

Abstract
Local production of IL-17 is a significant factor in effective host defense against Gram-negative bacteria. However, the proximal events mediating IL-17 elaboration by T cells remain unclear. In this study, we show in vivo that intact Toll-like receptor 4 signaling in the lung is required for induction of both the p19 transcript of IL-23 and IL-17 protein elaboration in response to Klebsiella pneumoniae. Although IL-17 is widely considered a CD4+ T cell product, we also demonstrate significant in vitro IL-17 production by CD8+ T cells after culture in medium from dendritic cells exposed to these bacteria. The dominant portion of this IL-17-inducing activity for both CD4+ and CD8+ T cells is IL-23. These data demonstrate the critical signaling pathway for IL-17 induction in the host response to Gram-negative pulmonary infection and suggest a direct role for IL-23 in CD8+ T cell IL-17 production.
Interleukin-17 was the first described member of an emerging family of cytokines with at least six species (IL-17 A–F) currently identified (1). The predominant source of IL-17 production in mice is reported to be CD4+ lymphocytes, particularly activated memory CD4+ T cells (2). IL-17 has been classified as a proinflammatory cytokine, because it induces inflammatory factors including G-CSF, GM-CSF, monocyte chemoattractant protein 1, macrophage-inflammatory protein-2 (MIP-2),3 keratinocyte-derived chemokine, IL-6, IL-8, ICAM-1, PGE2, and others (3). Studies have shown the importance of IL-17 in various physiologic and pathophysiologic processes including granulopoiesis, bacterial host defense, rheumatoid arthritis, allograft rejection, tumor modulation, and asthma (4–9).
Our group has previously described the importance of IL-17 signaling in a murine model of Klebsiella pneumoniae pulmonary infection (4). Mice lacking the receptor for IL-17 showed a blunted G-CSF and MIP-2 response, decreased neutrophil recruitment, greater bacterial burden, and worsened mortality. Conversely, IL-17 overexpression via intratracheal (i.t.) adenoviral vector administration resulted in enhanced chemokine production, neutrophil recruitment, and survival in the same model (10).
Although the importance of IL-17 signaling in host defense against K. pneumoniae infection seems evident, the specific physiologic trigger for its expression is unclear. Others have shown that certain microbial exotoxins, lipopeptides, and mycobacterial lysates can stimulate T cells to produce IL-17 in vitro (2). We hypothesized that LPS, the major superantigen of Gram-negative bacteria, may be responsible for IL-17 production in a murine model of pneumonia. Previous work has shown that LPS signaling is via Toll-like receptor (TLR)4, a pattern recognition receptor expressed on APC throughout a vast range of species (11). To test this hypothesis, we used C3H/HeJ mice, which have a mutation in the cytoplasmic tail of TLR4 and are hence unable to signal in response to LPS (11). In this study, we show that induction of IL-17 in the lung is TLR4 dependent. Using a dendritic cell (DC) and T cell coculture system, we demonstrate that DC-derived IL-23, a recently described heterodimer consisting of a p40 subunit identical with that of IL-12 and a unique p19 subunit (12), signals the induction of IL-17 in both CD4+ and CD8+ T cells.
Materials and Methods
Animals
C57BL/6, C3H/HeN (National Cancer Insitute, Frederick, MD), and C3H/HeJ, or IL-12 p35−/− (13) or p40−/− (14) mice on a C57BL/6 background (The Jackson Laboratory, Bar Harbor, ME), mice were received at 6–8 wk of age. K. pneumoniae strain 43816 (serotype 2) was from American Type Culture Collection (Manassas, VA). Mice were anesthetized with ketamine/xylazine, the trachea was cannulated with a 30-gauge needle, and 50 μl of bacteria (104 CFU) was injected. At 0, 4, and 16 h, animals were sacrificed, and bronchoalveolar lavage (BAL) was performed as described previously (10). Samples were frozen for later IL-17 ELISA (R&D Systems, Minneapolis, MN). Lungs were removed and homogenized in TRIzol (Life Technologies, Gaithersburg, MD), and total RNA was isolated. RNA was either subjected to real-time RT-PCR in an ABI 7700 thermocycler using TaqMan reagents (both from Applied Biosystems, Foster City, CA) for IL-17 or IL-23 p19 mRNA or analyzed on a murine genome array chip U74Av2 (Affymetrix, Santa Clara, CA). For T cell depletion experiments, animals were pretreated with i.p. anti-CD4+ (clone GK1.5; Taconic Labs, Germantown, NY) or anti-CD8+ Ab (clone 2.43; American Type Culture Collection) 4 days before bacterial challenge.
