

Abstract
Fluoroquinolones are antibacterial agents that attack DNA gyrase and topoisomerase IV on chromosomal DNA. The existence of two fluoroquinolone targets and stepwise accumulation of resistance suggested that new quinolones could be found that would require cells to obtain two topoisomerase mutations to display resistance. For wild-type cells to become resistant, the two mutations must be acquired concomitantly. That is expected to occur infrequently. To identify such compounds, fluoroquinolones were tested for the ability to kill a moderately resistant gyrase mutant. Compounds containing a C8-methoxyl group were particularly lethal, and incubation of wild-type cultures on agar containing C8-methoxyl fluoroquinolones produced no resistant mutant, whereas thousands arose during comparable treatment with control compounds lacking the C8 substituent. When the test strain contained a preexisting topoisomerase IV mutation, which by itself conferred no resistance, equally high numbers of resistant mutants were obtained for C8-methoxyl and control compounds. Thus C8-methoxyl fluoroquinolones required two mutations for expression of resistance. Although highly lethal, C8-methoxyl fluoroquinolones were not more effective than C8-H controls at blocking bacterial growth. Consequently, quinolone action involves two events, which we envision as formation of drug–enzyme–DNA complexes followed by release of lethal double-strand DNA breaks. Release of DNA breaks, which must occur less frequently than complex formation, is probably the process stimulated by the C8-methoxyl group. Understanding this stimulation should provide insight into intracellular quinolone action and contribute to development of fluoroquinolones that prevent selection of resistant bacteria.
The quinolones are exceptionally potent antibacterial agents that have long been known to trap DNA gyrase on DNA; recently it was established that DNA topoisomerase IV is also a target of the drugs (for review see ref. 1). Many bacteria are sensitive to the quinolones, and potent fluorine-containing derivatives have enjoyed considerable clinical success (for reviews, see ref. 2). However, some pathogenic species, such as Staphylococcus aureus, Pseudomonas aeruginosa, and Mycobacterium tuberculosis, readily acquire resistance mutations that severely limit fluoroquinolone usefulness (3–5). The growing problem of multidrug-resistant bacterial pathogens makes it important to find ways to reduce the emergence of mutants resistant to clinical doses of the compounds.
Topoisomerase-based resistance to the fluoroquinolones occurs stepwise, with moderate levels of resistance arising from single mutations in the primary target of the drug (gyrase or topoisomerase IV, depending on the bacterial species). Once reduced sensitivity to the drug arises from the first mutation, higher levels of resistance may occur from additional mutations in both the primary and secondary enzyme targets (6–11). If a compound could be found that would avidly attack a resistant, first-step mutant, then two mutations would be required for a bacterium to express resistance. A wild-type cell would have to acquire two concomitant mutations to resist such a compound, an event that would occur less frequently than a single mutation by orders of magnitude. Two general ideas underlie searches for new compounds that might prevent resistance. One is based on the existence of two fluoroquinolone targets. Compounds that attack both gyrase and topoisomerase IV equally would be ideal, because there would be no concentration at which a mutant having only one resistant target could arise (11, 12). For Escherichia coli, gyrase is the primary fluoroquinolone target, and so a goal with this organism would be to find compounds that are more effective at attacking topoisomerase IV. The second idea emerges from the stepwise nature of fluoroquinolone resistance: a first-step mutant protein may not completely resist attack by some fluoroquinolones, and so a second mutation would be necessary for resistance. For example, DU6859a, a fluoroquinolone containing a chlorine attached to position C8, is particularly effective at blocking growth of ciprofloxacin-resistant gyrase (gyrA CipR) mutants of P. aeruginosa and at inhibiting gyrase purified from these mutants (13). Such a compound should reduce the acquisition of resistance by bacteria in which gyrase is the primary target of the drug. According to this second idea, gyrase and topoisomerase IV need not be attacked equally to prevent resistance.
