells and the synthesis of the IE1 and IE2 immediate-early proteins were monitored. UV irradiation did not affect viral entry into the cells because the amount of pp65 delivered to the cells did not change with UV treatment (Fig. 1A). The IE1 and IE2 proteins were detected at 8 and 21 hr after infection in HCMV-infected cells, but not in UV HCMV-infected cells (Fig. 1A). Inhibition of viral RNA accumulation in UV HCMV-infected cells was also evident. The IE1 transcript could be detected at 8 hr after infection in HCMV-infected cells, but not in UV HCMV-infected cells (Fig. 1B). We also determined the location of a virion protein in cells infected with UV-treated virus. pp65 was visible in nuclei at 2 hr after infection with either HCMV or UV HCMV (Fig. 1C 1 and 2). As expected, nuclear IE1 protein was detected in HCMV-infected, but not in UV HCMV-infected, cells (Fig. 1C 3 and 4). These experiments show that UV irradiation of virions blocked the accumulation of detectable amounts of HCMV-encoded RNA without preventing the entrance of the virus into the cell or altering the intracellular localization of a virion protein.
Figure 1.
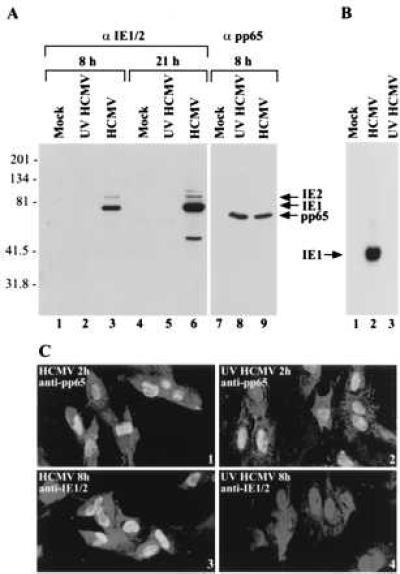
Characterization of UV HCMV. (A) Western blot showing that UV irradiation of virions blocks expression of the HCMV IE1 and IE2 RNAs, but has no effect on the delivery of a virion protein to cells. HF cells were mock-infected or infected, and extracts were prepared 8 or 21 hr later. Lanes 1–6 were reacted with antibody that binds to IE1 and IE2, whereas lanes 7–9 were reacted with antibody to pp65. The molecular weights of marker proteins are indicated on the left. (B) Northern blot showing that IE1 RNA is detected at 8 hr after infection of HF cells with HCMV but not after infection with UV HCMV. (C) Immunofluorescent localization of pp65 and IE1/2 within infected cells. HF cells were infected with HCMV or UV HCMV for 2 or 8 hr, reacted with antibody to pp65 or IE1/IE2 followed by a fluorescein-labeled secondary antibody (bright nuclear signal), and counterstained with ethidium homodimer-1 (dim whole cell signal).
We compared RNA levels by differential display (16, 17) at 8 hr after infection or mock infection. HCMV immediate-early proteins have accumulated to significant levels at this time (see Fig. 5), giving them an opportunity to influence host cell mRNA accumulation. PCR-generated bands that were evident in virus-infected, but not mock-infected, samples could be divided into two groups. One group contained an induced band that was present in the HCMV-infected sample, but not in the UV HCMV-infected sample. The induced bands in this group could be derived from either viral or cellular RNAs. The second group contained induced bands in both HCMV- and UV HCMV-infected samples. These bands should represent cellular RNAs that accumulate after infection, because viral mRNAs are not produced in UV HCMV-infected cells (Fig. 1B).
Figure 5.
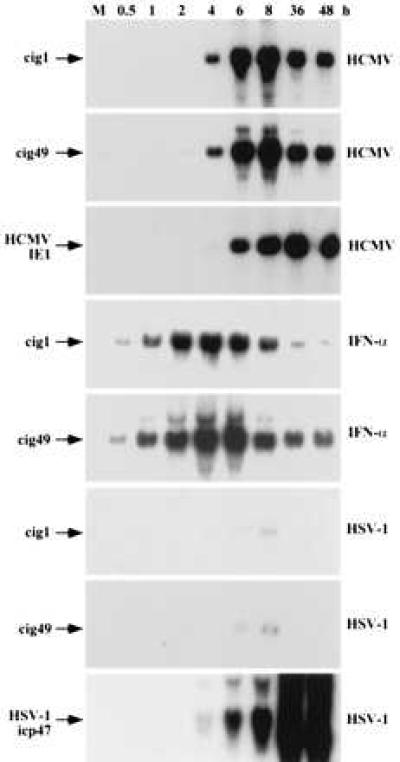
Kinetic analysis of cig RNA accumulation. HF cells were mock-infected (M) or treated with the inducers identified to the right (HCMV, HSV-1, and IFN-α), RNA was prepared at various times after treatment (indicated above lanes), and analyzed by Northern blot by using the probes indicated on the left (cig1, cig49, HCMV IE1, and HSV-1 icp47).
