

Abstract
It is now established that oxidative stress is one of the earliest, if not the earliest, change that occurs in the pathogenesis of Alzheimer's disease (AD). Consistent with this, mild cognitive impairment (MCI), the clinical precursor of AD, is also characterized by elevations in oxidative stress. Since such stress does not operate in vacuo, in this study we sought to determine whether redox-active iron, a potent source of free radicals, was elevated in MCI and preclinical AD as compared to cognitively-intact age-matched control patients. Increased iron was found at the highest levels both in the cortex and cerebellum from the pre-clinical AD/MCI cases. Interestingly, glial accumulations of redox-active iron in the cerebellum were also evident in preclinical AD patients and tend to increase as patients became progressively cognitively impaired. Our findings suggests that an imbalance in iron homeostasis is a precursor to the neurodegenerative processes leading to AD and that iron imbalance is not necessarily unique to affected regions. In fact, an understanding of iron deposition in other regions of the brain may provide insights into neuroprotective strategies. Iron deposition at the preclinical stage of AD may be useful as a diagnostic tool, using iron imaging methods, as well as a potential therapeutic target, through metal ion chelators.
Keywords: Alzheimer's disease, chelator, diagnostic, free radicals, iron, mild cognitive impairment (MCI), oxidative stress, pre-clinical, redox activity
Introduction
Alzheimer's disease (AD) is characterized by the accumulation of neurofibrillary pathology and amyloid plaques, occurring in great numbers throughout the cortex by the end of the disease [1, 2]. While considerable past efforts have focused on the role of such lesions in disease pathogenesis (e.g., [3, 4]), this is not without controversy [2, 5-7] and there is considerable evidence that such lesions may represent protective adaptations to the underlying disease [8, 9]. As such, there is an increasing awareness of other features of the disease such as mitochondrial abnormalities [10-13], aberrant protein phosphorylation [14], re-entry into the cell cycle by senescent neurons [15-17], metabolic dysfunction [18], and oxidative damage to cellular macromolecules including protein [19, 20], lipid [21], nucleic acids [22], and carbohydrates [23].
The role of oxidative stress in AD was originally thought of as nothing other than a “tombstone” end-stage manifestation [24] but is now treated as one of the earliest changes in disease pathogenesis [25, 26], occurring decades prior to more overt pathology [27, 28], and, as such, not surprisingly, is also found in the clinical precursors of AD such as mild cognitive impairment (MCI) [29, 30]. In fact, AD and MCI are indistinguishable on a number of levels related to oxidative stress including: i) plasma antioxidant status [31]; ii) peripheral DNA damage [32]; iii) antioxidant response [33]; and iv) oxidative damage [34, 35]. While these collective findings might advocate for the therapeutic value of prophylactic antioxidants [36, 37], it is of equal, if not greater, importance to attenuate the source of such free radicals and, in AD, such efforts have often focused on redox metals. In occult AD, iron, copper, and, consequently, redox active sites are all strikingly elevated [38-40].
In this study, our primary goal was to examine the cortical involvement of iron and redox active sites among preclinical and MCI subjects and compare with the cerebellum, an area often considered to be unaffected in the disease. Our findings support the extremely early contribution of iron dyshomeostasis, redox activity, and consequent oxidative stress in disease pathogenesis. Surprisingly, however, the cerebellum was also found to accumulate redox active iron in cases with mild cognitive decline and even in preclinical cases at higher levels than in normal individuals. Overall, our findings parallel the increased redox-active iron in cerebrospinal fluid (CSF) from cases of AD [41] and explain the increased level of free iron in controls in both cortex and cerebellum of AD patients [42].