Cell preparations
DC were derived from hemopoietic progenitors of mouse bone marrow. Cells were grown in RPMI 1640 medium with 10% FBS supplemented with 100 U/ml GM-CSF and 20 ng/ml IL-4 (R&D Systems). After 1 wk, all DC preparations were >90% positive for class II MHC (I-A), CD80, and CD11c, with <1% staining for CD4, CD8, CD19, or the NK cell marker DX-5. Spleens were processed through a 40-μm filter, and red cells were lysed. Total splenic T lymphocytes were purified using magnetic labeled anti-CD90 beads, or T cell subsets were positively selected using anti-CD4 or anti-CD8 beads (Miltenyi Biotec, Auburn, CA). Purified CD4+ or CD8+ subsets were at least 95% pure and contained <1% of the other T cell population based on FACS analysis.
DC/lymphocyte coculture system
After 1 wk of maturation, 105 DC were plated in 24-well tissue culture plates. K. pneumoniae (107 CFU; 100:1 bacteria/DC ratio) were added and incubated for 24 h. T cells (5 × 105; CD90+, CD4+, or CD8+ cells) were added to the system for an additional 24 h. For experiments designed to test the need for direct physical contact between DC and T cell, we placed the T cells into a Transwell 0.4 μm polyester membrane insert (Corning, Corning, NY). Also, T cells were exposed to conditioned medium from K. pneumoniae-pulsed DC. For experiments designed to test the role of IL-23, Ab directed against mouse p40, a subunit common to IL-12 and IL-23, or isotype control, was incubated for 1 h with conditioned medium before T cell addition (R&D Systems). To exclude IL-12 neutralization as a confounder, DC from p35−/−, p40−/−, or wild-type C57BL/6 animals were harvested and cultured as above in Cell preparations. After 24 h, samples were spun, and the supernatant was assayed for IL-17. Cells were resuspended in TRIzol, and RNA was isolated for IL-17 transcript analysis. Transcript copy number was determined by comparing the cycle threshold of each unknown sample to those of a standard curve of a known quantity of murine IL-17 or p19 cDNA standards, which we have previously cloned. All RT-PCR data were normalized to 18s rRNA content, also determined by realtime RT-PCR.
Statistical analysis
Data were analyzed using StatView statistical software (Brainpower, Calabasas, CA), and the statistical significance between means of data was determined by Student's t test. Significance was accepted at p < 0.05.
Results and Discussion
IL-17 protein in BAL fluid and mRNA expression in lung homogenate from C3H/HeN (LPS-sensitive) and C3H/HeJ (LPS-insensitive) mice were determined at 0, 4, and 16 h after i.t. infection (Fig. 1, A and B). IL-17 in BAL fluid was at the lower limit of detection by ELISA until 16 h. At this time point, C3H/HeJ mice demonstrated significantly lower IL-17 levels compared with C3H/HeN mice. We have previously observed that IL-17 in BAL fluid is reflective of IL-17 levels in lung homogenates (10). The lower IL-17 induction in C3H/HeJ mice was also confirmed by real-time RT-PCR for IL-17 transcripts. There was a highly statistically significant induction of IL-17 mRNA in C3H/HeN mice at 4 and 16 h compared with that of C3H/HeJ mice. Because IL-23 has been shown to induce IL-17 production in vitro (15), we investigated the time course of IL-23 p19 mRNA induction in this model. Compared with the low levels of transcripts present 0 h after infection in both strains, there was significant and earlier induction of p19 in C3H/HeN compared with C3H/HeJ mice as soon as 4 h after bacterial challenge (Fig. 1C). By 16 h, there was a delayed yet significant rise in p19 mRNA in the C3H/HeJ mice, although C3H/HeN p19 transcripts remained greater at this time point as well.
FIGURE 1.
