

Abstract
Epidemiologic studies have demonstrated that increased prolactin exposure raises the risk of invasive ERα positive breast cancer in women. However, the mechanism(s) whereby this occurs, and interactions with estrogen itself in this disease remain poorly understood. In order to investigate the role of ovarian hormones in the disease process, we employed a transgenic model (NRL-PRL) in which transgenic prolactin is directed to mammary epithelial cells by the prolactin- and estrogen-insensitive NRL promoter, mimicking the endogenous prolactin expression within the breast observed in women. This high local exposure leads to mammary lesion development and eventually carcinomas. Ovariectomy shortly after puberty did not alter the incidence or latency of prolactin-induced mammary carcinomas, consistent with the independence of prolactin from circulating estrogens as a risk factor for invasive breast cancer in women. However, chronic estrogen administration to ovariectomized NRL-PRL females decreased the latency of both ERα positive and negative tumors. We identified multiple mechanisms that may underlie this observation. Elevated estrogen exposure cooperated with prolactin to increase epithelial proliferation and myoepithelial abnormalities, increasing the incidence of preneoplastic lesions. Critical components of the extracellular matrix secreted by the myoepithelium were reduced with age, and transgenic prolactin raised transcripts for tenascin-C and maspin, both associated with tumor progression and poor prognosis in subclasses of clinical breast tumors. Mammary pERK1/2 and pAkt, but not pStat5, were markedly elevated by local prolactin. Together, these findings indicate that prolactin employs multiple mechanisms to promote mammary tumorigenesis.
Keywords: prolactin, ERα, breast cancer, transgenic mice, estrogen
Introduction
Although prolactin (PRL) is critical for physiologic development and differentiation of the mammary gland, its role in breast cancer remains poorly understood. Small early clinical studies and species differences in the sites of expression of this cytokine/ hormone, which include the mammary gland itself in women (Clevenger et al. 2003, Zinger et al. 2003), obscured its role in the human disease. However, recent epidemiologic studies and experimental models have pointed to an important role for PRL in the development and progression of breast cancer (for reviews, Arendt & Schuler 2008; Tworoger & Hankinson 2008). Moreover, the expression of the PRL receptor (PRLR) in a majority of clinical tumors (for reviews, Ginsburg & Vonderhaar 1995; Clevenger et al. 2003; Tworoger & Hankinson 2008), and limited phenotype of the PRL receptor knock-out mice apart from the mammary gland (Goffin et al. 2002), suggest that insight into the pathogenic activities of PRL may lead to preventative and therapeutic approaches with minimal side effects.
Epidemiologic studies have demonstrated a high correlation between circulating PRL and the risk of breast tumors that express estrogen receptor alpha (ERα), which is independent of levels of circulating estrogen (for review, Tworoger & Hankinson 2008). When assay variability is taken into account, PRL exposure confers a risk only slightly weaker than that for estrogen itself (Tworoger & Hankinson 2006). PRL promotes estrogenic signals in a variety of experimental systems: it increases ER expression (Edery et al. 1985; Frasor & Gibori 2003; Gutzman et al. 2004a), and cooperatively activates the AP-1 transcriptional enhancer (Gutzman et al. 2005). These activities suggest potential interactions between estrogen and PRL in the pathogenesis of this disease.
In order to study the dynamic processes whereby PRL contributes to breast cancer, we have developed a novel transgenic mouse in which PRL is directed to mammary epithelial cells by a PRL- and estrogen-independent promoter, neu-related lipocalin (NRL) (Rose-Hellekant et al. 2003; Arendt & Schuler 2008). This local expression mimics that in normal mammary tissue (Clevenger et al. 2003, Zinger et al.2003) and primary tumors in women (McHale et al.2008). NRL-PRL nonparous females develop preneoplastic lesions, and eventually diverse, aggressive tumors after a long latency, similar to the human disease. Unlike most mouse models, many of these tumors express ERα, like the majority of clinical tumors in women (Rose-Hellekant et al.2003). Both the ERα positive (ERα +) and ERα negative (ERα -) tumors that develop in these mice display transcript profiles similar to the luminal subtype of tumors observed in women (Arendt et al. ms in prep). This model permits us to investigate the effects of hormonal milieu on the multiple cell types and structures that comprise this complex tissue over time. Here we have examined the interplay of aging and estrogen exposure on PRL-induced lesions. Our findings demonstrate that PRL can induce mammary tumors even in the postpubertal absence of ovarian hormones, associated with loss of integrity of the myoepithelial layer. However, estrogen can contribute to the disease process via multiple mechanisms, including augmenting proliferation of epithelial cells, and promoting abnormalities in the myoepithelium with age. These studies elucidate important hormonal interactions in breast cancer development and progression.
Materials and methods
Reagents
5-bromo-2-deoxyuridine (BrdU) was obtained from Sigma Chemical Co. (St. Louis, MO), and 17β-estradiol (E2) was purchased from Steraloids, Inc. (Newport, RI). The following antibodies were used for immunohistochemical analyses: pStat 5 (AX1) from Advantex BioReagents, LLP (El Paso, TX), BrdU (MAS-250) from Accurate Scientific (Westbury, NY), estrogen receptor α (ERα; SC-542) from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), α-smooth muscle actin (α-SMA; A2547) from Sigma Chemical Co., ERK1/2 (9102) and phospho-ERK1/2 (Thr202/Tyr204, 9101) and Akt (9272) and pAkt S473 (9271) from Cell Signaling Technology (Beverly, MA), and cytokeratin 8 (K8; RB-9095), was obtained from Lab Vision (Fremont, CA).
Genotyping mice
FVB/N strain NRL-PRL mice (line 1647-13, TgN(Nrl-Prl)23EPS; line 1655-8, TgN(Nrl-Prl)24EPS) were generated and genotyped as described (Rose-Hellekant et al. 2003). Mice were housed and handled in accordance with the Guide for Care and Use of Laboratory Animals in AAALAC-accredited facilities, and all procedures were approved by the University of Wisconsin-Madison Animal Care and Use Committee.
Ovariectomy and treatment with 17β-estradiol
For some experiments, nonparous NRL-PRL and nontransgenic female mice were ovariectomized (ovx), subjected to sham surgery, or ovx and treated with E2 beginning at 12 weeks of age. For the latter, females received silastic capsules containing 20 μg E2, which were replaced every 6 weeks for the length of the experiment, maintaining circulating E2 levels at approximately those of estrus, as described (Medina et al. 2003). Animals were observed weekly for tumor development and considered to be end stage when tumor diameter reached 1.5 cm.
Histological examination of mammary tissue
Mammary tissue was fixed in 10% neutral buffered formalin for 18-24h, embedded in paraffin, and cut into 6 μm sections. Mice were injected with 200 mg/kg body weight BrdU 1h prior to sacrifice to label proliferating cells. Cells containing BrdU and ERα were detected using immunohistochemistry (IHC) and quantitated as described previously (Rose-Hellekant et al. 2003). For detection of pStat 5, deparaffinized slides were rehydrated, boiled in citrate buffer, pH=6.0, for antigen retrieval, blocked in 3% horse serum in PBS, and incubated with primary antibody diluted 1:750 overnight (Nevalainen et al. 2002). Signal was amplified using ImmPRESS Anti-Mouse Ig kit (MP-7402; Vector Laboratories, Burlingame, CA). Histological structures were identified as described (Rose-Hellekant et al. 2003). 1000 cells in morphologically normal ducts, alveoli, and epithelial hyperplasias were analyzed from 5 females of each genotype. Immunohistochemical indices were statistically analyzed using the Kruskal-Wallis test followed by Mann-Whitney post test.
Immunofluorescence
Deparaffinized slides were rehydrated and exposed to 0.5% H2O2 in H2O to block endogenous peroxidase activity, boiled for 15 min in 0.1 M Tris buffer, pH 9.0, for antigen retrieval, then blocked in 1:100 rabbit serum in TBST. Slides were incubated with ERα antibody (1:500) for 1 hour, rinsed, and incubated with Alexa Fluor 488-conjugated goat-anti-rabbit secondary antibody (1:400) overnight (Molecular Probes, Eugene, OR). Slides were rinsed, blocked, and incubated with BrdU (1:20), α-SMA (1:2500), or K8 (1:100) antibodies for 1 hour, rinsed, and incubated with Texas Red conjugated goat-anti-rat secondary antibody (1:400; Vector Laboratories, Burlingame, CA) for BrdU and K8 or Alexa Fluor 568-conjugated goat-anti-mouse secondary antibody (1:400; Molecular Probes, Eugene, OR) overnight. Slides were rinsed and mounted using VECTASHIELD mounting media with DAPI (Vector Laboratories, Burlingame, CA). Images were aligned and quantitated using ImageJ v. 1.36b (National Institutes of Health).