We have performed assays of fluoroquinolone action by using a first-step, nalidixic acid-resistant (NalR) gyrA mutant of E. coli to identify compounds that lower the acquisition of resistance. The present report focuses on fluoroquinolones having a methoxyl group attached to position C8 (C8-OMe; for structures, see Fig. 1). These compounds cause fewer side effects than those carrying C8 halogens (14, 15) while maintaining higher activity than fluoroquinolones lacking a C8 substituent (16). We first examined lethal action, because highly lethal compounds were expected to leave bacterial populations less able to acquire resistance arising from the mutagenic action of the SOS regulon, a DNA-damage response induced by fluoroquinolone treatment (17–19). The C8-OMe substituent increased lethal action, particularly against the gyrA mutant. When wild-type cells were then challenged with C8-OMe fluoroquinolones, recovery of resistant mutants was below detection and at least three orders of magnitude lower than observed with compounds lacking a methoxyl group at position C8. An unusual feature of this system was the failure of C8-OMe fluoroquinolones to block growth better than control compounds. This observation supports the idea that inhibition of growth and cell death arise from distinct events. As indicated in Fig. 2, the first event is a reversible formation of fluoroquinolone–gyrase–DNA complexes in which the DNA is broken but the ends are constrained. Release of broken DNA then leads to cell death. Within this context, the C8-OMe group increases lability rather than formation of quinolone–gyrase–DNA complexes.
Figure 1.
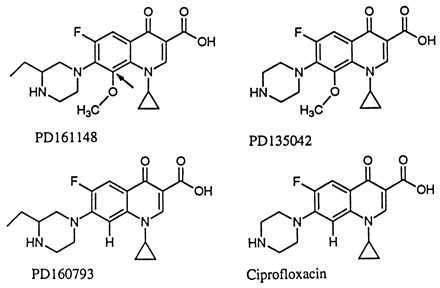
Fluoroquinolone structures. Arrow with PD161148 indicates C8 position.
Figure 2.

Intracellular action of fluoroquinolones. When DNA gyrase and DNA topoisomerase IV bind to DNA, they break both DNA strands at the interaction site. If fluoroquinolones are present, a reaction intermediate is trapped in which the DNA is broken and topoisomerase subunits are covalently bound to broken DNA. The trapped complexes block DNA synthesis and bacterial growth. Release of broken DNA in the complexes from the constraint imposed by the topoisomerases correlates with cell death (10).
METHODS AND MATERIALS
Bacterial strains were derivatives of E. coli K-12. Strain DM4100 (20) carries a cysB mutation; strain GP201 is a gyrA-307 (NalR Leu-83) transductant of DM4100 (21). Strains KD1366 [gyrA-307 (NalR Leu-83) parC (CipR Leu-80)], KD1368 [gyrA-307 (NalR Leu83) parC (CipR Lys-84)], and KD1373 [parC (CipR Leu-80)] were constructed by P1-mediated transduction (22) using GP201 or DM4100 as recipient. Strains A90 [AB1157 recBC sbcB kanamycin-resistant (KanR)], A83 [AB1157 recBC sbcB parC (CipR Leu-80) KanR], and A140 [AB1157 recBC sbcB parC (CipR Lys-84) KanR] served as donors in which KanR is closely linked to parC; they were obtained from A. Khodursky and N. Cozzarelli, University of California, Berkeley. parC transductants were selected for kanamycin resistance and screened for decreased susceptibility to ciprofloxacin (for KD1373, the gyrA-307 mutation was introduced by a subsequent transduction to test for the presence of the parC mutation, which by itself confers no resistance to fluoroquinolones). Transduction tests for second-step mutations mapping in gyrA, gyrB, and the par region used donor strains KD1600 (GP201 zfa-3145::Tn10 KanR), KO635 (tna::Tn10), and A90, respectively.
Ciprofloxacin was purchased from Miles Pharmaceutical; PD161148, PD160793, and PD135042 were synthesized by Parke–Davis Pharmaceutical (14). Stock solutions (10 mg/ml fluoroquinolone in 0.1 M NaOH) were stored at −70°C. Bacteriostatic action was measured by dilution of overnight cultures of E. coli into LB medium (23) containing various concentrations of fluoroquinolone followed by incubation at 37°C for 8 hr and measurement of culture turbidity. Under these conditions, untreated cultures reached stationary phase. The fluoroquinolone concentration needed to inhibit growth (increase in culture turbidity) to half that observed in the absence of drug was defined as ID50. Minimum inhibitory concentration (MIC) was determined by counting the number of colonies appearing on fluoroquinolone-containing agar plates spotted with 20-μl aliquots of diluted cultures and incubated overnight at 37°C. The fluoroquinolone concentration that allowed less than 1% of the cells to grow was defined as MIC. To test for acquisition of resistant mutants, E. coli strains DM4100 and KD1373 were first grown to stationary phase from single colonies. Cells were harvested by centrifugation at 2,000 × g, washed once with fresh LB medium, inoculated into LB medium having 20 times the original volume, and grown to stationary phase at 37°C with vigorous shaking. Cells were again harvested, resuspended in 0.1 vol of LB medium, mixed with 0.7% molten LB agar (0.5 ml of cells at about 1011 per ml plus 2 ml of agar and the appropriate amount of fluoroquinolone per plate), poured on LB-agar plates containing fluoroquinolone, and incubated at 37°C for 96 hr. With strain GP201, mutant selection was performed by spreading 50 μl of culture grown overnight in LB liquid medium onto LB agar plates containing PD135042 followed by incubation for 72 hr at 37°C. Quinolone resistance of all first-step and second-step mutants was confirmed by growth on LB agar plates containing 50 μg/ml nalidixic acid and 0.5 μg/ml ciprofloxacin, respectively.