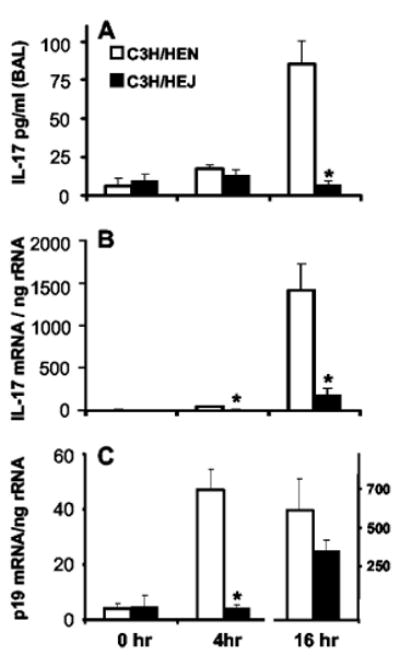
LPS-insensitive C3H/HeJ mice display markedly reduced IL-17 production and delayed p19 mRNA expression in response to i.t. challenge with K. pneumoniae. A, BAL fluid IL-17 protein content at the time of bacterial inoculation and 4 and 16 h later. B, Copy number of IL-17 mRNA transcripts in whole lung homogenate at the same time points, as measured by quantitative real-time RT-PCR. Copy numbers are normalized to 18s rRNA content for internal control. C, Lung homogenate mRNA transcripts for the p19 subunit of IL-23. C3H/HeJ mice show a blunted and delayed induction of p19 transcripts (note the separate y-axis for 16-h data for p19 mRNA; n = 6 per group; *, p < 0.05 compared with C3H/HeN group at the same time point for all three figures).
The putative cellular source of IL-17 is CD4+ T cells (2, 3, 16). Stimulated CD8+ CD45RO+ T cells from human preparations have also been demonstrated to generate significant IL-17 mRNA (17). We next depleted CD4+, CD8+, or both T cell subsets in C57BL/6 mice with Ab GK1.5 or 2.43, respectively, which deplete over 97% of lung and splenic CD4+ or CD8+ T cells (18, 19). CD4+ depletion resulted in 45% reduction in measurable IL-17 in BAL fluid 24 h after K. pneumoniae challenge (Fig. 2). However, the combination of CD4+ and CD8+ T cell depletion resulted in ∼ 90% reduction in IL-17 following K. pneumoniae challenge, suggesting that CD8+ T cells play a significant role in either mediating or directly producing IL-17 in this in vivo model of pneumonia.
FIGURE 2.
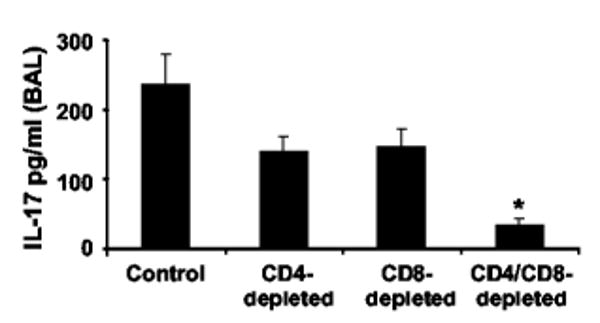
The presence of both CD4+ and CD8+ T cells is necessary for normal IL-17 production in response to K. pneumoniae. Ab depletion of CD4+ or CD8+ T cells reduces the IL-17 content in BAL fluid from C57/BL6 mice 24 h after i.t. challenge. Combined neutralization results in significant abrogation of detectable IL-17 (n = 6 per group; *, p < 0.05 compared with Ab-untreated animals).
As TLR4 is expressed on both macrophages and DC (20), we designed in vitro experiments to determine whether TLR4 expression on DC was required for IL-17 elaboration by T cells and whether this stimulation required cell contact. DC obtained from C3H/HeN or C3H/HeJ mice had similar levels of class II MHC, CD80, and CD86 expression by FACS (data not shown). Moreover, DC from both mice strains produced comparable IL-12 (data not shown) upon stimulation with 1 μg/ml soluble CD40 ligand (a gift from Dr. W. Fanslow (Immunex, Seattle, WA)). However, C3H/HeN DC pulsed with K. pneumoniae were capable of inducing much higher levels of IL-17 by total splenic T cells (CD90+) compared with C3H/HeJ DC (Fig. 3). There was no IL-17 induction in T cells incubated with unpulsed DC or T cells incubated directly with bacteria but not DC (data not shown).