We chose 71 of the most strongly induced PCR-generated bands for analysis. DNA fragments were reamplified by PCR, cloned, and used as probes for Northern blot analyses to confirm that the bands represented differentially expressed genes. Examples of these assays are displayed in Fig. 2A. Most of the cloned cDNA segments identified RNAs that were present at very low or nondetectable levels in mock-infected cells, but accumulated to a high level in infected cells. cDNA clones representing up-regulated RNAs were isolated from 57 of the 71 reamplified fragments. Each clone is termed a cig for cytomegalovirus inducible gene.
Figure 2.
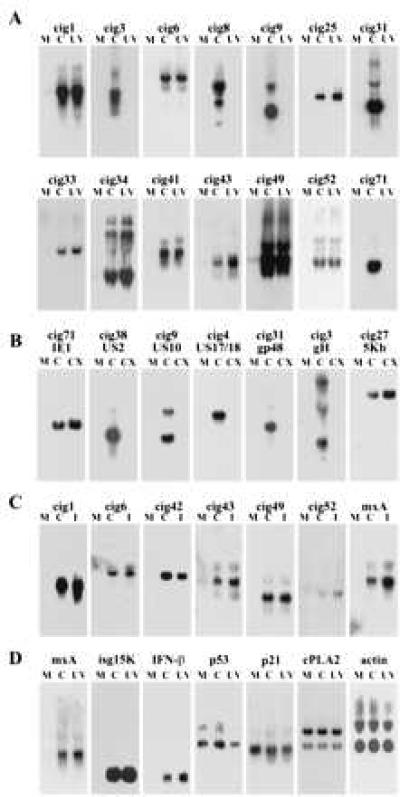
Differential expression of RNAs in HF cells assayed by Northern blot. (A) RNA was prepared from mock-infected (M), HCMV-infected cells (C), or UV HCMV-infected cells (UV) and assayed by using cloned cDNA segments. The different clones (cigs) are identified above the panels. (B) RNA was prepared from mock-infected (M), HCMV-infected (C), or HCMV-infected cells that were treated with cycloheximide (CX) and assayed as in A. (C) RNA was prepared from mock-infected cells (M), HCMV-infected cells (C), or cells treated with interferon α (I) and assayed by using probes corresponding to the cigs or the mxA gene. (D) RNA was prepared as in A and assayed with probes corresponding to interferon-inducible genes (mxA, isg15K, and IFN-β) or control genes that are not induced by interferon (p53, p21, cPLA2, and actin).
Thirty of 57 cig RNAs were induced by HCMV, but not UV HCMV, infection, and sequence analysis revealed that all of these clones corresponded to viral RNAs (data not shown). Two of the viral RNAs were produced after infection in the presence of cycloheximide, identifying them as immediate-early RNAs, and the synthesis of the others was inhibited by the drug, indicating that they are early RNAs (Fig. 2B and data not shown).
Infection with either HCMV or UV HCMV led to the accumulation of 27 of the 57 cig RNAs, and sequence analysis demonstrated that they correspond to as many as 15 different cellular genes (Table 1). Nine were previously identified, and the other six were not found in a blast search. Surprisingly, most of the known RNAs previously were shown to be induced by interferon α. The nine previously described RNAs were induced by interferon α in HF cells, as were the six new RNAs (Fig. 2C and data not shown). The RNAs were induced by both virus infection and interferon α in three lots of HF cells derived from different individuals (data not shown). Because the RNAs induced by infection corresponded to interferon-inducible genes, it seemed possible that other interferon-stimulated genes might be induced by HCMV. As expected, RNAs corresponding to mxA (33, 34), ISG15K (35, 36), and interferon β (37) also were induced (Fig. 2C). As controls, we tested the expression of p53, p21, cytosolic phospholipase A2 (cPLA2), and actin. The level of these RNAs did not change after infection (Fig. 2D and see Fig. 5).