RESULTS
Contribution of a C8-OMe Substituent to Fluoroquinolone Lethality.
Lethal action of fluoroquinolones was compared by treating cells with drug and then measuring the number of surviving cells as colony-forming units. With wild-type E. coli, we saw little difference between a C8-OMe and its C8-H control (Fig. 3A). At low concentration, the control was more effective, but the C8-OMe derivative was slightly more potent at the optimal bactericidal concentration [all quinolones display the unexplained property of being less bactericidal at very high concentrations than at moderate ones (24), making lethal action at the optimal concentration useful for comparing potency]. Against the gyrA mutant, the C8-OMe derivative was considerably more effective than the control. The dose required to kill 99.9% of the cells was 5-fold lower, and at the optimal concentration the C8-OMe compound was about 30 times more lethal (Fig. 3B). The C8-OMe compound also killed a gyrA parC mutant more effectively than the control fluoroquinolone (Fig. 3C). Thus, adding a C8-OMe substituent to fluoroquinolones increases lethality against resistant mutants.
Figure 3.
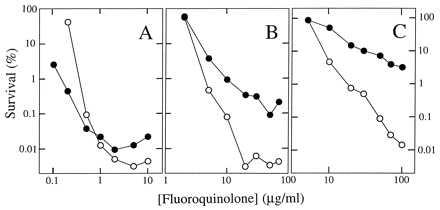
Bactericidal effects of quinolones on E. coli. Exponentially growing cultures of wild-type E. coli (A, strain DM4100), a gyrA mutant (B, strain GP201), or a gyrA parC mutant (C, strain KD1366) were incubated with the indicated concentrations of PD161148 (C8-OMe, ○) and PD160793 (C8-H, •) for 2 hr. Aliquots were then removed, diluted, and plated on drug-free agar for determination of viable cells. Similar results were obtained in two replicate experiments and with another pair of compounds, PD135042 and ciprofloxacin.
Inhibitors of RNA and protein synthesis interfere with the lethal action of fluoroquinolones (10, 25, 26). Interference was also observed in the present study, as cotreatment of wild-type cells with chloramphenicol, an inhibitor of protein synthesis, dramatically reduced the killing activity of both the C8-OMe compound and its control (compare Figs. 3A and 4). However, the action of the C8-OMe compound was less sensitive to chloramphenicol by a factor of about 10 (Fig. 4). Thus a C8-OMe substituent may enhance lethal activity against nongrowing wild-type cells. They are generally killed inefficiently by quinolones (27) and constitute a pool from which resistant mutants may emerge (28). This inference, along with the exceptional ability of C8-OMe fluoroquinolones to kill resistant mutants, encouraged us to examine acquisition of resistance in a wild-type population.
Figure 4.
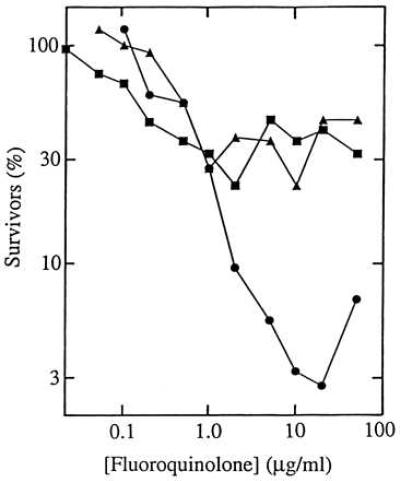
Effect of chloramphenicol on the lethal action of fluoroquinolones. Cultures of wild-type E. coli strain DM4100 were treated with PD161148 (C8-OMe, •), PD160793 (C8-H, ▴), and ciprofloxacin (▪) for 1 hr in the presence of 20 μg/ml chloramphenicol. Aliquots were then removed, diluted, and plated on drug-free agar for determination of viable cells. Similar results were obtained in two replicate experiments.
Acquisition of Fluoroquinolone Resistance by Wild-Type Cells.