FIGURE 3.
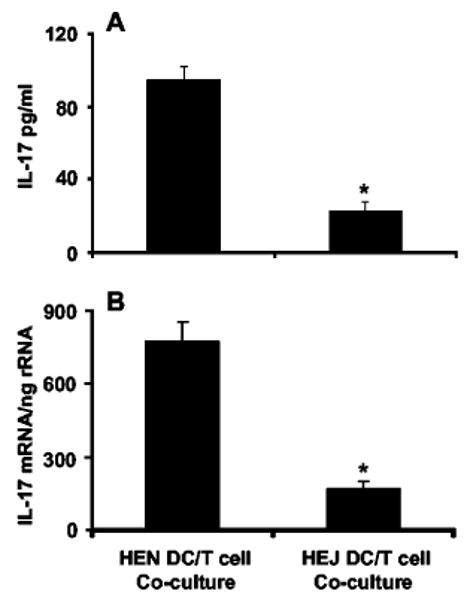
In vitro coculture of total spleen T cells (CD90+) with K. pneumoniae-pulsed DC. A, Twenty-four-hour cell culture supernatant analysis confirms that C3H/HeJ-derived cells exhibit diminished IL-17 expression compared with that of C3H/HeN cells. B, IL-17 mRNA transcripts by quantitative real-time RT-PCR of the same experiment (n = 6 per group; *, p < 0.05 compared with C3H/HeN mice).
Physical contact between DC and CD4+ or CD8+ T cells is not required for IL-17 production, because experiments using the Transwell membrane coculture system to separate DC and T cells showed no significant difference in IL-17 mRNA or protein expression compared with a nonseparated group (Fig. 4). In this system, CD8+ T cells from C3H/HeN mice were capable of expressing greater amounts of IL-17 mRNA transcripts and secreted protein after 24 h of culture compared with an equal number of CD4+ T cells. Similar IL-17 induction in CD4+ and CD8+ T cells was observed with cell-free conditioned medium from DC pulsed with K. pneumoniae. The same conditioned medium, when boiled, did not stimulate IL-17 (data not shown).
FIGURE 4.

In vitro DC/T cell physical contact is not required for IL-17 production in response to K. pneumoniae. CD8+ T cells produce more IL-17 protein and mRNA transcripts compared with those of CD4+ T cells in response to bacteria-pulsed DC. A, IL-17 in cell culture supernatant after 24 h of culture in each of the following conditions: A, T cell only plus bacteria; B, DC only plus bacteria; C, DC plus bacteria plus membrane barrier insert containing T cells; D, DC plus bacteria plus T cells without barrier; E, DC plus T cell without barrier or bacteria. B, IL-17 mRNA transcripts from total cell pellets of the same experimental groups (n = 4 per group; *, p < 0.05 compared with CD8+ cells of same exposure condition and to CD4+ T cells in condition A; ‡, p < 0.05 compared with CD8+ T cells in condition A).