Western analyses
Western analyses were performed as described previously (Arendt et al. 2006). In brief, 30 μg of mammary gland homogenate from 12 week old nonparous females was fractionated by standard Laemmli SDS-polyacrylamide gel electrophoresis, transferred to polyvinylidine fluoride membranes and then probed with antibodies as shown. Signals were visualized by enhanced chemiluminescence, followed by autoradiography.
Quantitative real-time PCR
cDNA was synthesized from 1 μg RNA using random hexamers (Amersham Biosciences, Piscataway, NJ) and MMLV Reverse Transcriptase (Promega, Madison, WI) as described (Gutzman et al. 2007). PCR was performed in a 25 μl reaction volume of 1× SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA), 3 μl cDNA sample, and 300 nM of each primer (120 nM for 18S primers), using the primers shown in Supplementary Table 1. Reactions were cycled for 15 min at 95°C for 15 sec/60° for 1 min on the Applied Biosystems 7300 Real-time PCR system. Results were calculated using the comparative CT method and normalized to 18S RNA, and analyzed for statistical significance using the nonparametric Mann-Whitney test.
Statistical analyses
Statistical analyses were performed using Prism v.4.03 (GraphPad Software, Inc., San Diego, CA). Differences were considered significant at p<0.05.
Results
Estrogen treatment enhances PRL-induced tumorigenesis
Studies in breast cancer cells in vitro have demonstrated cooperative interactions between prolactin and estrogen at many levels. To elucidate the contribution of circulating estrogen to PRL-induced mammary tumorigenesis in vivo, we altered the ovarian hormonal environment of nonparous 12 week old NRL-PRL females and nontransgenic littermates by either ovariectomy (ovx), sham surgery, or ovx followed by continuous administration of 17-β estradiol (E2) to about normal estrus levels. Sham-treated females developed mammary carcinomas of diverse histotypes with a latency similar to our previous study (Figure 1A, Supplementary Table 2, Rose-Hellekant et al. 2003). Ovx did not alter tumor incidence or latency. However, E2 significantly shortened the latency to tumor development, without altering incidence (Supplementary Table 2, Figure 1A, p=0.02). Ovariectomized females that were continuously treated with E2 for this prolonged period exhibited generally poorer health than age-matched sham or ovx females, particularly urinary tract abnormalities (Pearse et al. 2009). This resulted in earlier mortality independent of mammary tumors than the other treatments, which may have obscured an increase in incidence with estrogen administration. Ovarian hormonal status did not favor the development of a specific tumor histotype (Supplementary Table 2). Levels of ERα expression were highly variable among tumors in all treatment groups, and also were not correlated with specific carcinoma histotypes (Figure 1B). Tumor latency for ERα+ and ERα- tumors did not differ, regardless of treatment group; E2 treatment significantly decreased latency of both tumor subclasses (p<0.05; one way ANOVA, Tukey-Kramer post test). These results suggest that although postpubertal ovarian hormones are not necessary for PRL-induced tumor development, PRL and E2 can cooperatively enhance mammary oncogenesis. Further, this interaction does not appear to be mediated by ERα within epithelial cells of established tumors, although this does not preclude a role for this site of crosstalk at an earlier stage in oncogenesis.
Figure 1.

Estrogen decreases latency to tumor development. A. NRL-PRL female mice underwent ovariectomy (ovx), sham surgery, or ovx with 17β-estradiol (E2) administration after puberty, as described in Materials and methods. Ovx females had significantly decreased uterine weights (22±14 mg; mean±s.d.) compared to sham treated females (96±34 mg), while those from E2 treated females were significantly increased (208±47 mg). E2-treated NRL-PRL females developed mammary carcinomas with significantly shorter latencies compared to other treatment groups (p=0.02). B. Estrogen availability did not alter the proportion of ERα positive tumors. The percentage of ERα positive cells in mammary carcinomas from NRL-PRL females was determined as described in Materials and Methods. Each circle denotes an individual tumor.
Estrogen and PRL augment the development of preneoplastic lesions
To determine the interactions of these factors in lesion development, we histologically examined glands of nontransgenic and NRL-PRL females subjected to these prolonged manipulations of estrogen levels. Both NRL-PRL and nontransgenic females developed multiple abnormalities with age, with substantial variability among individuals (Table 1). Interestingly, epithelial hyperplasias (EH) were found in more intact females of both genotypes, suggesting a role for other ovarian factors, such as progesterone. We have previously shown that the EH that develop in NRL-PRL females express higher levels of ERα than those in nontransgenic females (Rose-Hellekant et al. 2003; Arendt et al. 2006). PRL-induced EH exposed to sustained E2 in the current study exhibited a higher rate of proliferation, as assessed by the proportion of cells exhibiting BrdU incorporation (ovx: 6.9±0.3%; ovx+E2: 9.9±0.6%; mean +/- s.e.m., p<0.0001 by Student's t test). In addition, mammary intraepithelial neoplasias (MIN), which resemble ductal carcinoma in situ (DCIS) in women, commonly developed in NRL-PRL females with all treatments, but not nontransgenic females. The incidence of these lesions was significantly enhanced by E2 (p=0.03; Table 1).
Table 1.
Histologic mammary abnormalities in aged1 NRL-PRL and nontransgenic FVB/N females.
| NRL-PRL | FVB/N | |||||
|---|---|---|---|---|---|---|
| ovx | sham | ovx-E2 | ovx | sham | ovx-E2 | |
| Epithelial Hyperplasias | 7/33 | 13/25* | 10/30 | 2/9 | 7/9* | 1/8 |
| MIN2 | 9/33† | 12/25† | 21/30*† | 0/9 | 0/9 | 0/8 |
| Squamous Changes | 3/33* | 9/25 | 9/30 | 2/9 | 5/9 | 1/8 |
| Carcinoma | 10/33† | 10/25† | 11/30† | 1/9 | 0/9 | 1/8 |
NRL-PRL females were 19.3±7.1 months old, when the tumor reached 1.5 cm in diameter; nontransgenic FVB/N females were examined at 24 months of age in the absence of tumor development.
MIN, mammary intraepithelial neoplasia.
, significantly different than in other treatment groups of the same genotype using the chi-square test, P<0.05.
, significantly higher in NRL-PRL females than in non-transgenic littermates in the same treatment groups using the chi-square test, p<0.05.
These data indicate that postpubertal ovarian hormones are not required for the development of PRL-induced mammary lesions, similar to effects on tumorigenesis. Consistent with previous reports in this species (Shimkin 1948; Nandi et al. 1995), estrogen alone did not induce lesions in nontransgenic females. However, E2 can enhance the development and/or proliferation of PRL-induced preneoplastic lesions, which may contribute to the reduction in tumor latency observed.
Epithelial proliferation increases with age and E2 exposure
Estrogen is believed to regulate proliferation via paracrine factors, since ERα-expressing cells in women or mouse models rarely contain evidence of this activity (Clarke et al. 1997; Shyamala et al. 2002). However, as women age, the proportion of cells that express ERα increases and colocalization of ERα with proliferation markers occurs more frequently (Shoker et al. 1999). In order to examine these events in the aging mouse and effects of long term alterations in PRL and E2 exposure on this process, we quantified mammary proliferation using BrdU incorporation, and ERα expression by immunohistochemistry in nontransgenic and NRL-PRL nonparous females at 6 and 24 months of age. As shown in Figure 2A, proliferation of ductal epithelium was significantly higher in older nontransgenic females, compared to those at 6 months of age. Manipulation of available estrogen by either ovx or ovx followed by continuous administration of E2 demonstrated a strong dependence on available estrogen. Consistent with our earlier reports (Arendt & Schuler 2008), transgenic PRL increased ductal proliferation (Figure 2A vs 2B). The rate in NRL-PRL glands was significantly greater than that in nontransgenic glands at both ages and treatments, except for the elderly sham-treated animals. The reasons for this are unclear; these data may reflect inhibitory factors produced by the intact aged ovary.
Figure 2.