Table 1.
Cellular cDNAs identified by differential display
| Clone (cig) | Gene | Reference |
|---|---|---|
| 1, 22, 51 | Interferon-stimulated gene 54K | 24 |
| 19 | KIAA0062 | 25 |
| 24, 70 | Glyceraldehyde-3-phosphate dehydrogenase | 26 |
| 25 | Guanylate binding protein I | 27 |
| 32 | Mn-superoxide dismutase | 28 |
| 34, 45, 46, 68 | Microtubular aggregate protein | 29 |
| 43 | IFP53 | 30 |
| 52 | (2′-5′) oligoadenylate synthetase | 31 |
| 53 | Guanylate binding protein II | 32 |
| 5–7, 15, 18, 44, 61, 69 | AF026941* | |
| 33 | AF026942* | |
| 41 | AF026943* | |
| 42 | AF026944* | |
| 49 | AF026939* | |
| 64 | AF026945* |
GenBank accession numbers are for newly described cDNA segments.
HCMV Particles Induce the Accumulation of cig RNAs Encoded by Cellular Genes.
The differential display analysis used the laboratory-adapted AD169 strain of HCMV. Towne, a second laboratory-adapted HCMV strain, and Toledo, a low passage clinical isolate of HCMV, also strongly activated the accumulation of cell-coded cig RNAs (Fig. 3A, lanes 10 and 11). Adenovirus did not activate the accumulation of cig RNAs and HSV-1 increased their expression to a very limited extent (Fig. 3A, lanes 8 and 9; see Fig. 5). The expression of an adenovirus and HSV-1 mRNA was monitored to be certain that cells were successfully infected (data not shown). Thus, whereas multiple HCMV strains strongly induced cig RNA accumulation, two other viruses did not.
Figure 3.
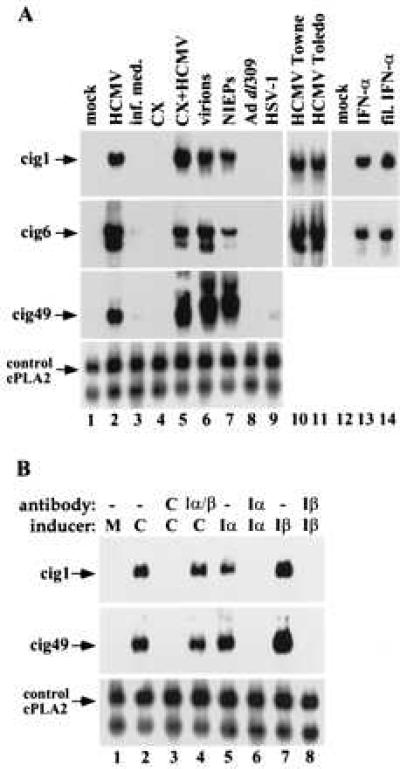
The HCMV particle mediates the induction of differentially expressed HF RNAs. (A) Requirements for induction monitored by Northern blot assay. The relative amounts of three cellular RNAs (cig1, cig6, and cig49) were monitored in mock-infected cells (mock), HCMV AD169-infected cells (HCMV), cells treated with medium from which virions were removed by filtration (inf. med.), mock-infected cells treated with cycloheximide (CX), HCMV AD169-infected cells treated with cycloheximide (CX+HCMV), cells infected with purified HCMV AD169 particles (virions), cells infected with purified noninfectious enveloped particles from HCMV AD169 (NIEPs), adenovirus-infected cells (Ad dl309), herpes simplex type 1-infected cells (HSV-1), HCMV Towne-infected cells, HCMV Toledo-infected cells, interferon α-treated cells (IFN-α), and cells treated with interferon that was added to medium and passed through the filter type used to exclude virus (fil. IFN-α). (B) Northern blot assay showing that antibody that neutralizes HCMV (antibody C) blocks the induction of cig RNA accumulation, whereas antibodies that neutralize interferon α or β (antibody Iα, Iβ) block the induction of cig RNAs by interferon α or β (inducer Iα, Iβ) but have no effect on the induction of cig RNAs by HCMV (C). RNA prepared from mock-infected control cells is designated M. The cellular cytosolic phospholipase A2 RNA was assayed as a loading control (control, cPLA2).