The ability of a fluoroquinolone to allow resistant mutants to arise was assessed by counting the number of colonies that formed when large numbers of cells were incubated on agar plates containing the drug. To minimize differences between fluoroquinolones with respect to nontopoisomerase factors, such as drug uptake and efflux, we first compared the ability of the compounds to select resistant mutants at concentrations that were equally effective at blocking cell growth (10 times MIC for the strain). From three independent experiments, a total of 1,307 colonies were obtained on plates containing the C8-H control; none were found on plates containing the C8-OMe compound (Table 1, lines 1 and 2). Thus by this assay resistance mutations arise at least 1,300 times less frequently with the C8-OMe derivative.
Table 1.
Acquisition of spontaneous fluoroquinoloneresistant mutants
| Line | Fluoroquinolone | Conc., μg/ml | Resistant mutants recovered
|
|||
|---|---|---|---|---|---|---|
| Exp. 1 | Exp. 2 | Exp. 3 | Total | |||
| 1 | C8-H (PD160793) | 0.34* | 300 | 444 | 563 | 1,307 |
| 2 | C8-OMe (PD161148) | 1* | 0 | 0 | 0 | 0 |
| 3 | C8-H (ciprofloxacin) | 0.1* | 287 | 175 | 770 | 1,232 |
| 4 | C8-OMe (PD135042) | 0.23* | 0 | 0 | 0 | 0 |
| 5 | C8-H (ciprofloxacin) | 0.14 | 159 | 593 | 1,032 | 1,784 |
| 6 | C8-OMe (PD135042) | 0.14 | 0 | 0 | 0 | 0 |
Concentrations used represented 10 times the MIC for wild-type E. coli (strain DM4100).
As a general strategy it is desirable to place a C8-OMe group on a fluoroquinolone already known to be effective against a particular bacterium, since other portions of the compound may make some fluoroquinolones better able to penetrate cells or to resist efflux systems. For example, ciprofloxacin, a C8-H compound, is 3 times better able to block growth of E. coli than is the C8-H compound examined above (data not shown). When we tested a C8-OMe derivative of ciprofloxacin (PD135042) at a concentration 10 times the MIC, more than 1,200 mutants were obtained with ciprofloxacin, whereas none were recovered with its C8-OMe derivative (Table 1, lines 3 and 4). Thus it is likely that many fluoroquinolones can be rendered more effective against E. coli by adding a methoxyl moiety at the C8 position. As a further test, we used the same concentration (0.14 μg/ml) of ciprofloxacin and its C8-OMe derivative on agar plates. From three independent experiments more than 1,700 mutants were obtained with ciprofloxacin and none with the C8-OMe compound (Table 1, lines 5 and 6).
Acquisition of Resistance by a Preexisting Mutant.
Since a parC (CipR) mutation lowered sensitivity to both C8-OMe and control compounds (compare Fig. 3 B and C), topoisomerase IV is a target of the fluoroquinolones examined in this study. To determine whether attack of topoisomerase IV is important to the prevention of resistance, we challenged a parC (CipR) mutant with ciprofloxacin and its C8-OMe derivative. Mutations in the genes encoding topoisomerase IV confer no resistance by themselves (resistance is seen only in combination with a gyrA mutation), and so the same fluoroquinolone concentrations used with wild-type cells could be used with the parC mutant. When 5.5 × 1011 cells of strain KD1373 (parC CipR) were plated on LB agar plates containing 0.23 μg/ml C8-OMe compound or 0.1 μg/ml ciprofloxacin (10 times MIC), equal numbers of mutants were obtained with the two compounds (212 with ciprofloxacin and 192 with the C8-OMe compound). Finding equal numbers of mutants with the two compounds contrasted dramatically with results obtained with wild-type cells (Table 1). Mutation of topoisomerase IV increased the recovery of mutants, indicating that attack of this enzyme is involved in the ability of the C8-OMe group to reduce selection of resistant mutants.