Aging significantly enhances proliferation and ERα expression in morphologically normal structures in glands of mice. A. Proliferation was significantly enhanced with aging in ducts of nontransgenic FVB/N (FVB) mice regardless of treatment. B. Ducts in glands from 6 month old sham treated NRL-PRL (PRL) females demonstrated similar levels of proliferation as those of 2 year old females. C. Long-term ovx or ovx plus E2 significantly enhanced ERα expression in ducts of nontransgenic females. D. ERα expression was enhanced with E2 treatment in NRL-PRL females, similar to nontransgenic females. BrdU and ERα labeled cells were detected and quantitated as described in Materials and methods, and expressed as mean ± s.d. Data were analyzed by the Kruskal-Wallis test followed by Mann-Whitney post test. Different lower and uppercase letters denote statistical differences among treatments, within 6 month old and end stage animals, respectively, for each genotype (p<0.05). Asterisks denote differences with age for the same treatment of the same genotype (*p<0.05, **p<0.01, ***p<0.001). Proliferation of ducts in NRL-PRL females was higher than all age-matched FVB/N animals with the same treatment, with the exception of the sham-treated elderly females (p<0.005).
Aging alters ERα expression
Although proliferation was enhanced, ERα levels were not significantly altered with aging in sham-treated nontransgenic (Figure 2C) or NRL-PRL females (Figure 2D). Exposure to ligand downregulates ERα (Alarid 2006); consistently, E2-treated ovx females of both genotypes exhibited lower numbers of ERα-expressing cells than ovx females at both 6 months and 2 years of age. However, the extent of this reduction was substantially greater in the younger females: E2-treatment reduced ERα by about 50% at 6 months in both genotypes, but less than 25% in aged females. This dysregulation of proliferation and ERα expression with age resembles reports in women (Clarke et al. 1997), and suggests that ERα-expressing cells may also proliferate more frequently in aged mice. To directly examine this question, we examined colocalization of BrdU with ERα expression using immunofluorescence. However, any effect was obscured by high variability among individuals (data not shown).
Prolonged E2 administration alters the pattern of ERα expression in elderly mice, and reduces the integrity of the myoepithelial layer
In light of the reduced downregulation of ERα in the elderly ovx mice receiving E2, we investigated the location of this receptor. In the normal mammary epithelium, ERα and progesterone receptor (PR) are expressed together within luminal epithelial cells (for review, Anderson et al. 2000; Shyamala et al. 2002). In glands of elderly ovx and sham-treated females of both genotypes, as well as six month old females regardless of genotype or treatment, ERα-labeling was detected in a subset of ductal cells (Figure 3A). In these glands, ERα-labeled nuclei (green) were centrally located in cells expressing the luminal marker, cytokeratin 8 (K8; red, Figure 3C). PR was similarly located (not shown). The ducts in these females were surrounded by a continuous layer of myoepithelial cells, detected with the marker, α-smooth muscle actin (α-SMA; Figure 3E). However, long-term E2 administration to ovx females shifted ERα (Figure 3B) expression to a more basal pattern by 2 years of age. Glands of NRL-PRL females exhibited a similar pattern (data not shown), indicating that this was independent of PRL status. Despite this altered position, ERα-staining cells also expressed K8, indicating that these cells were of the luminal lineage (Figure 3D). However, in these aged E2-treated females, the α-SMA staining was no longer continuous around the ducts, suggesting abnormalities in the myoepithelial layer (Figure 3F).
Figure 3.
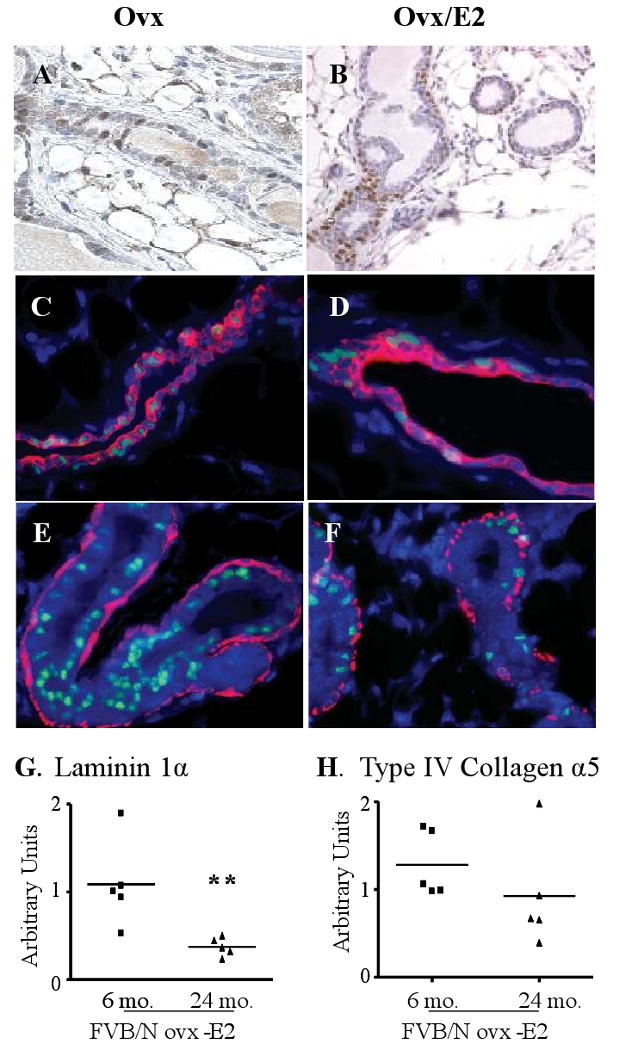
Lifetime administration of 17β-estradiol alters the pattern of ERα expression in nontransgenic ovx nonparous aged females. A. Luminal ERα expression in morphologically normal ducts of a 2 year old ovariectomized (ovx) nontransgenic female (similar to 6 month old ovx, and ovx-E2 treated females). B. Basilar ERα expression in morphologically normal ducts of a 2 year old E2-treated ovx nontransgenic female. ERα-labeled cells (green) colocalize with cytokeratin 8 (K8; red) expression in ducts of ovx (C) and ovx/E2-treated (D) nontransgenic females. ERα labeled cells do not colocalize with cells expressing α-smooth muscle actin (α-SMA; red) in ducts of ovx (E) and ovx/ E2-treated (F) nontransgenic females. Representative micrographs. Original magnification: A, B, 400×; C-F, 600×. Prolonged 17β-estradiol administration reduces laminin 1α (G) and collagen IV α5 (H) transcripts with age. Nontransgenic FVB/N nonparous females were ovx post-pubertally, and thereafter were administered E2, as described in the Materials and Methods. RNA from whole mammary glands of 6 and 24 month old animals was examined for the myoepithelial ECM products by qRT-PCR as described in the Materials and methods. Each symbol denotes a single animal. Asterisks indicate the significant difference in laminin 1α mRNA between the 6 month and 24 month old animals (p=0.007; Student's t test).
Myoepithelial cells surround the ducts and alveoli, forming a structural barrier between the luminal epithelial cells and surrounding stroma. They secrete components of the basement membrane, which contribute to signals maintaining epithelial integrity and hormonal responsiveness, including ERα expression (Gudjonsson et al. 2002; Novaro et al. 2003; Haslam & Woodward 2003; Naylor et al. 2005). In order to determine if the apparent abnormalities in the myoepithelial layer of E2-treated aged females were associated with altered levels of key components of the extracellular matrix (ECM), we compared levels of transcripts for some of the molecules implicated in these activities using qRT-PCR. Laminin 1α mRNA, which is expressed exclusively by myoepithelial cells (Kenny & Bissell 2003), was significantly decreased in glands of E2-treated 2 year old nontransgenic females, compared to those of 6 month old females with the same treatment, consistent with the morphologic changes (Figure 3G). Similarly, collagen type IV 5a mRNA was strikingly reduced in 4/5 of the glands of E2-treated nontransgenic females with age (Figure 3H).
PRL alone accelerates loss of the myoepithelial layer, associated with increases in transcripts of genes implicated in tumor invasion
In order to determine if PRL can modulate the integrity of the myoepithelial layer, we compared the effect of age and E2 availability in nontransgenic females and NRL-PRL mice. Nontransgenic mice at 6 months of age with all treatments demonstrated continuous α-SMA staining (Figure 4A-C), which was only slightly reduced in the 24 month old ovx and sham-treated females (Figure 4D, E). However, as noted above, α-SMA staining in E2-treated ovx aged FVB/N females demonstrated generalized discontinuities (Figure 3F, 4F). In contrast, although glands of 6 month old ovx NRL-PRL mice displayed normal α-SMA distribution (Figure 4G), glands of both sham- and E2-treated NRL-PRL females were already abnormal at this age (Figure 4H, I). In the older NRL-PRL females, α-SMA staining became markedly abnormal with all treatments (Figure 4J-L). The reduction in the myoepithelial α-SMA-expressing cells relative to K8-expressing luminal cells was quantitatively confirmed by western analyses (Figure 5A).