To ask if cellular protein synthesis was required for the induction of cellular interferon responsive RNAs, cells were infected in the presence of cycloheximide. It did not block the induction of cig RNAs by HCMV, and the drug itself had no effect on cig RNA expression (Fig. 3A, lanes 4 and 5). This result indicates that the accumulation of cig RNAs does not require the synthesis of viral or cellular proteins after infection. It also rules out the possibility that a protein factor, such as a cytokine, is synthesized in response to the infection, and released from the cell so that it can interact with a cell surface receptor to induce cig RNAs.
Because infected cell lysates were used as virus stocks in our initial experiments, it was possible that soluble signaling molecules were present that could mediate the induction of RNAs encoded by the cell. We therefore performed a series of experiments to identify the component in HCMV stocks that was responsible for the induction. Initially, an HCMV stock was separated into two fractions by filtration through a 100-kDa cutoff membrane. The virus fraction was further purified by rate-velocity centrifugation, separating infectious virions and noninfectious enveloped particles (NIEPs, lacking viral DNA). The filtered lysate, purified virions, and NIEPs were used to treat cells, and their abilities to induce the accumulation of cig RNAs were assayed. Purified virions and NIEPs activated cig RNA accumulation (Fig. 3A, lanes 6 and 7), whereas the filtered lysate had little effect (Fig. 3A, lane 3). To prove that small molecules could pass through the filter, interferon α (500 units/ml) was added to the infected cell lysate, and there was no detectable loss of interferon activity after filtration (Fig. 3A, lanes 13 and 14).
We used neutralizing antibodies to demonstrate that the activation of cig RNA accumulation is mediated by HCMV particles and not by interferon. When the virus stock was incubated with polyclonal antibody to virions (pooled human sera), its ability to induce cig RNAs was blocked, whereas antibody to interferon α or β had no effect (Fig. 3B). The same amounts of interferon-specific antibodies were sufficient to block interferon α or β activity in uninfected cultures (Fig. 3B). We conclude that the HCMV particle or a molecule tightly associated with the particle initiates the induction of cellular cig RNAs. Expression of viral genes is not required, because purified NIEPs and UV HCMV can induce cig RNAs.
We next explored the possibility that interferon might be carried within the HCMV particle. Purified viral particles were treated with Triton X-100 (0.5%) and deoxycholate (0.5%) and subjected to centrifugation to produce a supernatant fraction containing HCMV membrane proteins and a pellet containing internal virion constituents. With detergent treatment, pp65 (a marker for the tegument/capsid fraction) was in the pellet fraction and gB (a marker for the membrane fraction) was in the supernatant fraction. Without detergent treatment, the particle remained intact, and both pp65 and gB were in the pellet fraction (data not shown). As expected, without detergent treatment, the pellet fraction, but not supernatant fraction, activated cig RNA accumulation; with detergent treatment, neither the pellet fraction, nor supernatant the fraction activated the accumulation (Fig. 4A). When interferon α was treated with the detergent mixture, its activity was not affected (Fig. 4A). This experiment indicates that the intact virus particle is required for the induction of cig RNAs, and further argues that this induction is not because of contaminating interferons.
Figure 4.
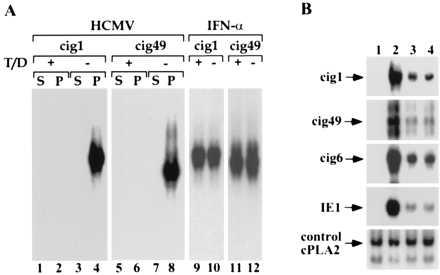
Requirements for the induction of cig RNA accumulation. (A) An intact HCMV particle is required. Purified virions were treated with a mixture of Triton X-100 and deoxycholate (T/C) and separated by centrifugation into supernatant (S) and pellet (P) fractions. Northern blot assays show the effect of detergent treatment on the induction of two cig RNAs (cig1 and cig49) by virions (HCMV) or interferon α (IFN-α). (B) The induction of cig RNAs does not involve the release of mediators stored within infected HF cells. At 8 hr after treatment, RNA was prepared from mock-infected cells (lane 1), HCMV-infected cells (lane 2), or a 9:1 mixture of mock and infected cells (lanes 3 and 4). The two mixed cultures differed in the time of mixing. In lane 3, cells were mixed at 1 hr after infection; in lane 4, RNAs were prepared and mixed from 8 hr mock- and HCMV-infected cells. RNAs were analyzed by Northern blot by using cellular (cig1, cig6, cig49, and cPLA2) and viral (IE1) probes.