As TLR4 signaling appears to be critical for the timely expression of IL-23 p19 induction in vivo, we investigated whether IL-23 elaboration by DC is critical for IL-17 induction by T cells. Neutralization experiments were performed in vitro on bacteria-pulsed C57BL/6 DC-conditioned medium using an anti-IL-12 p40 Ab which blocks IL-23 signaling (15). We observed a significant reduction in IL-17 protein and mRNA in both CD4+ and CD8+ T cells exposed to the p40 Ab-treated medium but not isotype control (Fig. 5, A and B). Gene knockout experiments, using DC from mice on a C57 background, suggest that this activity is not due to IL-12 neutralization, because DC from p35−/− mice (capable of making IL-23 but not IL-12) pulsed with K. pneumoniae actually induced higher levels of IL-17 protein and mRNA. This finding is consistent with published data that rIL-12 can, in fact, inhibit IL-17 production in a dose-dependent manner (15). Conversely, DC from p40−/− mice (capable of making neither IL-23 nor IL-12) showed a significantly lower level of IL-17 induction compared with wild-type or p35−/− mice, supporting the conclusion that IL-23, not IL-12, is responsible for T cell IL-17 expression in this in vitro system. We observed a greater percentage of inhibition in CD8+ T cell IL-17 production using anti-p40 Ab or supernatant from p40−/− mice compared with that of CD4+ T cells. These data suggest that IL-23 is critical for both CD4+ and CD8+ T cell IL-17 production in response to TLR4 signaling, although other soluble factors may be operative in CD4+ T cell elaboration of IL-17. Toward this end, we performed in vitro Ab neutralization of TNF-α, IL-β, IL-6, and IL-15. All of these manipulations failed to inhibit IL-17 elaboration by CD4+ T cells (data not shown). Given the proven cause and effect of IL-23 on in vitro IL-17 elaboration combined with the earlier rise in p19 mRNA transcripts in our in vivo pneumonia model, we speculate that IL-23 is likely a key proximal mediator of the early, physiologically important induction of IL-17 in Gram-negative pneumonia. The previous finding that only APCs concomitantly express p40 and p19 (12) and, hence, are the only cells believed capable of producing functional IL-23 underscores the importance of intact antigenic signaling via cognate receptors on APC (21). Indeed, other studies have already proven the vital role of TLR2 on IL-23 expression (22). The combination of our neutralization experiments and the time course of p19 induction in vivo supports the hypothesis that IL-23 in response to K. pneumoniae is a key proximal event leading to IL-17 production in vivo. Future experiments using p19 knockout animals will enable us to answer this question more directly.
FIGURE 5.
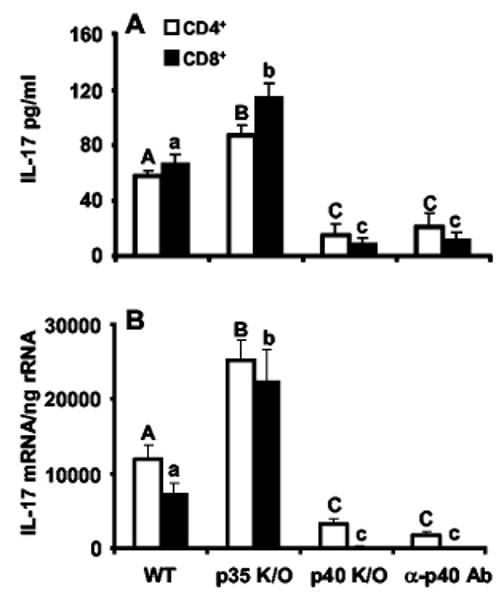
CD4+ and CD8+ T cell IL-17 response to conditioned medium from K. pneumoniae-pulsed DC requires biologically active IL-23. A, Conditioned medium from p35−/− (knockout (K/O)) DC (devoid of IL-12) elicits an augmented IL-17 response from CD4+ and CD8+ T cells, whereas medium from p40−/− DC (devoid of IL-12 and IL-23) shows marked attenuation in comparison to wild type (WT). Neutralization of conditioned medium with anti-p40 also showed significantly less IL-17 induction in both CD4+ and CD8+ T cells. B, IL-17 mRNA copy number from T cells of the same experiment (n = 6 per group; columns with different uppercase (CD4+) or lowercase (CD8+) letters denote statistical significance (p < 0.05) compared with different letters of the same case).
These results significantly extend those recently obtained by Aggarwal et al. (15). Our data demonstrate that TLR4 signaling is the principal mechanism to ultimately induce IL-17 in response to K. pneumoniae infection both in vitro and in vivo. We also show CD8+ T cells are a significant source of IL-17, through an IL-23-dependent mechanism. C3H/HeN-derived CD8+ T cells were capable of greater in vitro IL-production compared with that of CD4+ T cells, whereas C57/BL6-derived CD8+ and CD4+ populations showed equivalent expression, suggesting strain differences may exist in the ability of T cell subsets to elaborate IL-17. Also, whereas the work of Aggarwal et al. focused on memory T cells that were activated and cultured for 3 days (hence representing a substantially different culture system than ours), we specifically focused on early (≤24 h) IL-17 elaboration in primary T cells, because IL-17 is induced rapidly in vivo in the lung after K. pneumoniae challenge (10).