If gyrase is also a target of the C8-OMe fluoroquinolones, then mutations recovered by challenging the parC mutant with fluoroquinolone should map in one of the gyrase genes. We determined the nucleotide sequence of the quinolone-resistance region of gyrA [amino acids 67–106 (29)] in four mutants generated with strain KD1373 (parC CipR) by treatment with ciprofloxacin or its C8-OMe derivative. In the case of ciprofloxacin, the two mutants examined had a TCG to TTG mutation of gyrA that was expected to change amino acid 83 from serine to leucine. Such a mutation is commonly associated with quinolone resistance (1). The two mutants obtained with the C8-OMe compound also exhibited changes in gyrA. Both had a GAC to GCC mutation that was expected to change amino acid 82 of the GyrA protein from aspartic acid to alanine [mutation of position 82 is not commonly associated with quinolone resistance, although another example, an aspartic acid to glycine change, has been recently reported (30)]. We also carried out a complementation experiment with the two GyrA Ala-82 mutants by introducing a gyrA+-expressing plasmid, pRM386 (31). The plasmid conferred fluoroquinolone sensitivity (data not shown), as expected for GyrA-mediated resistance. These data indicate that high-level resistance to the C8-OMe compound can arise stepwise from two mutations, one in parC and a second in gyrA. The reciprocal experiment, acquisition of a second-site mutation in a gyrA (NalR) background, produced mutants only at low concentrations of the C8-OMe compound (below 0.5 μg/ml, 2.5 times MIC). We selected mutants on agar containing 0.23 μg/ml and 0.4 μg/ml PD135042; two mutants obtained at each concentration were then examined by transduction. Surprisingly, none of the mutations mapped in the gyrA, gyrB, or par regions of the chromosome (data not shown). For two mutants obtained after challenge with 0.23 μg/ml, the nucleotide sequences of the quinolone-resistance regions of parC and parE were identical to those of wild-type cells. Additional work is required to determine whether these low-level, second-step mutations map in genes involved in drug uptake, efflux, and/or detoxification.
Contribution of a C8-OMe Substituent to Growth Inhibition by Fluoroquinolones.
Increased lethality due to a C8-OMe group also made it possible to test the idea that inhibition of growth and cell death arise from distinct events (Fig. 2). Such a model can accommodate fluoroquinolone substituents having different effects on growth inhibition and lethality, whereas a single-step model cannot. To determine whether a C8-OMe group enhances growth inhibition as it does lethality, we measured bacteriostatic activity. The C8-OMe compound was less active than its C8-H control, as judged by higher values of ID50 (Table 2, lines 1 and 2) and MIC (data not shown). By the same criteria, the C8-OMe compound PD135042 was also less bacteriostatic than its C8-H control, ciprofloxacin (data not shown). Thus the C8-OMe group interfered with blocking growth, but it stimulated killing (Fig. 3). By measuring survival at drug concentrations that gave the same bacteriostatic effect (5 times ID50), we were able to estimate stimulation of lethal action: one C8-OMe compound was 16 and 57 times more lethal than its C8-H control for wild-type and gyrA (NalR) strains, respectively (Table 2, lines 6 and 7). The other C8-OMe compound (PD135042) was 76 and 140 times more effective than its C8-H control (ciprofloxacin) against wild-type and gyrA mutant cells, respectively (data not shown). Taken together, these data support the model shown in Fig. 2 and argue against C8-OMe-mediated lethality being due to enhanced drug uptake or reduced efflux, processes that should strongly affect inhibition of growth.
Table 2.
Activity of fluoroquinolones against E. coli mutants
| Line | Strain* | C8-OMe† | C8-H† | C8-OMe/C8-H |
|---|---|---|---|---|
| ID50, μg/ml | ||||
| 1 | DM4100 (WT) | 0.12 ± 0.03 | 0.039 ± 0.006 | 3 |
| 2 | GP201 (gyrAR) | 1.2 ± 0.07 | 0.82 ± 0.039 | 1.5 |
| 3 | KD1366 (gyrAR parCR)‡ | 2.1 ± 0.27 | 3.1 ± 0.13 | 0.67 |
| ID50 ratio | ||||
| 4 | GP201/DM4100 | 10 | 21 | |
| 5 | KD1366/GP201 | 1.8 | 3.8 | |
| % survivors at 5-fold ID50§ | ||||
| 6 | DM4100 (WT) | 0.07 | 1.15 | 1/16 |
| 7 | GP201 (gyrAR) | 0.11 | 6.3 | 1/57 |
WT, wild type; gyrAR, NalR; parCR, CipR.
Nearly identical results were obtained for the gyrAR parCR strain KD1368.
Cells were treated with fluoroquinolone for 1 hr, after which they were diluted and plated on LB agar. Colonies were counted after 16 hr of incubation at 37°C.
DISCUSSION
The experiments described above establish a general strategy for identifying fluoroquinolones that reduce the ability of E. coli populations to acquire resistance: new compounds are sought that effectively kill first-step, quinolone-resistant gyrase mutants and require wild-type cells to obtain two mutations to become resistant. In the cases studied, a C8-OMe group markedly improved lethal action against a gyrA mutant (Fig. 3B; Table 2, line 7); even a gyrA parC double mutant was extensively killed by a C8-OMe fluoroquinolone (Fig. 3C). When tested against wild-type cells, C8-OMe fluoroquinolones reduced the ability of E. coli to acquire resistance by at least three orders of magnitude relative to C8-H controls (Table 1), one of which (ciprofloxacin) is in wide clinical use.