Our results argue that the induction of cell-coded cig RNAs does not result from contaminants in HCMV preparations or from newly synthesized signaling proteins. Nevertheless, one might propose that a trace amount of a signaling molecule is stored in the cell, secreted after infection, and then acts at the surface of neighboring cells to induce cig RNAs. Accordingly, we performed an experiment in which uninfected cells and cells infected 1 hr earlier were mixed in a ratio of 9:1, and a sufficient number of cells were plated to generate a confluent monolayer. At the same time, 100% infected cells or 100% noninfected cells were plated at the same density. RNA was prepared at 8 hr after infection, and the expression of cig RNAs and the HCMV IE1 RNA were assayed. The viral and cig RNAs were induced in the infected culture, but not in the uninfected culture (Fig. 4B). The RNA levels were induced to the same extent in the mixed culture as was seen for an uninfected/infected (ratio, 9:1) cell mixture prepared immediately before the extraction of RNA (Fig. 4B). Infected cells did not significantly induce the accumulation of cig RNAs in their uninfected neighbors.
Kinetics of cig RNA Induction by HCMV as Compared with Interferon α.
The kinetics of cig RNA accumulation varied when cells were treated with different inducers (Fig. 5). Accumulation was first evident at 4–6 hr after infection with HCMV, cig RNA levels peaked at about 8 hr, and remained at high levels for the duration of the experiment (48 hr). The HCMV IE1 gene showed a similar expression pattern. The induction of cig RNA expression in cells treated with interferon α was more rapid and transient. The cig RNAs were detected at 30 min and reached their peak at 2–4 hr before declining. The marked difference in the kinetics of cig RNA accumulation in HCMV-infected cells as compared with interferon-treated cells further supports the conclusion that the induction observed subsequent to HCMV infection is not the result of contaminating interferon in virus preparations.
In HSV-1-infected cells, the induction of cig RNAs was very limited (Fig. 5), consistent with the view that the strong induction of cig RNA accumulation observed in HCMV-infected cells is not a common cellular response to all herpes viruses. As a control, the HSV-1 icp47 immediate-early gene was shown to be expressed at a high level, demonstrating that the culture was successfully infected.
DISCUSSION
We cloned 57 cDNA segments corresponding to RNAs that are present at a higher concentration in HCMV-infected human fibroblasts as compared with mock-infected human fibroblasts. The 57 clones represent no more than 26 different mRNAs because some of the RNAs corresponded to more than one cDNA fragment generated by different primer sets. Six of the partial cellular cDNAs were not found in a blast search, and we have determined the complete sequence of one of the newly discovered RNAs. Because the others are only partially sequenced, more than one of the remaining five sequences might be contained within the same RNA molecule. However, only two of the five partially sequenced clones appear to recognize RNAs of similar size (Fig. 2 and data not shown).
Of the 26 cDNA clones, 11 were virus-coded. All of the immediate-early and some early HCMV mRNAs should have accumulated to detectable levels when cells were harvested at 8 hr after infection; and partial cDNA clones corresponding to both classes of viral RNA were isolated. The screen identified two of approximately 10 immediate-early mRNAs. One cannot accurately estimate the total number of HCMV early mRNAs expressed at 8 hr because the number increases continually from about 4–24 hr after infection (15). Given the uncertainties about the number of different viral mRNAs present, it is difficult to estimate accurately the proportion of HCMV RNAs that were identified in the differential display analysis. However, because we identified two of about 10 immediate-early mRNAs, it seems likely that the screen identified substantially less than half of the viral mRNAs that were present, even though multiple clones were isolated that corresponded to several of the viral transcripts. Partial cDNA clones corresponding to the most abundant immediate-early (IE1/IE2: refs. 38 and 39) and early (TRL4: ref. 40) mRNAs were isolated, so our screen likely favored the identification of the more plentiful species.
Given the proportion of immediate-early viral mRNAs that were identified in the screen, it seems likely that we also identified substantially less than half of the cellular RNAs that were induced at 8 hr after infection. Nevertheless, multiple partial cDNA clones corresponding to some of the cellular transcripts were isolated (Table 1). In fact, eight overlapping clones were isolated that corresponded to one of the cellular RNAs whose sequence was not found in a blast search.