In this model of Gram-negative pneumonia, a critical early host response is brisk neutrophil recruitment to quickly phagocytize invading pathogens before they begin geometric multiplication. Animals unable to mount this neutrophilic response to pulmonary K. pneumoniae infection display greater lung bacterial burden, bacteremia, and death (23). Earlier work has shown a critical role of IL-17 in neutrophil chemoattraction via its up-regulation of CXC chemokines and G-CSF (3, 10, 24). Given the ability of IL-17 to induce neutrophil chemokine induction as well as granulopoiesis, it stands to reason that this cytokine likely functions as an important signal from T lymphocytes in orchestrating an augmented innate immune response to enhance pathogen clearance.
Our work proves that both CD4+ and CD8+ T cells can produce IL-17 in response to Gram-negative bacteria via TLR4 signaling, presumably by APC in the lung. It will be important to further define subpopulations of CD4+ and CD8+ T cells responsible for in vivo IL-17 production. Because IL-17 expression is not clearly dichotomized into Th1- or Th2-like expression profiles (2), it is quite possible that IL-17-producing T cells do not represent either entity of this paradigm of acquired immunity.
Despite their lacking TLR4 signaling ability, the C3H/HeJ mice nevertheless demonstrate an increase, albeit attenuated and delayed, in both IL-17 and p19 mRNA. Based on our microarray data, this finding is consistent with a delayed expression of other proinflammatory genes, including MIP-2 and IL-6, in the C3H/HeJ strain. These later time points of gene induction may be due to LPS/TLR4-independent mechanisms, such as lipopeptide (TLR2) or unmethylated CpG motif (TLR9) signaling, which remain intact in C3H/HeJ mice. In addition, by 16 h, C3H/HeJ mice suffer a greater bacterial burden in the lung compared with that of controls. This differential inflammatory stimulus may invoke other, less potent signaling pathways to become operant in the expression of IL-17. As stated, this C3H/HeJ catch-up phenomenon is seen in many other proinflammatory signals, but likely represents too little too late, given the aforementioned increase in bacterial load and earlier occurrence of death in the C3H/HeJ strain compared with the C3H/HeN.
Our data show that intact TLR4 signaling represents the early and physiologically requisite signaling pathway in the timely expression of IL-23 and IL-17 in this Gram-negative pneumonia model. Further characterization of IL-23 and other downstream T cell mediators will greatly enlighten our understanding of the early communication between innate and acquired immunity in response to bacterial challenge.
Footnotes
This work was supported by Public Health Service Grants P50AA009803 (to J.K.K., S.N., G.J.B., and K.I.H.), R01HL061271-04 (to J.K.K., J.E.S., and M.Z.), and R01AI051677 (to J.E.S. and J.K.K.).
Abbreviations used in this paper: MIP-2, macrophage-inflammatory protein 2; i.t., intratracheal; TLR, Toll-like receptor; DC, dendritic cell; BAL, bronchoalveolar lavage.
References
- 1.Aggarwal S, Gurney AL. IL-17: prototype member of an emerging cytokine family. J Leukocyte Biol. 2002;71:1. [PubMed] [Google Scholar]
- 2.Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial lipopeptides induce the production of IL-17 in Th cells. J Immunol. 2000;165:6107. doi: 10.4049/jimmunol.165.11.6107. [DOI] [PubMed] [Google Scholar]
- 3.Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, Pin JJ, Garrone P, Garcia E, Saeland S, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chabaud M, Fossiez F, Taupin JL, Miossec P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. 1998;161:409. [PubMed] [Google Scholar]
- 6.Schwarzenberger P, La Russa V, Miller A, Ye P, Huang W, Zieske A, Nelson S, Bagby GJ, Stoltz D, Mynatt RL, et al. IL-17 stimulates granulopoiesis in mice: use of an alternate, novel gene therapy-derived method for in vivo evaluation of cytokines. J Immunol. 1998;161:6383. [PubMed] [Google Scholar]