An important characteristic of the C8-OMe moiety is its more pronounced effect on fluoroquinolone lethality than on inhibition of growth. For example, C8-OMe compounds exhibited bacteriostatic activity the same as or lower than C8-H derivatives against the gyrase mutant (Table 2, line 2), but the C8-OMe compounds were 10- to 100-fold more lethal (Fig. 3 and data not shown). Our interpretation of these data rests on the idea that quinolone action occurs as two sequential but distinct events in which formation of bacteriostatic quinolone–topoisomerase–DNA complexes precedes release of lethal DNA breaks (Fig. 2). Preferential stimulation of lethal events suggests that many complexes form without releasing lethal DNA breaks. The C8-OMe group appears to render the complexes more labile.
We have proposed that the release of lethal double-strand DNA breaks from gyrase-containing complexes occurs by two pathways (10). One is blocked by inhibitors of protein synthesis, such as chloramphenicol, and so it probably involves induction of a protein factor that exposes DNA breaks. A second, chloramphenicol-insensitive, mode of killing may arise from gyrase subunit dissociation, as if the quinolones pry the enzyme subunits apart. With wild-type E. coli, the latter process occurred more with the C8-OMe derivatives than with their C8-H controls (Fig. 4). The chloramphenicol-insensitive killing mode was absent when gyrA was mutant (data not shown), suggesting that in this case enhanced lethal action (Table 2, line 7) arose from the chloramphenicol-sensitive, protein-dependent pathway. We are now investigating this change in killing mode elicited by mutation of gyrA.
As pointed out above, an expected consequence of enhanced lethality against first-step resistance mutants is the need for two mutations to confer resistance. The frequency that two independent mutations will occur concomitantly is roughly the product of single mutation frequencies. Because approximately 108 to 109 cells had to be spread on agar plates to obtain a single mutant with a C8-H fluoroquinolone (data not shown), more than 1016 cells would need to be examined to obtain a double mutant. Thus the difference between the C8-OMe and C8-H compounds, with respect to acquisition of resistance, could be many orders of magnitude. Under the conditions of our experiments, that difference was at least 1,000-fold (Table 1). Even when 2 × 1013 cells were plated on agar containing PD135042 (0.14 μg/ml), no mutant was recovered. As a test for the two-mutation requirement, we compared the ability of C8-OMe and C8-H compounds to allow resistance to arise in a preexisting, first-step parC (CipR) mutant that exhibited no quinolone resistance. Equal numbers of resistant, second-step mutants were readily recovered following challenge with the two compounds, and with each fluoroquinolone the additional mutation mapped in gyrA, as expected. In the reciprocal experiment, challenge of a first-step gyrase mutant with a C8-H compound (ciprofloxacin) leads to the expected mutation in topoisomerase IV [par region by transduction (10); parE by sequence analysis (32)]. When we challenged the gyrA (NalR) strain GP201 with the C8-OMe compound, second-step mutations failed to map in genes encoding gyrase and topoisomerase IV. The absence of second-step par mutations suggests that topoisomerase IV may be only slightly more sensitive than resistant gyrase to C8-OMe fluoroquinolones. A small difference would make it difficult to obtain a drug concentration that would allow attack of topoisomerase IV but not resistant gyrase, the situation needed to select a resistant topoisomerase IV mutant. The map position of the mutations that did arise is not yet known.
Because we did not obtain a resistant mutant when wild-type cells were challenged with moderate concentrations of C8-OMe fluoroquinolones, we were unable to determine whether resistance would arise from a double mutation in gyrase or a pair of single mutations in gyrase and topoisomerase IV. We suspect that both cases occur. With respect to gyrase, the protective effect of a gyrA (NalR) mutation was less for C8-OMe than for C8-H derivatives (Fig. 3; Table 2, line 4), consistent with resistant gyrase being more vulnerable to C8-OMe fluoroquinolones than to C8-H derivatives. Topoisomerase IV is also a target, because a preexisting parC mutation eliminated differences between C8-OMe and C8-H compounds with respect to mutant acquisition. Surprisingly, addition of a parC mutation to a gyrA (NalR) mutant was about twice as protective for a C8-H compound as for its C8-OMe derivative (Table 2, line 5; compare Fig. 3 B and C, concentration required to kill 99% of cells). These data are consistent with resistant topoisomerase IV being more sensitive to C8-OMe fluoroquinolones than to C8-H derivatives.