Materials and Methods
Tissue
To explore iron and redox active sites in early cases of probable AD, a series of paraffin sections of the cortex and cerebellum were obtained from the Neuropathology Core Laboratory of the Washington University Alzheimer's Disease Research Center (ADRC), St. Louis. Cases representing four distinct diagnostic categories using the nomenclature and parameters as described previously (control, preclinical, CDR=0.5 and CDR=1.0) [43-45] were examined. The control cases represent cognitively normal individuals that are generally free of amyloid plaques and neurofibrillary tangles in the brain. Cognitively normal individuals that meet current pathological criteria for AD are classified as preclinical AD. Those cases with very mild dementia had CDR scores equal to 0.5, and mild dementia cases had CDR scores equal to 1.0. Included in the analysis were five control cases (ages 80-95 yr), four pre-clinical cases (ages 83-93 yr), six cases with CDR=0.5 (ages 90-102 yr), and five cases with CDR=1.0 (ages 74-96 yr). Following autopsy, brain samples of the frontal cortex and cerebellum were fixed in routine formalin and embedded in paraffin. Sections were cut at a thickness of 6 μm and placed on coated slides.
Iron histochemistry and redox analysis
Following deparaffinization and rehydration to Tris buffer (50 mM Tris, pH 7.6), sections were incubated overnight at 4°C in a solution of 7% potassium ferrocyanide in 3% HCl. This method involves binding of iron(II) cyanide (ferrocyanide) to iron(III) in situ. After rinsing three times in Tris buffer, the mixed-valence iron(II/III) complexes were detected by incubation in 0.75 mg/ml 3′-3′-diaminobenzidine in Tris buffer, with H2O2 as previously described [38].
On adjacent sections, sites of catalytic redox activity were visualized by incubation only with 0.75 mg/ml 3′-3′-diaminobenzidine in Tris buffer, with 3% H2O2 as previously described [39].
Immunocytochemistry
Amyloid-β (Aβ) deposits were detected by immunocytochemistry with the antibody 4G8 (Covance, Princeton, NJ), which detects Aβ1-42, using the peroxidase anti-peroxidase method with DAB as chromogen [46]. On some sections, glial cells were visualized with anti-glial fibrillary acidic protein (GFAP) using Alexa fluor 488 detection with fluorescence microscopy. As controls for the specificity of our techniques, some adjacent sections were incubated with chelators, EDTA, or deferroxamine (0.1M, 16 hours) as described previously [38, 39] prior to iron histochemistry or redox analysis. For each technique, all cases were stained simultaneously and developed for the same amount of time to allow for direct comparison for image analysis.
Quantification
Quantification of histology and redox activity was determined using computer assisted image analysis as described previously [47]. Briefly, images of five fields were obtained of either the iron or amyloid stained tissue sections with an Axiocam (Zeiss, Thornwood, NJ), and either the percent area stained, number of structures stained, or staining intensity was measured with the Axiovision software (Zeiss, Thornwood, NJ). For measuring staining intensity densitometrically, all stained structures within each field were measured and the background staining levels of the surrounding neuropil were subtracted. Correlation coefficients and statistical differences between groups were determined using the Student's t-test.
Results
To ensure reliability and consistency, all sections from the well-characterized control, preclinical, and MCI cases were stained for redox metals in parallel. Overall staining levels were found to be qualitatively different between the groups. In the cortical sections, iron was localized to amyloid plaques, a small number of NFT, and other intracellular locations. Representative images of the intracellular metal accumulations in sections from the different groups are shown (Figure 1 A-C). Quantification of the staining levels (mean % area covered from five representative fields) shows dramatic increases correlating with disease progression. Control patients display significantly lower amounts of cortical redox iron than the other groups (Figure 1 D; p<0.05).
Figure 1.