- 7.van Kooten C, Boonstra JG, Paape ME, Fossiez F, Banchereau J, Lebecque S, Bruijn JA, De Fijter JW, Van Es LA, Daha MR. Interleukin-17 activates human renal epithelial cells in vitro and is expressed during renal allograft rejection. J Am Soc Nephrol. 1998;9:1526. doi: 10.1681/ASN.V981526. [DOI] [PubMed] [Google Scholar]
- 8.Linden A. Role of interleukin-17 and the neutrophil in asthma. Int Arch Allergy Immunol. 2001;126:179. doi: 10.1159/000049511. [DOI] [PubMed] [Google Scholar]
- 9.Benchetrit F, Ciree A, Vives V, Warnier G, Gey A, Sautes-Fridman C, Fossiez F, Haicheur N, Fridman WH, Tartour E. Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood. 2002;99:2114. doi: 10.1182/blood.v99.6.2114. [DOI] [PubMed] [Google Scholar]
- 10.Ye P, Garvey PB, Zhang P, Nelson S, Bagby G, Summer WR, Schwarzenberger P, Shellito JE, Kolls JK. Interleukin-17 and lung host defense against K pneumoniae infection. Am J Respir Cell Mol Biol. 2001;25:335. doi: 10.1165/ajrcmb.25.3.4424. [DOI] [PubMed] [Google Scholar]
- 11.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 12.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 13.Mattner F, Magram J, Ferrante J, Launois P, Di Padova K, Behin R, Gately MK, Louis JA, Alber G. Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania major and mount a polarized Th2 cell response. Eur J Immunol. 1996;26:1553. doi: 10.1002/eji.1830260722. [DOI] [PubMed] [Google Scholar]
- 14.Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu CY, Ferrante J, Stewart C, Sarmiento U, Faherty DA, Gately MK. IL-12-deficient mice are defective in IFN-γ production and type 1 cytokine responses. Immunity. 1996;4:471. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- 15.Aggarwal S, Ghilardi N, Xie MH, De Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 16.Rouvier E, Luciani MF, Mattei MG, Denizot F, Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J Immunol. 1993;150:5445. [PubMed] [Google Scholar]
- 17.Shin HC, Benbernou N, Fekkar H, Esnault S, Guenounou M. Regulation of IL-17, IFN-γ and IL-10 in human CD8+ T cells by cyclic AMP-dependent signal transduction pathway. Cytokine. 1998;10:841. doi: 10.1006/cyto.1998.0375. [DOI] [PubMed] [Google Scholar]
- 18.D'Souza NB, Mandujano FJ, Nelson S, Summer WR, Shellito JE. CD4+ T lymphocyte depletion attenuates lipopolysaccharide-induced tumor necrosis factor secretion by alveolar macrophages in the mouse. Lymphokine Cytokine Res. 1994;13:359. [PubMed] [Google Scholar]
- 19.Steele C, Zheng M, Young E, Marrero L, Shellito JE, Kolls JK. Increased host resistance against Pneumocystis carinii pneumonia in γδ T-cell-deficient mice: protective role of γ-interferon and CD8+ T cells. Infect Immun. 2002;70:5208. doi: 10.1128/IAI.70.9.5208-5215.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoshino K, Kaisho T, Iwabe T, Takeuchi O, Akira S. Differential involvement of IFN-β in Toll-like receptor-stimulated dendritic cell activation. Int Immunol. 2002;14:1225. doi: 10.1093/intimm/dxf089. [DOI] [PubMed] [Google Scholar]
- 21.Lankford CS, Frucht DM. A unique role for IL-23 in promoting cellular immunity. J Leukocyte Biol. 2003;73:49. doi: 10.1189/jlb.0602326. [DOI] [PubMed] [Google Scholar]
- 22.Re F, Strominger JL. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J Biol Chem. 2001;276:37692. doi: 10.1074/jbc.M105927200. [DOI] [PubMed] [Google Scholar]
- 23.Greenberger MJ, Strieter RM, Kunkel SL, Danforth JM, Laichalk LL, McGillicuddy DC, Standiford TJ. Neutralization of macrophage inflammatory protein-2 attenuates neutrophil recruitment and bacterial clearance in murine Klebsiella pneumonia. J Infect Dis. 1996;173:159. doi: 10.1093/infdis/173.1.159. [DOI] [PubMed] [Google Scholar]
- 24.Witowski J, Pawlaczyk K, Breborowicz A, Scheuren A, Kuzlan-Pawlaczyk M, Wisniewska J, Polubinska A, Friess H, Gahl GM, Frei U, Jorres A. IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GROα chemokine from mesothelial cells. J Immunol. 2000;165:5814. doi: 10.4049/jimmunol.165.10.5814. [DOI] [PubMed] [Google Scholar]