Figure 4.
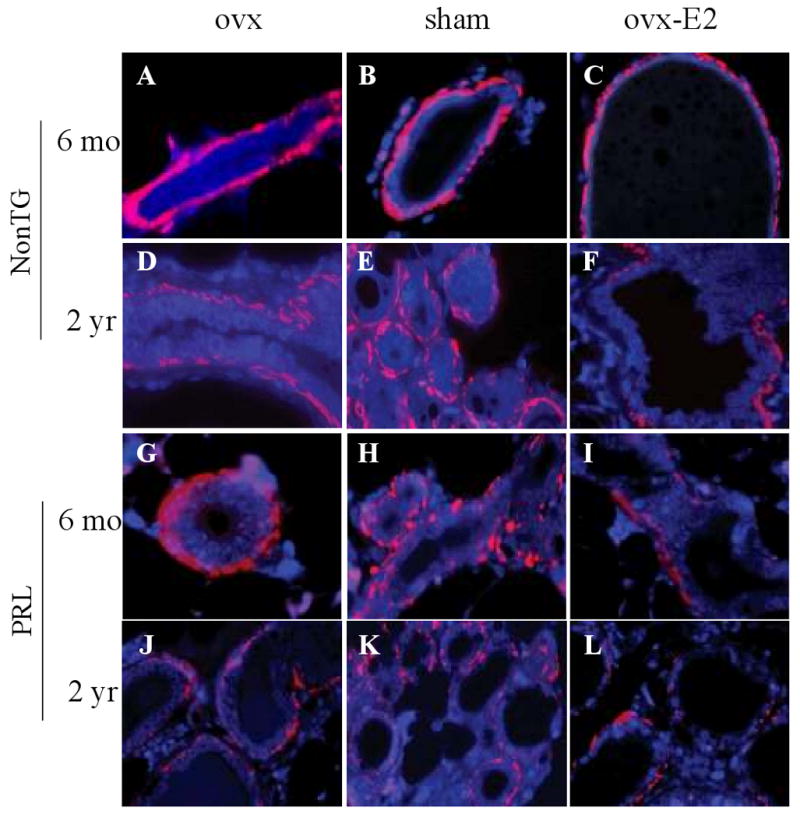
PRL and 17β-estradiol diminish the myoepithelial layer surrounding morphologically normal structures. At 6 months of age, ovx (A), sham treated (B), and ovx/E2-treated (C) nontransgenic mice demonstrated normal α-SMA staining around ducts. At 2 years of age, ovx/E2-treated (F) nontransgenic females exhibited decreased α-SMA staining, compared to those subjected to ovx alone (D) or sham surgery (E). Abnormalities in α-SMA staining were present in sham- (H) and ovx/E2- (I) treated NRL-PRL females at 6 months of age, compared to ovx NRL-PRL females (G) of the same age. Pronounced abnormalities were present in all treatment groups at 2 years of age (J, K, L) in NRL-PRL females, with the greatest discontinuities in the E2-treated group (L). Morphologically normal structures were stained for α-SMA (red) and counterstained with DAPI (blue) as described in Materials and methods. Representative micrographs. Original magnification of all images: 600×.
Figure 5.

Transgenic PRL reduces the relative level of α-SMA: keratin 8 protein expression, and raises mammary tenascin C and maspin transcripts, without altering fibronectin 1. Protein homogenates and RNA from whole mammary glands of sham-treated 2 year old nontransgenic (FVB) and NRL-PRL nonparous females were examined for levels of α-SMA and keratin 8 protein by western analyses (A), and myoepithelial products by qRT-PCR (B, C, D), as described in the Materials and methods. A. Data are expressed as the ratio between α-SMA and keratin 8 staining (mean ± s.d., N=3). B, C, D. Data are expressed as mean ± s.e.m., N=5. Asterisk indicate a significant difference between genotypes by Student's t test (*, p<0.05; ***, p<0.0001).
Myoepithelial cells also secrete many factors including protease and angiogenic inhibitors which inhibit invasion and vascularization, contributing to their natural tumor suppressor activity (for reviews, (Polyak & Hu 2005, Gudjonsson et al. 2005, Faraldo et al. 2005, Barsky & Karlin 2005). Because of the pronounced PRL-induced disruption of this layer, we compared levels of transcripts for some of the implicated mediators using qRT-PCR in aged sham-treated NRL-PRL and nontransgenic females. Tenascin-C is a component of the ECM which interacts with integrins. Its expression is increased upon invasion through the basement membrane, and therefore it is used as a stromal marker for epithelial malignancies of various organs, including the breast (for reviews, Jones 2001; Orend & Chiquet-Ehrismann 2006). As shown in Figure 5B, transcripts for this glycoprotein were elevated in aged NRL-PRL glands. Maspin is a serine protease inhibitor secreted by myoepithelial cells. Maspin is considered to be a component of myepithelial tumor suppressor activity because it can inhibit invasion and angiogenesis and promote apoptosis (Barsky & Karlin 2005; Bailey et al. 2006). Surprisingly, maspin transcripts were also elevated in the NRL-PRL glands compared to nontransgenic glands (Figure 5C). In contrast, levels of fibronectin mRNA, another ECM component synthesized by the stroma rather than myoepithelial cells, were not altered by PRL (Figure 5D).
PRL increases phosphorylation of multiple signaling mediators
In order to better understand the signaling pathways whereby local PRL may contribute to these pathogenic processes, we examined mammary glands of 12 week old nonparous females, prior to the development of detectable lesions. Stat5 is a critical mediator of PRL in alveologenesis, and mammary overexpression of Stat5 in mouse models reduces apoptosis at involution, and eventually results in tumors. Furthermore, high levels of tyrosine phosphorylated Stat5 (pStat5) are found in a subset of well-differentiated clinical tumors (Wagner & Rui 2008). To determine if transgenic local PRL elevates pStat5, we examined glands of 12 week old nonparous intact females, prior to the development of any visible pathology. Surprisingly, nontransgenic and NRL-PRL females exhibited similar levels of pStat5 in mammary lysates by immunoblotting (data not shown). Even at 6 months of age, nuclear pStat5 was detectable in similar proportions of MECs in nontransgenic and NRL-PRL glands by IHC (Figure 6A). However, when glands of aged (18-24 month old) NRL-PRL females containing plentiful preneoplastic lesions were examined, levels of pStat5 were significantly higher in EH than morphologically normal structures (Figure 6B), suggesting a role for this pathway in the development of some lesions.
Figure 6.

NRL-PRL glands exhibit elevated pStat5, pERK1/2 and pAkt. A, B. Phosphorylated Stat5 (pStat5) was examined in morphologically normal structures of age-matched 6 month old nontransgenic and NRL-PRL intact nonparous females (A), and normal structures and epithelial hyperplasias (EH) in glands of 2 year old NRL-PRL females (B). pStat5-labeled cells were detected and quantitated as described in Materials and methods and expressed as mean ± s.d. Lower case letters denote significant differences using ANOVA followed by Student-Newman-Keuls Multiple Comparison test (p<0.05). C. Glands of NRL-PRL females at 12 weeks of age contain elevated pERK1/2 and pAkt, compared to nontransgenic littermates. Each lane represents mammary homogenate from a single 12 week old NRL-PRL female or nontransgenic littermate (FVB), examined by immunoblotting as shown. D. The ratio of phosphorylated: total kinase for each animal shown in (C) was calculated as described in the methods. An asterisk indicates a significant difference between genotypes by a one-tailed Student's t test (*, p<0.05).
PRL also activates other downstream mediators that have been implicated in breast cancer, and displays positive crosstalk with growth factors to these signaling cascades (Arendt et al. 2006; Carver & Schuler 2008; Arendt et al. 2009). Approximately half of primary breast tumors in women express more activated ERK1/2 than surrounding tissue, although the relationship to clinical outcome is not clear (Santen et al. 2002; Milde-Langosch et al. 2005). Activation of the PI3K-Akt pathway also has been associated with breast cancer and anti-estrogen resistance (for reviews, Liu et al. 2007; Dillon et al. 2007). As shown in Figure 6C, D, glands from 12 week old NRL-PRL mice exhibited enhanced levels of both pERK1/2 and pAkt, compared to nontransgenic littermates. Both pERK1/2 and pAkt were detected by immunohistochemistry in both epithelial cells and stroma, although pERK1/2 tended to be stronger in stroma (data not shown). These data suggest that PRL signals to these non-Jak2/ Stat5 pathways also may contribute to early changes leading to tumor development.