All of the cellular RNAs that were induced at 8 hr after infection proved to be interferon-inducible (Table 1 and Fig. 2C). We presume that they are induced by HCMV infection at the level of transcription as is the case when their accumulation is induced by interferon, but we have not yet determined that. A complete cDNA corresponding to one of the interferon-inducible RNAs (cig49) has been cloned and sequenced. It is related to ISG54K (24). One of the partial cDNA sequences (cig42) also appears to be related to ISG54K, and the other four are not related in their primary sequence to known genes.
We were concerned that the cellular RNAs identified in the screen might be induced by interferon or another contaminant of the virus preparations, but a variety of observations argue that the induction is mediated by virus particles. The most direct evidence supporting this view derives from neutralization experiments (Fig. 3B), and the timing of the induction is not consistent with a role for interferon (Fig. 5). Further, it is unlikely that the induction involves a small molecule other than interferon in the virus preparations because the inducing activity fractionated with the virions (Fig. 3A). We have ruled out the possibility that interferon or another signaling molecule is synthesized by infected cells and secreted to act at the cell surface, because the interferon-responsive mRNAs are induced in the presence of cycloheximide (Fig. 3A). Finally, experiments in which infected cells were mixed with uninfected cells (Fig. 4B) argue that pre-existing stores of a signaling molecule are not released after infection with HCMV to act at the cell surface and initiate a signal cascade.
A constituent of the virus particle, rather than a viral gene product synthesized after infection, mediates the induction because UV-irradiated particles that fail to express immediate-early mRNAs (Fig. 1) can sponsor the accumulation (Fig. 2A). We currently are working to identify the inducer and its mode of action.
Three different strains of HCMV strongly induced the accumulation of interferon response RNAs (Fig. 3A), and the AD169 strain was shown to induce these RNAs in HF cells prepared from three different tissue samples (data not shown). Adenovirus did not induce and HSV-1 generated a very weak induction (Figs. 3A and 5). Thus, the relatively strong HCMV-mediated induction is not a general feature of infection by DNA viruses. Adenovirus has been shown to block the induction of interferon response genes through the action of its E1A proteins (41–43). However, an E1A-deficient adenovirus mutant, dl312 (21), also failed to induce the genes (data not shown). In contrast HSV-1 has been shown to induce the production of interferon α in human peripheral mononuclear cells (44–46). So the weak induction observed in HSV-1-infected HF cells might result from a direct induction of interferon-responsive genes, from the production of double-stranded RNA that can induce the genes or from the initial induction of interferon β with a subsequent general induction of interferon-response genes as the secreted interferon acts at the cell surface. Besides the strength of induction, the HSV-1- and HCMV-mediated reactions differ in another important respect. HCMV induces interferon-response mRNAs very early during its replication cycle in HF cells (Fig. 5), beginning about 20 hr before the onset of viral DNA replication. In contrast, the induction observed for HSV-1 occurs later during its more rapid replication cycle (47).
Apparently, in contrast to some other viruses, HCMV has not evolved the means to block the induction of many interferon-inducible mRNAs. The antiviral actions of the induced cellular products might be antagonized by viral products at a posttranscriptional level. Alternatively, HCMV might activate these genes as part of a strategy to slow and minimize the extent of its replication within an infected host. Such a strategy, together with the ability to undergo latency, could facilitate the long-term association of the pathogen with its host. It is also possible that the virus uses a component of the interferon-response pathway to activate its own genes.
Acknowledgments
We thank J. Nelson for HCMV neutralizing antibody; X. Chen for anti-interferon antibodies; B. Banfield for HSV-1; F. Liu for a plasmid carrying the HSV-1 icp47 coding region; and L. Enquist, A. Hirsch, A. Marchini, and C. Patterson for commenting on the manuscript. H.Z. is a research associate and J.-P.C. is a technical assistant of the Howard Hughes Medical Institute. T.S. is an American Cancer Society Professor and Investigator of the Howard Hughes Medical Institute.