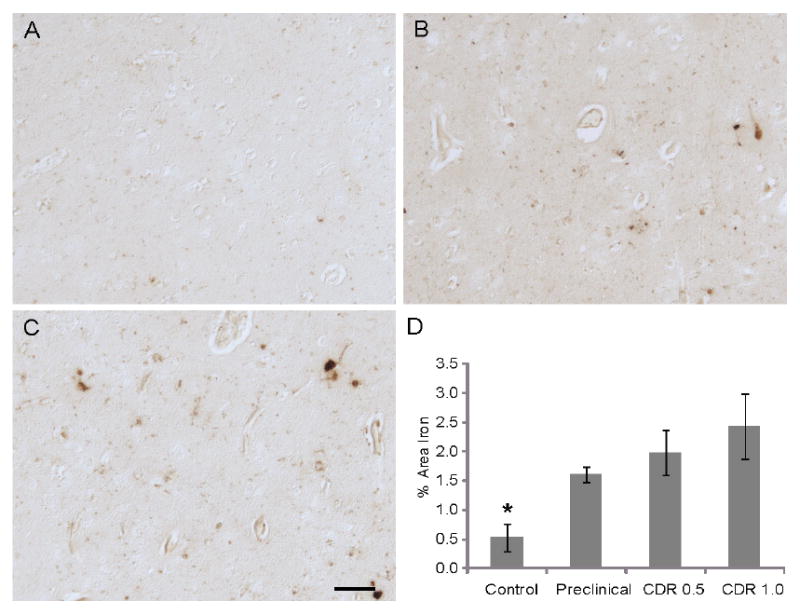
Redox metals accumulate within the cortex in preclinical/MCI cases. Cognitively normal patients (n=5) have very low levels (A), while preclinical cases (n=4) (B), and cases with either CDR scores of 0.5 (n-6) (C) or 1.0 (n=5) have substantially higher levels of cellular accumulation of redox metals. Quantification of all the cases in the study of the metal deposition of 5 fields of representatively stained cortical regions, show that metal deposition increases with disease progression and that the control cases have significantly lower levels (* p<0.05, D). Scale bar = 50 μm.
In the cerebellum, qualitatively, the controls also showed markedly less iron accumulation, but there was striking metal deposition found in the Purkinje cell layer, but rarely within the Purkinje cells in the MCI cases. Instead, smaller spherical structures were found to be positive in most of the pre-AD cases in greater numbers than the controls (Figure 2 A-D). Quantification of 5 representative fields taken at 20× magnification focusing on the Purkinje cell layer revealed the MCI cases, with CDR scores of 0.5, showed significantly greater amount of redox metal accumulation than the controls (Figure 2 E). These structures were found to be associated with glial cells, detected by immunofluorescence with antibody to GFAP (Figure 3 A-C).