Discussion
While increased PRL exposure raises the risk of invasive ERα+ breast cancer in women, the mechanism(s) whereby this occurs, and interactions with estrogen itself in this disease remain poorly understood. Using our mouse model of augmented mammary PRL exposure, we demonstrated that post-pubertal ovarian hormones are not necessary for PRL-induced lesion and tumor development, consistent with the independence of PRL as a risk factor for invasive breast cancer independent of circulating estrogens in women (Tworoger & Hankinson 2006). However, elevated estrogen exposure cooperated with PRL to increase epithelial proliferation and myoepithelial abnormalities, increasing the incidence of preneoplastic lesions, and decreasing the latency of both ERα+ and ERα- mammary tumors. Critical components of the ECM secreted by the myoepithelium were reduced with age, and transgenic PRL raised transcripts for tenascin-C and maspin, both associated with tumor progression and poor prognosis in subclasses of clinical breast tumors. These studies suggest that myoepithelial cells may be important and understudied effectors of hormonal contributions to breast cancer, which may become increasingly important with age.
The potent oncogenic activity of PRL at the mammary gland is underscored by the ability of local overexposure to promote tumors even in the absence of postpubertal ovarian hormones. However, chronic estrogen exposure significantly reduced the latency of PRL-induced tumors. Lifetime estrogen exposure is one of the strongest risk factors for development of invasive breast cancer in women (The Endogenous Hormones and Breast Cancer Collaborative Group 2002,Writing Group for the Women's Health Initiative Investigators 2002; Eliassen et al. 2006). Although E2 alone could not elicit preneoplastic hyperplasias or MINs, as previously observed in this species (Shimkin 1948; Nandi et al. 1995), our studies revealed multiple mechanisms by which E2 can accelerate PRL-induced lesion development. ERα and PRLR are expressed in both mammary epithelial and stromal cells (Bera et al. 1994; Hovey et al. 2001; Mueller et al. 2002); their coexpression in many epithelial cells (Grimm et al. 2002) suggests that intracellular crosstalk may contribute to this outcome (Gutzman et al. 2005; Silva & Shupnik 2007). The ability of E2 to reduce the latency of both PRL-induced ERα+ and ERα- tumors indicates that direct action on established tumor cells themselves is not required. However, the E2-induced increases in lesions and proliferation of normal and hyperplastic structures suggest that intracellular crosstalk may be important early, but may be reduced with progression and loss of ERα expression in some tumors. The significance of the E2-induced restriction of ERα expression to a distinct subpopulation of luminal cells with age is not known. Green and colleagues noted a similar location of ERα in MINs induced by transgenic C3(1)SV40TAg (Yoshidome et al. 2000). Prolonged chronic exposure to estrogen may promote expansion and/or stability of a distinct epithelial subpopulation. Some evidence suggests that a subpopulation of basally located cells may have progenitor cell characteristics (for reviews, Smith et al. 1984; Smith & Chepko 2001), although any relationship to ERα expression is controversial (Booth & Smith 2006; Asselin-Labat et al. 2006; Sleeman et al. 2007). The growing repertoire of tools to characterize these discrete cell subpopulations will enable further characterization of the ERα+ cells identified here, and implications for actions of other neoplastic factors, such as PRL. E2 also enhanced the development of age-dependent abnormalities in the myoepithelial layer, as discussed further below. Although myoepithelial cells do not express ERα, they do express ERβ (Barsky & Karlin 2005), suggesting possible direct and/or indirect effects on these cells. Finally, E2 may promote mammary lesions via targets outside the mammary gland. Emerging evidence from several laboratories has demonstrated that elevated systemic E2 can mobilize bone marrow-derived stromal and endothelial progenitors to enhance the growth, stromal infiltration, and vascularization of transplanted mammary tumors, including those that do not express ER (Gupta et al. 2007; Suriano et al. 2008).
The myoepithelial layer is an important determinant of the mammary microenvironment, secreting components of the ECM that modulate responsiveness to many endocrine/ paracrine factors, including hormones. The importance of these cells in maintaining epithelial organization and preventing invasion and migration has earned them recognition as natural tumor suppressors (Polyak & Hu 2005; Barsky & Karlin 2005; Adriance et al. 2005). We found that the structural integrity of this layer diminished with age, associated with lower expression of laminin-1α and collagen IV. These key components of the ECM not only regulate hormonal responsiveness of the breast (Novaro et al. 2003; Haslam & Woodward 2003; Naylor et al. 2005), but also decline with tumorigenesis (Nakano et al. 1999, Maatta et al. 2001; Gudjonsson et al. 2002). Local PRL dramatically hastened the loss of this layer. PRL elicited these changes even in ovx females, although estrogen could enhance the loss. WAP-PRL Balb/c females, in which transgenic PRL expression is elevated during pregnancy, also develop myoepithelial abnormalities (Manhes et al. 2006). The ability of PRL to induce this pathologic feature in markedly different hormonal milieus in distinct mouse strains confirms the potency of this action.
In the current study, PRL-induced elevation of the myoepithelial transcripts, tenascin C and maspin, was readily apparent even in highly heterogeneous whole mammary glands. Tenascin-C is strongly expressed in the stroma in areas of wound healing, inflammation and neoplasia (for reviews, (Jones 2001; Orend & Chiquet-Ehrismann 2006). Its activation of integrin-mediated signals influences growth factor receptor expression and activation, and the cytoskeleton. It can also augment vascularization, associated with increased expression of proteases. These activities are consistent with its association with increased metastases in mouse models (Calvo et al. 2008) and clinical tumors (Minn et al. 2005). The PRL-induced increase in its expression in our studies indicates functional breeches in the barrier imposed by the myoepithelium and basement membrane, and suggests that this may contribute to lesion progression. Our finding that maspin transcripts also were elevated in NRL-PRL glands is in seeming conflict with the portrait of this gene as a tumor suppressor (Barsky & Karlin 2005; Bailey et al. 2006). However, recent reports indicate that elevated maspin mRNA may be a negative prognostic indicator for patients with node negative breast cancer (Bieche et al. 2003; Tsoli et al. 2007), suggesting a more complicated biological role for this protein. Interestingly, other transcripts elevated in myoepithelial cells of DCIS lesions in women (Polyak & Hu 2005), including IGF-2, SOCS-3 and the AP-1 proteins, JunD and c-Fos, have been reported to be elevated by PRL at mammary targets (Tam et al. 2001; Brisken et al. 2002; Hovey et al. 2003; Gutzman et al. 2004b). Together, our data suggest that effects on myoepithelial cells may contribute to the oncogenic actions of PRL. The exacerbation of these myoepithelial abnormalities with age and estrogen exposure supports further investigation of the effect of post-menopausal hormonal therapies on this mammary subpopulation.
Our examination of activated mediators revealed that prolonged exposure to elevated PRL enhances a complex matrix of signaling pathways. Surprisingly, morphologically normal structures in NRL-PRL glands did not display elevated pStat5, the well-characterized mediator of most PRL-induced physiologic mammary growth and differentiation (Wagner & Rui 2008). This difference from the response to acute administration (Nevalainen et al. 2002) may result from the temporally constant exposure in this transgenic model, or a shift in the spectrum of pathways activated by autocrine PRL. The modest rise in pStat5 in HE in elderly NRL-PRL females suggests that this pathway may play a role in the proliferation of these preneoplastic lesions (Wagner & Rui 2008). However, our observation that postpubertal ovarian hormones are not required for PRL-induced lesions, in combination with the recent report that ovarian hormones, but not PRL, are necessary for mammary Stat5 expression (Santos et al. 2008), suggests that Stat5 is not the primary mediator of PRL-promoted oncogenesis. In contrast, transgenic PRL markedly elevated pERK1/2 and pAkt in glands of relatively young females (12 weeks old). These kinases are major downstream effectors of PRL in some breast cancer cell lines in vitro (Gutzman et al. 2007); in the intact mammary gland, the elevated phosphorylation that we observed may indicate direct activation via the PRLR, or indirect stimulation via secondary paracrine factors. Of note, elevated tenacsin-C facilitates ERK1/2 phosphorylation, which can itself stimulate tenascin-C expression (Orend & Chiquet-Ehrismann 2006). Although myoepithelial PRLR has not been reported, identification of target cells that may express low levels of this receptor has been hampered by the lack of antibodies which recognize the murine PRLR. Both ERK1/2 and Akt are nodes of potent crosstalk between PRL and other oncogenic growth factors, such as TGFα/EGF (Arendt et al. 2006; Huang et al. 2006; Arendt et al. 2009) and IGF-1 (Carver & Schuler 2008) in breast cancer cells in vitro. These kinases are implicated in many processes in mammary tumorigenesis and progression, including proliferation, survival, the epithelial to mesenchymal transition and altered relationship with the ECM, and invasion (Vivanco & Sawyers 2002; Sebolt-Leopold & Herrera 2004; Turley et al. 2008), as well as treatment resistance (Svensson et al. 2005; Liu et al. 2007).