To date, most evaluations of quinolone potency have focused on complex formation with purified topoisomerases and/or on inhibition of bacterial growth. Using these assays should make it possible to identify new quinolones that exhibit high activity against resistant gyrase or against both wild-type gyrase and topoisomerase IV. The present work shows that searches should also be directed at compounds that facilitate the release of lethal double-strand DNA breaks from complexes, because the absence of mutants cannot be explained by the C8-OMe group increasing bacteriostatic activity (Table 2, lines 1 and 2). The standard biochemical and bacteriostasis assays are likely to overlook many effective compounds. Moreover, attention should be paid to derivatives that kill nongrowing cells, because those cells can mutate and form a pool from which resistance might arise (for a recent example, see ref. 28). Addition of a C8-OMe group to fluoroquinolones is a step in the right direction with respect to the latter two considerations. However, the C8-OMe modification is imperfect: single-step gyrA resistance mutants can arise from wild-type cells when challenged with low enough concentrations (0.05 μg/ml, 2 times MIC) of PD135042. Thus moderate concentrations of this compound are required to kill single-step mutants avidly and avoid the acquisition of resistance.
In preliminary experiments we have found that C8-OMe fluoroquinolones kill a parC mutant of S. aureus and a gyrA mutant of Mycobacterium bovis BCG much more effectively than their C8-H controls. Studies with quinolone-resistant clinical isolates of several bacterial species also show that C8-modified fluoroquinolones have superior activity (16, 33–35). Thus the principles discussed in the present work probably apply to most bacterial species that are sensitive to fluoroquinolones. Indeed, the approach of using a partially resistant mutant to identify compounds that reduce selection of resistance mutations is likely to be useful with any type of antimicrobial agent for which resistance is acquired in a stepwise fashion.
Acknowledgments
We thank S. M. Friedman, M. Gellert, M. Gennaro, S. Kayman, and J.-Y. Wang for critical comments on the manuscript; L. El-Bash, P. Feng, Y.-H. Wang, and J.-F. Zhou for excellent technical assistance; A. Khodursky and N. Cozzarelli for parC mutants; and R. Menzel for a gyrA plasmid. This work was supported by National Institutes of Health Grant AI 35257.
ABBREVIATIONS
- CipR
ciprofloxacin-resistant
- NalR
nalidixic acid-resistant
- MIC
minimum inhibitory concentration
References
- 1.Drlica K, Zhao X. Microbiol Mol Biol Rev. 1997;61:377–392. doi: 10.1128/mmbr.61.3.377-392.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hooper D, Wolfson J. Quinolone Antimicrobial Agents. Washington, DC: Am. Soc. Microbiol.; 1993. [Google Scholar]
- 3.Sullivan E A, Kreiswirth B N, Palumbo L, Kapur V, Musser J M, Ebrahimzadeh A, Frieden T R. Lancet. 1995;345:1148–1150. doi: 10.1016/s0140-6736(95)90980-x. [DOI] [PubMed] [Google Scholar]
- 4.Fisher L M, Oram M, Sreedharan S. In: Molecular Biology of DNA Topoisomerases. Andoh T, editor. Boca Raton, FL: CRC; 1993. pp. 145–155. [Google Scholar]
- 5.Yoshida T, Muratani T, Iyobe S, Mituhashi S. Antimicrob Agents Chemother. 1994;38:1466–1469. doi: 10.1128/aac.38.7.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belland R J, Morrison S G, Ison C, Huang W M. Mol Microbiol. 1994;14:371–380. doi: 10.1111/j.1365-2958.1994.tb01297.x. [DOI] [PubMed] [Google Scholar]
- 7.Ferrero L, Cameron B, Manse B, Lagneaux D, Crouzet J, Famechon A, Blanche F. Mol Microbiol. 1994;13:641–653. doi: 10.1111/j.1365-2958.1994.tb00458.x. [DOI] [PubMed] [Google Scholar]
- 8.Ferrero L, Cameron B, Crouzet J. Antimicrob Agents Chemother. 1995;39:1554–1558. doi: 10.1128/aac.39.7.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khodursky A B, Zechiedrich E L, Cozzarelli N R. Proc Natl Acad Sci USA. 1995;92:11801–11805. doi: 10.1073/pnas.92.25.11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen C-R, Malik M, Snyder M, Drlica K. J Mol Biol. 1996;258:627–637. doi: 10.1006/jmbi.1996.0274. [DOI] [PubMed] [Google Scholar]