Figure 2.
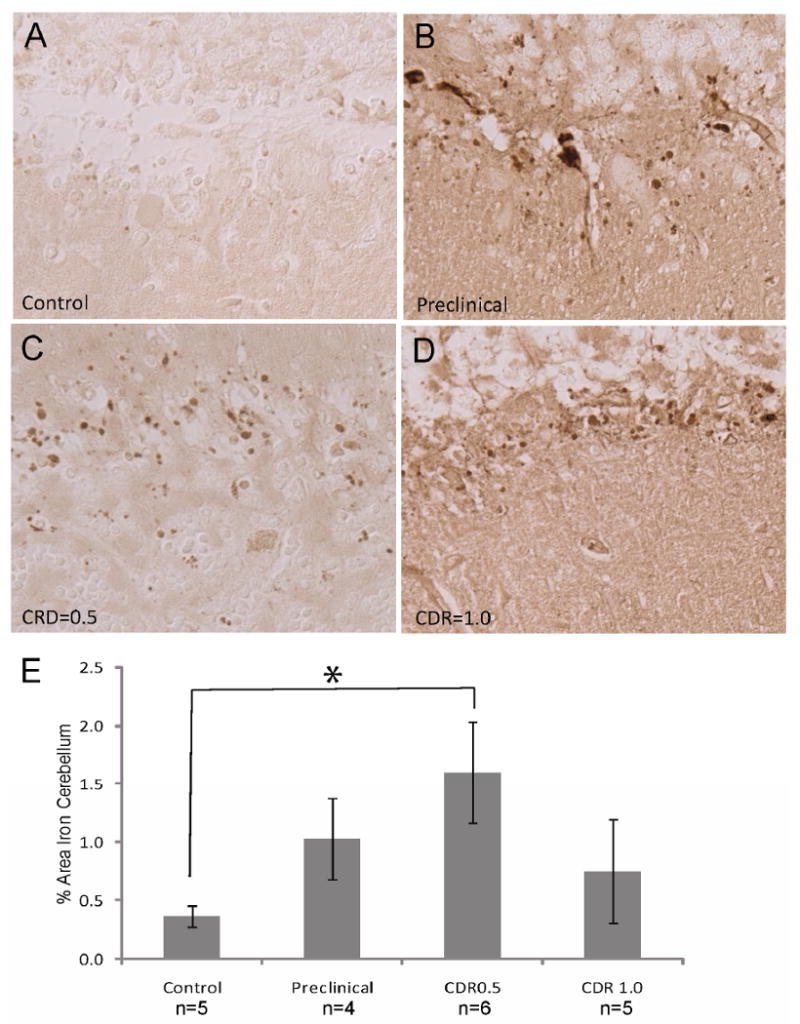
Redox metals accumulate within the cerebellum in preclinical/MCI cases. In preclinical cases (B), CDR=0.5 cases (C), and CDR=1.0 cases (D), redox metals accumulate within spherical structures within the Purkinje cell layer of the cerebellum at much higher levels than normal controls (A). Quantification reveals that only the MCI cases with CDR=0.5 show significantly greater levels of redox metal accumulation than normal controls (*p<0.05; E).
Figure 3.
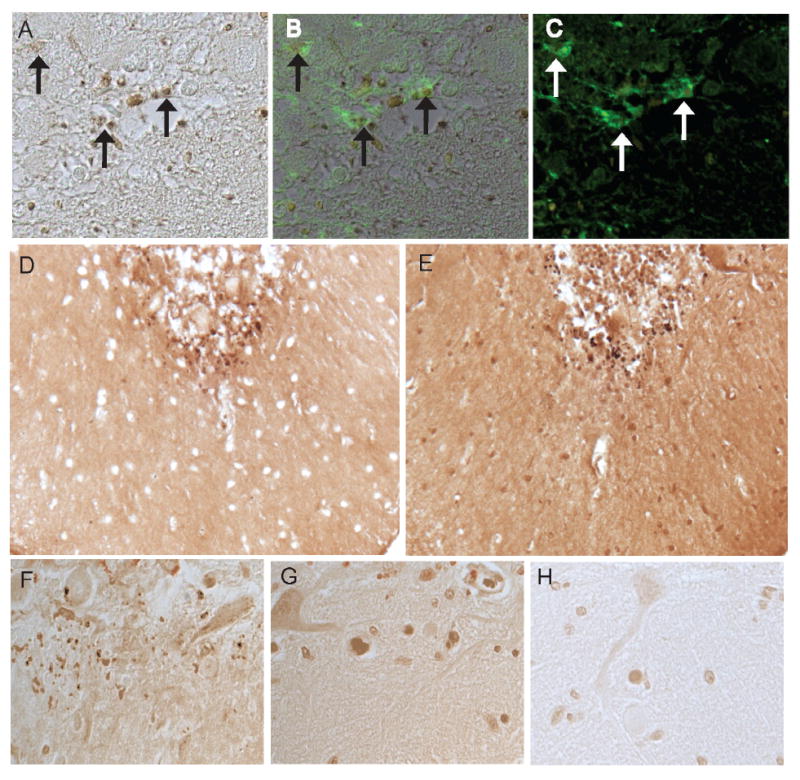
In a representative MCI case, many of the iron-positive structures (A) are associated with glial cells recognized by fluorescence microscopy using GFAP as the antibody (C). B is a merged image showing the iron-stained bodies associated with GFAP-positive glia (arrows). The same structures on adjacent serial sections are labeled using both the detection method with potassium ferrocyanide (D) as well as incubation with only DAB/ H2O2 to detect sites of metal-catalyzed redox activity (E). Redox metal positive structures within the cerebellum of an MCI case (F) are sensitive to chelation treatment with EDTA (G) and deferroxamine (H).