Transgenic PRL promotes development of murine mammary tumors that resemble the luminal-type tumors in women (Arendt et al., ms in prep). Its ability to potently stimulate proliferation of luminal epithelial cells (Rose-Hellekant et al. 2003; Arendt et al. 2006; Oakes et al. 2007) suggests one component of its oncogenic activity. Our data herein indicate that effects on nonluminal components, including the myoepithelial cell layer, may also play roles in disease processes. In clinical oncogenesis, PRL is likely to cooperate with other neoplastic factors, including estrogen, as shown herein, as well as growth factors (Arendt et al. 2006). Interestingly, PRL in combination with TGFα reduces responsiveness to E2 in mouse models in vivo (Arendt et al. 2009), pointing to ill-understood complexities among these factors. Understanding the actions of PRL and the interplay with other factors implicated in mammary tumorigenesis will lead to improved preventative and therapeutic strategies for this pervasive and devastating disease.
Supplementary Material
Supplementary Table 1. Primers employed for qRT-PCR analyses
Supplementary Table 2. Mammary tumor latency and histotype in nonparous NRL-PRL mice.
Acknowledgments
Funding: This publication was made possible by Grant Number K01-RR021858 (L.M.A.) from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), RO1- CA78312 (L.A.S.) and R01-CA101841 and R01-CA118740 (H.R.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NCRR or NIH.
The authors thank Tara Grafwallner-Huseth for assistance with sample collection and analysis, and Drs. Kathleen O'Leary and Caroline Alexander for helpful discussions.
Footnotes
Declarations of interest: The authors have no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Publisher's Disclaimer: Disclaimer. This is not the definitive version of record of this article. This manuscript has been accepted for publication in Journal of Endocrinology, but the version presented here has not yet been copy edited, formatted or proofed. Consequently, the Society for Endocrinology accepts no responsibility for any errors or omissions it may contain.
References
- Adriance MC, Inman JL, Petersen OW, Bissell MJ. Myoepithelial cells: good fences make good neighbors. Breast Cancer Research. 2005;7:190–197. doi: 10.1186/bcr1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarid ET. Lives and times of nuclear receptors. Molecular Endocrinology. 2006;20:1972–1981. doi: 10.1210/me.2005-0481. [DOI] [PubMed] [Google Scholar]
- Anderson E, Clarke RB, Howell A. Estrogen receptor in mammary gland physiology. In: Ethier SP, editor. Contemporary Endocrinology: Endocrine Oncology. Totowa, NJ: Humana Press Inc; 2000. pp. 1–16. [Google Scholar]
- Arendt LM, Grafwallner-Huseth TL, Schuler LA. Prolactin-growth factor crosstalk reduces mammary estrogen responsiveness despite elevated ERα expression. American Journal of Pathology. 2009;174:1065–1074. doi: 10.2353/ajpath.2009.080719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt LM, Rose-Hellekant TA, Sandgren EP, Schuler LA. Prolactin potentiates TGFα induction of mammary neoplasia in transgenic mice. American Journal of Pathology. 2006;168:1365–1374. doi: 10.2353/ajpath.2006.050861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt LM, Schuler LA. Transgenic models to study actions of prolactin in mammary neoplasia. Journal of Mammary Gland Biology and Neoplasia. 2008;13:29–40. doi: 10.1007/s10911-008-9073-9. [DOI] [PubMed] [Google Scholar]
- Asselin-Labat ML, Shackleton M, Stingl J, Vaillant F, Forrest NC, Eaves CJ, Visvader JE, Lindeman GJ. Steroid hormone receptor status of mouse mammary stem cells. Journal of the National Cancer Institute. 2006;98:1011–1014. doi: 10.1093/jnci/djj267. [DOI] [PubMed] [Google Scholar]
- Bailey CM, Khalkhali-Ellis Z, Seftor EA, Hendrix MJ. Biological functions of maspin. Journal of Cellular Physiology. 2006;209:617–624. doi: 10.1002/jcp.20782. [DOI] [PubMed] [Google Scholar]
- Barsky SH, Karlin NJ. Myoepithelial cells: autocrine and paracrine suppressors of breast cancer progression. Journal of Mammary Gland Biology and Neoplasia. 2005;10:249–260. doi: 10.1007/s10911-005-9585-5. [DOI] [PubMed] [Google Scholar]
- Bera TK, Hwang S, Swanson SM, Guzman RC, Edery M, Nandi S. In situ localization of prolactin receptor message in the mammary glands of pituitary-isografted mice. Molecular and Cellular Biochemistry. 1994;132:145–149. doi: 10.1007/BF00926923. [DOI] [PubMed] [Google Scholar]
- Bieche I, Girault I, Sabourin JC, Tozlu S, Driouch K, Vidaud M, Lidereau R. Prognostic value of maspin mRNA expression in ER alpha-positive postmenopausal breast carcinomas. British Journal of Cancer. 2003;88:863–870. doi: 10.1038/sj.bjc.6600812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth BW, Smith GH. Estrogen receptor-alpha and progesterone receptor are expressed in label-retaining mammary epithelial cells that divide asymmetrically and retain their template DNA strands. Breast Cancer Research. 2006;8:R49. doi: 10.1186/bcr1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brisken C, Ayyannan A, Nguyen C, Heineman A, Reinhardt F, Tan J, Dey SK, Dotto GP, Weinberg RA. IGF-2 is a mediator of prolactin-induced morphogenesis in the breast. Developmental Cell. 2002;3:877–887. doi: 10.1016/s1534-5807(02)00365-9. [DOI] [PubMed] [Google Scholar]
- Calvo A, Catena R, Noble MS, Carbott D, Gil-Bazo I, Gonzalez-Moreno O, Huh JI, Sharp R, Qiu TH, Anver MR, Merlino G, Dickson RB, Johnson MD, Green JE. Identification of VEGF-regulated genes associated with increased lung metastatic potential: functional involvement of tenascin-C in tumor growth and lung metastasis. Oncogene. 2008;27:5373–5384. doi: 10.1038/onc.2008.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver KC, Schuler LA. Prolactin does not require insulin-like growth factor (IGF) intermediates, but synergizes with IGF-1 in human breast cancer cells. Molecular Cancer Research. 2008;6:634–643. doi: 10.1158/1541-7786.MCR-07-2069. [DOI] [PubMed] [Google Scholar]
- Clarke RB, Howell A, Potten CS, Anderson E. Dissociation between steroid receptor expression and cell proliferation in the human breast. Cancer Research. 1997;57:4987–4991. [PubMed] [Google Scholar]
- Clevenger CV, Furth PA, Hankinson SE, Schuler LA. Role of prolactin in mammary carcinoma. Endocrine Reviews. 2003;24:1–27. doi: 10.1210/er.2001-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon RL, White DE, Muller WJ. The phosphatidyl inositol 3-kinase signaling network: implications for human breast cancer. Oncogene. 2007;26:1338–1345. doi: 10.1038/sj.onc.1210202. [DOI] [PubMed] [Google Scholar]
- Edery M, Imagawa W, Larson L, Nandi S. Regulation of estrogen and progesterone receptor levels in mouse mammary epithelial cells grown in serum-free collagen gel cultures. Endocrinology. 1985;116:105–112. doi: 10.1210/endo-116-1-105. [DOI] [PubMed] [Google Scholar]
- Eliassen AH, Missmer SA, Tworoger SS, Spiegelman D, Barbieri RL, Dowsett M, Hankinson SE. Endogenous steroid hormone concentrations and risk of breast cancer among premenopausal women. Journal of the National Cancer Institute. 2006;98:1406–1415. doi: 10.1093/jnci/djj376. [DOI] [PubMed] [Google Scholar]
- Faraldo MM, Teuliere J, Deugnier MA, Taddei-De La Hosseraye I, Thiery JP, Glukhova MA. Myoepithelial cells in the control of mammary development and tumorigenesis: data from genetically modified mice. Journal of Mammary Gland Biolology and Neoplasia. 2005;10:211–219. doi: 10.1007/s10911-005-9582-8. [DOI] [PubMed] [Google Scholar]