ABBREVIATIONS
- HCMV
human cytomegalovirus
- HF
human foreskin
- HSV-1
herpes simplex virus type 1
- NIEPs
noninfectious enveloped particles
- UV HCMV
UV-inactivated HCMV
Footnotes
References
- 1.Yurochko A D, Hwang E-S, Rasmussen L, Keay S, Pereira L, Huang E-S. J Virol. 1997;71:5051–5059. doi: 10.1128/jvi.71.7.5051-5059.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nowak B, Gmiener A, Sarnow P, Levine A J, Fleckenstein B. Virology. 1984;134:91–102. doi: 10.1016/0042-6822(84)90275-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roby C, Gibson W. J Virol. 1986;59:714–727. doi: 10.1128/jvi.59.3.714-727.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruger B, Klages S, Walla B, Albrecht J, Fleckenstein B, Tomlinson P, Barrell B. J Virol. 1987;61:446–453. doi: 10.1128/jvi.61.2.446-453.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu B, Stinski M F. J Virol. 1992;66:4434–4444. doi: 10.1128/jvi.66.7.4434-4444.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pizzorno M C, O’Hare P, Sha L, LaFemina R L, Hayward G S. J Virol. 1988;62:1167–1179. doi: 10.1128/jvi.62.4.1167-1179.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malone C L, Vesole D H, Stinski M F. J Virol. 1990;64:1498–1506. doi: 10.1128/jvi.64.4.1498-1506.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stenberg R M, Fortney J, Barlow S W, Magrane B P, Nelson J A, Ghazal P. J Virol. 1990;64:1556–1565. doi: 10.1128/jvi.64.4.1556-1565.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klucher K, Sommer M, Kadonaga J T, Spector D H. Mol Cell Biol. 1993;13:1238–1250. doi: 10.1128/mcb.13.2.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lukac D M, Manuppello J R, Alwine J C. J Virol. 1994;68:5184–5193. doi: 10.1128/jvi.68.8.5184-5193.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jault F M, Jault J-M, Ruchti F, Fortunato E A, Clark C, Corbeil J, Richman D D, Spector D H. J Virol. 1995;69:6697–6704. doi: 10.1128/jvi.69.11.6697-6704.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bresnahan W A, Boldogh I, Thompson E A, Albrecht T. Virology. 1996;224:150–160. doi: 10.1006/viro.1996.0516. [DOI] [PubMed] [Google Scholar]
- 13.Lu M, Shenk T. J Virol. 1996;70:8850–8857. doi: 10.1128/jvi.70.12.8850-8857.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dittmer D, Mocarski E S. J Virol. 1997;71:1629–1634. doi: 10.1128/jvi.71.2.1629-1634.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mocarski E S., Jr . In: Fields Virology. 3rd Ed. Fields B N, Knipe D M, Howley P M, editors. Philadelphia: Lippincott; 1995. pp. 2447–2492. [Google Scholar]
- 16.Liang P, Pardee A B. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- 17.Liang P, Bauer D, Averboukh L, Warthoe P, Rohrwild M, Muller H, Strauss M, Pardee A B. Methods Enzymol. 1995;254:304–321. doi: 10.1016/0076-6879(95)54022-9. [DOI] [PubMed] [Google Scholar]
- 18.Elek S D, Stern H. Lancet. 1974;1:1–5. doi: 10.1016/s0140-6736(74)92997-3. [DOI] [PubMed] [Google Scholar]
- 19.Plotkin S A, Farquhar J, Hornberger E. J Infect Dis. 1976;134:470–475. doi: 10.1093/infdis/134.5.470. [DOI] [PubMed] [Google Scholar]
- 20.Quinnan G V, Delery M, Rook A H, Frederick W R, Epstein J S, Manischewitz J F, Jackson L, Ramsey K M, Mittal K, Plotkin S A, Hilleman M R. Ann Intern Med. 1984;101:478–483. doi: 10.7326/0003-4819-101-4-478. [DOI] [PubMed] [Google Scholar]
- 21.Jones N C, Shenk T. Cell. 1979;17:683–689. doi: 10.1016/0092-8674(79)90275-7. [DOI] [PubMed] [Google Scholar]