It should be noted that differential sites of accumulation were noted between those cases classified as controls compared to those with measurable cognitive decline or significant pathology. In some of the control cases, Purkinje cells showed a diffuse accumulation of redox metals, yet in the MCI cases, most of the metal accumulation was within the spherical glial-associated structures. The histochemical staining of these structures was abolished following chelation (Figure 3 F-H). Also, the same structures, as seen on adjacent serial sections (Figure 3 D,E) were detected by both incubation with the potassium ferrocyanide/HCl method, and with incubation of only DAB/ H2O2 for recognizing sites of metal-catalyzed redox activity. These results verify that the sites of redox activity are in fact identical to sites of metal accumulation within the brain and are evident at higher levels in the preclinical/MCI cases.
Comparisons were then made between the levels of cortical accumulation detected immunhistochemically using the monoclonal antibody 4G8, with the measured levels of redox metal accumulation. The cognitively and pathologically normal cases had no Aβ deposition, while all other cases (classified as either preclinical, CDR=0.5 or CDR=1.0) showed significant numbers of Aβ plaques and areas of diffuse amyloid deposition, similar to previously reported Aβ levels in these cases [43, 44]. The cortical Aβ density correlated significantly with the levels of cerebellar redox metal accumulation (R=0.81; p=0.02), but did not reach significance when compared to the cortical redox metal accumulation.
Discussion
In this study, we provide evidence that iron and associated iron-mediated redox activity is significantly elevated at the very earliest preclinical and MCI precursor stages of AD. Moreover, we show that iron dyshomeostasis is not restricted to cortical “affected” regions but is also evident in the cerebellum, an area often considered “unaffected”. That subsequent neurodegeneration is mainly restricted to cortical regions in occult AD individuals suggests either that iron-dyshomeostasis is necessary but not sufficient [48, 49] or that specific neuronal populations are selectively affected by a more systemic pathogenic factor. In the latter regard, it is notable that cells as diverse as fibroblasts [50] and lymphocytes [51] show evidence of systemic oxidative stress.
Mitochondria are regarded as the major repository and, as such, contributor of redox active metals in living systems, and perhaps not surprisingly given the major disturbances in iron regulation in AD [52-56], mitochondria and cellular metabolism are profoundly compromised, both structurally and chemically, in AD [57-60]. Of particular note, fewer mitochondria exhibiting normal morphology are found within AD neurons, as well as an overall reduction in size, shape, and intact cristae [10]. An abnormal distribution within cells is found in AD patients, such that mitochondria are often found restricted to the neuronal cell body, accumulated around the nucleus, as opposed to the even distribution, throughout the cell body and extending down the axon and dendrite, seen in neurons in control patients [61]. This phenomenon is likely explained by changes in mitochondrial fission and fusion proteins [13, 61] and increased mitochondria turnover [11]. The net consequence of excess iron, coupled with alterations in mitochondrial structure, function, metabolism, and dynamics, which like the iron alterations reported here, also occur at the very earliest stages of disease [62-64], undoubtedly provides an ideal environment for the generation of reactive oxygen species (ROS) through Fenton chemistry. Thus, this continued cycle of events over time could result in the clinical and pathological hallmarks known as AD.
In this study, the preclinical, CDR0.5 and CDR 1.0, cases displayed significant levels of Aβ in the cortex in accordance with the described Aβ loads previously shown to occur in cases from this series of disease classifications [43, 44]. Of note, oligomeric forms of Aβ are found at higher levels in the cortex, as well as in the cerebellum, in AD cases [65] and may directly affect mitochondrial function [13, 61]. On the other hand, cellular defense mechanisms counteract ROS to maintain adequate redox balance. When this equilibrium is lost, cellular systems are then susceptible to oxidative damage [66, 67]. As such, Aβ may be a direct sequelae of oxidative stress since both amyloid-β protein precursor and Aβ [68, 69], as well as β- and γ-secretases [70-72], are markedly upregulated under conditions of oxidative stress. While such Aβ may provide a vicious cycle scenario, Aβ may also serve a protective role through sequestration [73-75] and redox-silencing [8, 9] of metals, including iron.