- Frasor J, Gibori G. Prolactin regulation of estrogen receptor expression. Trends in Endocrinology and Metabolism. 2003;14:118–123. doi: 10.1016/s1043-2760(03)00030-4. [DOI] [PubMed] [Google Scholar]
- Ginsburg E, Vonderhaar BK. Prolactin synthesis and secretion by human breast cancer cells. Cancer Research. 1995;55:2591–2595. [PubMed] [Google Scholar]
- Goffin V, Binart N, Touraine P, Kelly PA. Prolactin: The new biology of an old hormone. Annual Review of Physiology. 2002;64:47–67. doi: 10.1146/annurev.physiol.64.081501.131049. [DOI] [PubMed] [Google Scholar]
- Grimm SL, Seagroves TN, Kabotyanski EB, Hovey RC, Vonderhaar BK, Lydon JP, Miyoshi K, Hennighausen L, Ormandy CJ, Lee AV, Stull MA, Wood TL, Rosen JM. Disruption of steroid and prolactin receptor patterning in the mammary gland correlates with a block in lobuloalveolar development. Molecular Endocrinology. 2002;16:2675–2691. doi: 10.1210/me.2002-0239. [DOI] [PubMed] [Google Scholar]
- Gudjonsson T, Adriance MC, Sternlicht MD, Petersen OW, Bissell MJ. Myoepithelial cells: their origin and function in breast morphogenesis and neoplasia. Journal of Mammary Gland Biology and Neoplasia. 2005;10:261–272. doi: 10.1007/s10911-005-9586-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudjonsson T, Ronnov-Jessen L, Villadsen R, Rank F, Bissell MJ, Petersen OW. Normal and tumor-derived myoepithelial cells differ in their ability to interact with luminal breast epithelial cells for polarity and basement membrane deposition. Journal of Cell Science. 2002;115:39–50. doi: 10.1242/jcs.115.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PB, Proia D, Cingoz O, Weremowicz J, Naber SP, Weinberg RA, Kuperwasser C. Systemic stromal effects of estrogen promote the growth of estrogen receptor-negative cancers. Cancer Research. 2007;67:2062–2071. doi: 10.1158/0008-5472.CAN-06-3895. [DOI] [PubMed] [Google Scholar]
- Gutzman JH, Miller KK, Schuler LA. Endogenous hPRL and not exogenous hPRL induces ERα and PRLR expression and increases estrogen responsiveness in breast cancer cells. Journal of Steroid Biochemistry and Molecular Biology. 2004a;88:69–77. doi: 10.1016/j.jsbmb.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Gutzman JH, Nikolai SE, Rugowski DE, Watters JJ, Schuler LA. Prolactin and estrogen enhance the activity of Activating Protein-1 in breast cancer cells: role of ERK1/2-mediated signals to c-fos. Molecular Endocrinology. 2005;19:1765–1778. doi: 10.1210/me.2004-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutzman JH, Rugowski DE, Nikolai SE, Schuler LA. Stat5 activation inhibits prolactin-induced AP-1 activity: distinct prolactin initiated signals in tumorigenesis dependent on cell context. Oncogene. 2007;26:6341–6348. doi: 10.1038/sj.onc.1210454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutzman JH, Rugowski DE, Schroeder MD, Watters JJ, Schuler LA. Multiple kinase cascades mediate prolactin signals to activating protein-1 in breast cancer cells. Molecular Endocrinology. 2004b;18:3064–3075. doi: 10.1210/me.2004-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslam SZ, Woodward TL. Host microenvironment in breast cancer development: epithelial-cell-stromal-cell interactions and steroid hormone action in normal and cancerous mammary gland. Breast Cancer Research. 2003;5:208–215. doi: 10.1186/bcr615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovey RC, Harris J, Hadsell DL, Lee AV, Ormandy CJ, Vonderhaar BK. Local insulin-like growth factor-II mediates prolactin-induced mammary gland development. Molecular Endocrinology. 2003;17:460–471. doi: 10.1210/me.2002-0214. [DOI] [PubMed] [Google Scholar]
- Hovey RC, Trott JF, Ginsburg E, Goldhar A, Sasaki MM, Fountain SJ, Sundararajan K, Vonderhaar BK. Transcriptional and spatiotemporal regulation of prolactin receptor mRNA and cooperativity with progesterone receptor function during ductal branch growth in the mammary gland. Developmental Dynamics. 2001;222:192–205. doi: 10.1002/dvdy.1179. [DOI] [PubMed] [Google Scholar]
- Huang Y, Li X, Jiang J, Frank SJ. Prolactin modulates phosphorylation, signaling and trafficking of epidermal growth factor receptor in human T47D breast cancer cells. Oncogene. 2006;25:7565–7576. doi: 10.1038/sj.onc.1209740. [DOI] [PubMed] [Google Scholar]
- Jones PL. Extracellular matrix and tenascin-C in pathogenesis of breast cancer. Lancet. 2001;357:1992–1994. doi: 10.1016/S0140-6736(00)05133-3. [DOI] [PubMed] [Google Scholar]
- Kenny PA, Bissell MJ. Tumor reversion: Correction of malignant behavior by microenvironmental cues. International Journal of Cancer. 2003;107:688–695. doi: 10.1002/ijc.11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Bagaitkar J, Watabe K. Roles of AKT signal in breast cancer. Frontiers of Bioscience. 2007;12:4011–4019. doi: 10.2741/2367. [DOI] [PubMed] [Google Scholar]
- Maatta M, Virtanen I, Burgeson R, Autio-Harmainen H. Comparative analysis of the distribution of laminin chains in the basement membranes in some malignant epithelial tumors: the alpha1 chain of laminin shows a selected expression pattern in human carcinomas. Journal of Histochemistry and Cytochemistry. 2001;49:711–726. doi: 10.1177/002215540104900605. [DOI] [PubMed] [Google Scholar]
- Manhes C, Kayser C, Bertheau P, Kelder B, Kopchick JJ, Kelly PA, Touraine P, Goffin V. Local over-expression of prolactin in differentiating mouse mammary gland induces functional defects and benign lesions, but no carcinoma. Journal of Endocrinology. 2006;190:271–285. doi: 10.1677/joe.1.06829. [DOI] [PubMed] [Google Scholar]
- McHale K, Tomaszewski JE, Puthiyaveettil R, Livolsi VA, Clevenger CV. Altered expression of prolactin receptor-associated signaling proteins in human breast carcinoma. Modern Pathology. 2008;21:565–571. doi: 10.1038/modpathol.2008.7. [DOI] [PubMed] [Google Scholar]
- Medina D, Kittrell FS, Shepard A, Contreras A, Rosen JM, Lydon J. Hormone dependence in premalignant mammary progression. Cancer Research. 2003;63:1067–1072. [PubMed] [Google Scholar]
- Milde-Langosch K, Bamberger AM, Rieck G, Grund D, Hemminger G, Muller V, Loning T. Expression and prognostic relevance of activated extracellular-regulated kinases (ERK1/2) in breast cancer. British Journal of Cancer. 2005;92:2206–2215. doi: 10.1038/sj.bjc.6602655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SO, Clark JA, Myers PH, Korach KS. Mammary gland development in adult mice requires epithelial and stromal estrogen receptor α. Endocrinology. 2002;143:2357–2365. doi: 10.1210/endo.143.6.8836. [DOI] [PubMed] [Google Scholar]
- Nakano S, Iyama K, Ogawa M, Yoshioka H, Sado Y, Oohashi T, Ninomiya Y. Differential tissular expression and localization of type IV collagen alpha1(IV), alpha2(IV), alpha5(IV), and alpha6(IV) chains and their mRNA in normal breast and in benign and malignant breast tumors. Laboratory Investigation. 1999;79:281–292. [PubMed] [Google Scholar]
- Nandi S, Guzman RC, Yang J. Hormones and mammary carcinogenesis in mice, rats, and humans: a unifying hypothesis. Proceedings of the National Academy of Science,USA. 1995;92:3650–3657. doi: 10.1073/pnas.92.9.3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor MJ, Li N, Cheung J, Lowe ET, Lambert E, Marlow R, Wang P, Schatzmann F, Wintermantel T, Schuetz G, Clarke AR, Mueller U, Hynes NE, Streuli CH. Ablation of beta1 integrin in mammary epithelium reveals a key role for integrin in glandular morphogenesis and differentiation. Journal of Cellular Biology. 2005;171:717–728. doi: 10.1083/jcb.200503144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevalainen MT, Xie JW, Bubendorf L, Wagner KU, Rui H. Basal activation of transcription factor Stat5 in nonpregnant mouse and human breast epithelium. Molecular Endocrinology. 2002;16:1108–1124. doi: 10.1210/mend.16.5.0839. [DOI] [PubMed] [Google Scholar]