- 22.Baldick C J, Shenk T. J Virol. 1996;70:6097–6105. doi: 10.1128/jvi.70.9.6097-6105.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao F, Courtney R. J Virol. 1992;66:2709–2716. doi: 10.1128/jvi.66.5.2709-2716.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levy D, Larner A, Chaudhuri A, Babiss L E, Darnell J E., Jr Proc Natl Acad Sci USA. 1986;83:8929–8933. doi: 10.1073/pnas.83.23.8929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nomura N, Nagase T, Sazuka T, Tanaka A, Sato S, Seki N, Kawarabayasi Y, Ishikawa K, Tabata S. DNA Res. 1994;1:223–229. doi: 10.1093/dnares/1.5.223. [DOI] [PubMed] [Google Scholar]
- 26.Bereta J, Bereta M. Biochem Biophys Res Comm. 1995;217:363–369. doi: 10.1006/bbrc.1995.2785. [DOI] [PubMed] [Google Scholar]
- 27.Cheng Y S, Becker-Manley M F, Chow T P, Horan D C. J Biol Chem. 1985;260:15834–15835. [PubMed] [Google Scholar]
- 28.Church S L. Biochim Biophys Acta. 1990;1087:250–252. doi: 10.1016/0167-4781(90)90213-l. [DOI] [PubMed] [Google Scholar]
- 29.Kitamura A, Takahashi K, Okajima A, Kitamura N. Eur J Biochem. 1994;224:877–883. doi: 10.1111/j.1432-1033.1994.00877.x. [DOI] [PubMed] [Google Scholar]
- 30.Buwitt U, Flohr T, Bottger E C. EMBO J. 1992;11:489–496. doi: 10.1002/j.1460-2075.1992.tb05079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shiojiri S, Fukunaga R, Ichii Y, Sokawa Y. J Biochem. 1986;99:1455–1464. doi: 10.1093/oxfordjournals.jbchem.a135615. [DOI] [PubMed] [Google Scholar]
- 32.Cheng Y-S, Patterson C E, Staeheli P. Mol Cell Biol. 1991;11:4717–4725. doi: 10.1128/mcb.11.9.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aebi M, Fah J, Hurt N, Samuel C E, Thomis D, Bazzigher L, Pavlovic J, Haller O, Staeheli P. Mol Cell Biol. 1989;9:5062–5072. doi: 10.1128/mcb.9.11.5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horisberger M A, McMaster G K, Zeller H, Wathelet M G, Content J. J Virol. 1990;64:1171–1181. doi: 10.1128/jvi.64.3.1171-1181.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blomstrom D C, Fatey D, Kutny R, Korant D, Knight E. J Biol Chem. 1986;261:8811–8816. [PubMed] [Google Scholar]
- 36.Reich N, Evans B, Levy D, Fatey D, Knight E, Darnell J E., Jr Proc Natl Acad Sci USA. 1987;84:6394–6398. doi: 10.1073/pnas.84.18.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hiscott J, Nguyen H, Lin R. Semin Virol. 1995;6:161–173. [Google Scholar]
- 38.Wathen M W, Stinski M F. J Virol. 1982;41:462–477. doi: 10.1128/jvi.41.2.462-477.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDonough S H, Spector D H. Virology. 1983;125:31–46. doi: 10.1016/0042-6822(83)90061-2. [DOI] [PubMed] [Google Scholar]
- 40.McDonough S H, Staprans S I, Spector D H. J Virol. 1985;53:711–718. doi: 10.1128/jvi.53.3.711-718.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reich N, Pine R, Levy D, Darnell J E., Jr J Virol. 1988;62:114–119. doi: 10.1128/jvi.62.1.114-119.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gutch M J, Reich N C. Proc Natl Acad Sci USA. 1991;88:7913–7917. doi: 10.1073/pnas.88.18.7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalvakolanu D V R, Bandyopadhyay S K, Harter M L, Sen G C. Proc Natl Acad Sci USA. 1991;88:7459–7463. doi: 10.1073/pnas.88.17.7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fitzgerald P A, Von Wussow P, Lopez C. J Immunol. 1982;129:819–824. [PubMed] [Google Scholar]
- 45.Feldman M, Fitzgerald-Bocarsly P. J Interferon Res. 1990;10:435–446. doi: 10.1089/jir.1990.10.435. [DOI] [PubMed] [Google Scholar]
- 46.Li Q, Feldman M, Harmon C, Fitzgerald-Bocarsly P. J Interferon Cytokine Res. 1996;1:109–118. doi: 10.1089/jir.1996.16.109. [DOI] [PubMed] [Google Scholar]
- 47.Roizman B, Sears A E. In: Fields Virology. 3rd Ed. Fields B N, Knipe D M, Howley P M, editors. Philadelphia: Lippincott; 1995. pp. 2231–2295. [Google Scholar]