While the cerebellum is often said to be unaffected by the neurodegenerative progression of AD, this erroneous belief is likely a consequence of the lack of a cerebellar clinical phenotype. Both diffuse (aka “streak”) and cored (aka “stellate”) non-neuritic tau-negative senile plaques are found in all three cerebellar layers in CDR stages 0.5 through 3 AD [76]. Indeed, there is a significant decrease in synapses [77], abnormalities in mitochondria [78], and structural abnormalities [79], as well as oxidative stress markers [80] in the cerebellum in patients with AD and MCI. Such findings are consistent with spectrophotometric analysis using bathophenanthroline, a ferrous binding chromogen, where a determination of non-heme and non-ferritin “loosely” bound iron found significantly higher levels of total iron and ferric iron in the cortex and in the cerebellum of AD cases compared to controls [42]. This pool of iron is presumably responsible for free radical formation through the Fenton reaction. The fact that in the current study, high levels of iron are found in both the cortex and cerebellum even in the preclinical cases, suggests the brain as the source of the redox active metals. In another recent study, it was found that in the CSF, while total iron was not significantly different between normal patients and those with either mild MCI, moderate MCI or AD, redox active iron in the CSF was significantly higher in the MCI cases, but not in those with definite AD [41]. In AD it has been shown that both iron and copper binding sites are present in neurofibrillary tangles and senile plaques [39], thus, while iron has been extensively studied using a variety of methodologies, the role of copper in AD development and progression cannot be ruled out. Further studies on the role of copper in preclinical cases should provide these answers.
In the current study, the large Purkinje cells do not appear to be a target of redox metal accumulation, but rather other cell populations within the cerebellum, including glial cells, appear more important. Perhaps this differential cellular attack reflects the different redox environment within the cerebellum, one that rarely shows a similar pathological profile compared to the cortical and hippocampal areas in the brain. Conversely, rather than try to fit the altered redox profile into the same mold as the rest of the brain where Aβ plaques and neurofibrillary tangles take center stage, the fact that the cerebellum in fact shows increased redox imbalance as early in the disease as the other brain areas, suggests a more systemic problem as previously reported by us [50, 81] and others [82]. In any event, the current work, taken together with other studies showing altered brain chemistry in the cerebellum, will hopefully promote more intensive research into the extent of cerebellar damage during AD and the antioxidant capabilities of cellular populations of the cerebellum. Such studies could prove useful in the development of neuroprotective strategies for regions of the brain clearly more affected by the neurodegenerative process.
In conclusion, our findings reported here demonstrate an altered redox iron state in MCI and preclinical cases of AD suggestive of an important and early contribution to the disease process prior to significant cognitive decline. Notably, cortical regions and cerebellum, as well as CSF, all display evidence of iron dyshomeostasis and oxidative imbalances early in the disease and a more comprehensive analysis of the cerebellar involvement in AD development is warranted. Given that at the onset of the earliest demonstrable disease, altered metal homeostasis is evident throughout the brain, continued research into using chelators or ionophores as potential treatment options for AD is critical. As such, ongoing efforts looking at PBT2, clioquinol, nanoparticles, and other iron chelators appear to have great potential [83-88]. Indeed, while protecting hippocampal and other cortical functions is critical for maintaining cognitive function, developing therapies focused on global brain protection, and initiating such therapies at the very earliest pre-stages of disease may prove more efficacious than targeted late treatment approaches.
Acknowledgments
Work in the authors' laboratories is supported by the National Institutes of Health (R01 AG026151, R01 AG031852) and the Alzheimer's Association.
Additionally, the National Institute on Aging (P50AG05681 and P01AG03991) supported the provision of postmortem tissue from individuals who were well-characterized during life by the Washington University Alzheimer's Disease Research Center (Dr. John C. Morris, Director).