- 11.Ng E Y, Trucksis M, Hooper D C. Antimicrob Agents Chemother. 1996;40:1881–1888. doi: 10.1128/aac.40.8.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pan X-S, Ambler J, Mehtar S, Fisher L M. Antimicrob Agents Chemother. 1996;40:2321–2326. doi: 10.1128/aac.40.10.2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitamura A, Hoshino K, Kimura Y, Hayakawa I, Sato K. Antimicrob Agents Chemother. 1995;39:1467–1471. doi: 10.1128/aac.39.7.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanchez J P, Gogliotti R D, Domagala J M, Gracheck S J, Huband M D, Sesnie J A, Cohen M A, Shapiro M A. J Med Chem. 1995;38:4478–4487. doi: 10.1021/jm00022a013. [DOI] [PubMed] [Google Scholar]
- 15.Marutani K, Matsumoto M, Otabe Y, Nagamuta M, Tanaka K, Miyoshi A, Hasegawa T, Nagano H, Matsubara S, Kamide R. Antimicrob Agents Chemother. 1993;37:2217–2223. doi: 10.1128/aac.37.10.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito T, Matsumoto M, Nishino T. Antimicrob Agents Chemother. 1995;39:1522–1525. doi: 10.1128/aac.39.7.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phillips I, Culebras E, Moreno F, Baquero F. J Antimicrob Chemother. 1987;20:631–638. doi: 10.1093/jac/20.5.631. [DOI] [PubMed] [Google Scholar]
- 18.Piddock L, Wise R. FEMS Microbiol Lett. 1987;41:289–294. [Google Scholar]
- 19.Piddock L J, Walters R N, Diver J M. Antimicrob Agents Chemother. 1990;34:2331–2336. doi: 10.1128/aac.34.12.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sternglanz R, DiNardo S, Voelkel K A, Nishimura Y, Hirota Y, Becherer A K, Zumstein L, Wang J C. Proc Natl Acad Sci USA. 1981;78:2747–2751. doi: 10.1073/pnas.78.5.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pruss G, Franco R, Chevalier S, Manes S, Drlica K. J Bacteriol. 1986;168:276–282. doi: 10.1128/jb.168.1.276-282.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wall J D, Harriman P D. Virology. 1974;59:532–544. doi: 10.1016/0042-6822(74)90463-2. [DOI] [PubMed] [Google Scholar]
- 23.Miller J. Experiments in Molecular Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1972. [Google Scholar]
- 24.Crumplin G C, Smith J T. Antimicrob Agents Chemother. 1975;8:251–261. doi: 10.1128/aac.8.3.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deitz W H, Cook T M, Goss W A. J Bacteriol. 1966;91:768–773. doi: 10.1128/jb.91.2.768-773.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewin C, Howard B, Smith J. J Med Microbiol. 1991;34:19–22. doi: 10.1099/00222615-34-1-19. [DOI] [PubMed] [Google Scholar]
- 27.Howard B M, Pinney R J, Smith J T. Arzneim-Forsch. 1993;43:1125–1129. [PubMed] [Google Scholar]
- 28.Foster P. J Bacteriol. 1997;179:1550–1554. doi: 10.1128/jb.179.5.1550-1554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshida H, Bogaki M, Nakamura M, Nakamura S. Antimicrob Agents Chemother. 1990;34:1271–1272. doi: 10.1128/aac.34.6.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Truong Q, Van J-C, Shlaes D, Gutman L, Moreau N. Antimicrob Agents Chemother. 1997;41:85–90. doi: 10.1128/aac.41.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rudd K, Menzel R. Proc Natl Acad Sci USA. 1987;84:517–521. doi: 10.1073/pnas.84.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Breines D M, Ouabdesselam S, Ng E Y, Tankovic J, Shah S, Soussy C J, Hooper D C. Antimicrob Agents Chemother. 1997;41:175–179. doi: 10.1128/aac.41.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ito T, Otsuk M, Nishino T. Antimicrob Agents Chemother. 1992;36:1708–1714. doi: 10.1128/aac.36.8.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carlyn C J, Doyle L J, Knapp C C, Ludwig M D, Washington J A. Antimicrob Agents Chemother. 1995;39:1606–1608. doi: 10.1128/aac.39.7.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen M, Yoder S, Huband M, Roland G, Courtney C. Antimicrob Agents Chemother. 1995;39:2123–2127. doi: 10.1128/aac.39.9.2123. [DOI] [PMC free article] [PubMed] [Google Scholar]