- Novaro V, Roskelley CD, Bissell MJ. Collagen-IV and laminin-1 regulate estrogen receptor α expression and function in mouse mammary epithelial cells. Journal of Cell Science. 2003;116:2975–2986. doi: 10.1242/jcs.00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes SR, Robertson FG, Kench JG, Gardiner-Garden M, Wand MP, Green JE, Ormandy CJ. Loss of mammary epithelial prolactin receptor delays tumor formation by reducing cell proliferation in low-grade preinvasive lesions. Oncogene. 2007;26:543–553. doi: 10.1038/sj.onc.1209838. [DOI] [PubMed] [Google Scholar]
- Orend G, Chiquet-Ehrismann R. Tenascin-C induced signaling in cancer. Cancer Letters. 2006;244:143–163. doi: 10.1016/j.canlet.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Pearse G, Frith J, Randall KJ, Klinowska T. Urinary retention and cystitis associated with subcutaneous estradiol pellets in female nude mice. Toxicologic Pathology. 2009;37:227–234. doi: 10.1177/0192623308329281. [DOI] [PubMed] [Google Scholar]
- Polyak K, Hu M. Do myoepithelial cells hold the key for breast tumor progression. Journal of Mammary Gland Biology and Neoplasia. 2005;10:231–247. doi: 10.1007/s10911-005-9584-6. [DOI] [PubMed] [Google Scholar]
- Rose-Hellekant TA, Arendt LM, Schroeder MD, Gilchrist K, Sandgren EP, Schuler LA. Prolactin induces ERα-positive and ERα-negative mammary cancer in transgenic mice. Oncogene. 2003;22:4664–4674. doi: 10.1038/sj.onc.1206619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen RJ, Song RX, McPherson R, Kumar R, Adam L, Jeng MH, Yue W. The role of mitogen-activated protein (MAP) kinase in breast cancer. Journal of Steroid Biochemistry and Molecular Biology. 2002;80:239–256. doi: 10.1016/s0960-0760(01)00189-3. [DOI] [PubMed] [Google Scholar]
- Santos SJ, Haslam SZ, Conrad SE. Estrogen and progesterone are critical regulators of Stat5a expression in the mouse mammary gland. Endocrinology. 2008;149:329–338. doi: 10.1210/en.2007-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nature Reviews Cancer. 2004;4:937–947. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- Shimkin MB. Breast cancer in mice. California Medicine. 1948;68:264–269. [PMC free article] [PubMed] [Google Scholar]
- Shoker BS, Jarvis C, Clarke RB, Anderson E, Hewlett J, Davies MP, Sibson DR, Sloane JP. Estrogen receptor-positive proliferating cells in the normal and precancerous breast. American Journal of Pathology. 1999;155:1811–1815. doi: 10.1016/S0002-9440(10)65498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyamala G, Chou YC, Louie SG, Guzman RC, Smith GH, Nandi S. Cellular expression of estrogen and progesterone receptors in mammary glands: regulation by hormones, development and aging. Journal of Steroid Biochemistry and Molecular Biology. 2002;80:137–148. doi: 10.1016/s0960-0760(01)00182-0. [DOI] [PubMed] [Google Scholar]
- Silva CM, Shupnik MA. Integration of steroid and growth factor pathways in breast cancer: focus on signal transducers and activators of transcription and their potential role in resistance. Molecular Endocrinology. 2007;21:1499–1512. doi: 10.1210/me.2007-0109. [DOI] [PubMed] [Google Scholar]
- Sleeman KE, Kendrick H, Robertson D, Isacke CM, Ashworth A, Smalley MJ. Dissociation of estrogen receptor expression and in vivo stem cell activity in the mammary gland. Journal of Cell Biology. 2007;176:19–26. doi: 10.1083/jcb.200604065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, Monaghan P, Neville AM. Basal clear cells of the normal human breast. Virchows Archiv A: Pathology Anatomy Histopathology. 1984;402:319–329. doi: 10.1007/BF00695085. [DOI] [PubMed] [Google Scholar]
- Smith GH, Chepko G. Mammary epithelial stem cells. Microscopy Research and Technique. 2001;52:190–203. doi: 10.1002/1097-0029(20010115)52:2<190::AID-JEMT1005>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Suriano R, Chaudhuri D, Johnson RS, Lambers E, Ashok BT, Kishore R, Tiwari RK. 17Beta-estradiol mobilizes bone marrow-derived endothelial progenitor cells to tumors. Cancer Research. 2008;68:6038–6042. doi: 10.1158/0008-5472.CAN-08-1009. [DOI] [PubMed] [Google Scholar]
- Svensson S, Jirstrom K, Ryden L, Roos G, Emdin S, Ostrowski MC, Landberg G. ERK phosphorylation is linked to VEGFR2 expression and Ets-2 phosphorylation in breast cancer and is associated with tamoxifen treatment resistance and small tumours with good prognosis. Oncogene. 2005;24:4370–4379. doi: 10.1038/sj.onc.1208626. [DOI] [PubMed] [Google Scholar]
- Tam SP, Lau P, Djiane J, Hilton DJ, Waters MJ. Tissue-specific induction of SOCS gene expression by PRL. Endocrinology. 2001;142:5015–5026. doi: 10.1210/endo.142.11.8466. [DOI] [PubMed] [Google Scholar]
- The Endogenous Hormones and Breast Cancer Collaborative Group. Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. Journal of the National Cancer Institute. 2002;94:606–616. doi: 10.1093/jnci/94.8.606. [DOI] [PubMed] [Google Scholar]
- Tsoli E, Tsantoulis PK, Papalambros A, Perunovic B, England D, Rawlands DA, Reynolds GM, Vlachodimitropoulos D, Morgan SL, Spiliopoulou CA, Athanasiou T, Gorgoulis VG. Simultaneous evaluation of maspin and CXCR4 in patients with breast cancer. Journal of Clinical Pathology. 2007;60:261–266. doi: 10.1136/jcp.2006.037887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turley EA, Veiseh M, Radisky DC, Bissell MJ. Mechanisms of disease: epithelial-mesenchymal transition--does cellular plasticity fuel neoplastic progression. Nature Clinical Practice in Oncology. 2008;5:280–290. doi: 10.1038/ncponc1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tworoger SS, Hankinson SE. Prolactin and breast cancer risk. Cancer Letters. 2006;243:160–169. doi: 10.1016/j.canlet.2006.01.032. [DOI] [PubMed] [Google Scholar]
- Tworoger SS, Hankinson SE. Prolactin and breast cancer etiology: an epidemiologic perspective. Journal of Mammary Gland Biology and Neoplasia. 2008;13:41–53. doi: 10.1007/s10911-008-9063-y. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nature Reviews Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Rui H. Jak2/Stat5 signaling in mammogenesis, breast cancer initiation and progression. Journal of Mammary Gland Biology and Neoplasia. 2008;13:93–103. doi: 10.1007/s10911-008-9062-z. [DOI] [PubMed] [Google Scholar]
- Writing Group for the Women's Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principle results from the women's health initiative randomized controlled trial. Journal of the American Medical Association. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- Yoshidome K, Shibata MA, Couldrey C, Korach KS, Green JE. Estrogen promotes mammary tumor development in C3(1)/SV40 large T- antigen transgenic mice: paradoxical loss of estrogen receptor alpha expression during tumor progression. Cancer Research. 2000;60:6901–6910. [PubMed] [Google Scholar]
- Zinger M, McFarland M, Ben Jonathan N. Prolactin expression and secretion by human breast glandular and adipose tissue explants. J Clin Endocrinol Metab. 2003;88:689–696. doi: 10.1210/jc.2002-021255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Primers employed for qRT-PCR analyses
Supplementary Table 2. Mammary tumor latency and histotype in nonparous NRL-PRL mice.