Footnotes
Authors' disclosures available online (http://www.j-alz.com/disclosures/view.php?id=135).
References
- 1.Smith MA. Alzheimer disease. Int Rev Neurobiol. 1998;42:1–54. doi: 10.1016/s0074-7742(08)60607-8. [DOI] [PubMed] [Google Scholar]
- 2.Castellani RJ, Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Neuropathology of Alzheimer disease: pathognomonic but not pathogenic. Acta Neuropathol (Berl) 2006;111:503–509. doi: 10.1007/s00401-006-0071-y. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J. Alzheimer's disease: the amyloid cascade hypothesis: an update and reappraisal. J Alzheimers Dis. 2006;9:151–153. doi: 10.3233/jad-2006-9s317. [DOI] [PubMed] [Google Scholar]
- 4.Lee VM, Trojanowski JQ. Progress from Alzheimer's tangles to pathological tau points towards more effective therapies now. J Alzheimers Dis. 2006;9:257–262. doi: 10.3233/jad-2006-9s328. [DOI] [PubMed] [Google Scholar]
- 5.Lee HG, Perry G, Moreira PI, Garrett MR, Liu Q, Zhu X, Takeda A, Nunomura A, Smith MA. Tau phosphorylation in Alzheimer's disease: pathogen or protector? Trends Mol Med. 2005;11:164–169. doi: 10.1016/j.molmed.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 6.Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Amyloid beta: the alternate hypothesis. Curr Alzheimer Res. 2006;3:75–80. doi: 10.2174/156720506775697124. [DOI] [PubMed] [Google Scholar]
- 7.Smith MA, Casadesus G, Joseph JA, Perry G. Amyloid-beta and tau serve antioxidant functions in the aging and Alzheimer brain. Free Radic Biol Med. 2002;33:1194–1199. doi: 10.1016/s0891-5849(02)01021-3. [DOI] [PubMed] [Google Scholar]
- 8.Hayashi T, Shishido N, Nakayama K, Nunomura A, Smith MA, Perry G, Nakamura M. Lipid peroxidation and 4-hydroxy-2-nonenal formation by copper ion bound to amyloid-beta peptide. Free Radic Biol Med. 2007;43:1552–1559. doi: 10.1016/j.freeradbiomed.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 9.Nakamura M, Shishido N, Nunomura A, Smith MA, Perry G, Hayashi Y, Nakayama K, Hayashi T. Three histidine residues of amyloid-beta peptide control the redox activity of copper and iron. Biochemistry (Mosc) 2007;46:12737–12743. doi: 10.1021/bi701079z. [DOI] [PubMed] [Google Scholar]
- 10.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M, Nunomura A, Szweda LI, Aliev G, Smith MA, Zhu X, Perry G. Autophagocytosis of mitochondria is prominent in Alzheimer disease. J Neuropathol Exp Neurol. 2007;66:525–532. doi: 10.1097/01.jnen.0000240476.73532.b0. [DOI] [PubMed] [Google Scholar]
- 12.Reddy PH, Beal MF. Are mitochondria critical in the pathogenesis of Alzheimer's disease? Brain Res Brain Res Rev. 2005;49:618–632. doi: 10.1016/j.brainresrev.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 13.Wang X, Su B, Fujioka H, Zhu X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients. Am J Pathol. 2008 doi: 10.2353/ajpath.2008.071208. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Avila J. Tau protein, the main component of paired helical filaments. J Alzheimers Dis. 2006;9:171–175. doi: 10.3233/jad-2006-9s320. [DOI] [PubMed] [Google Scholar]
- 15.McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol. 1997;150:1933–1939. [PMC free article] [PubMed] [Google Scholar]
- 16.Bowser R, Smith MA. Cell cycle proteins in Alzheimer's disease: plenty of wheels but no cycle. J Alzheimers Dis. 2002;4:249–254. doi: 10.3233/jad-2002-4316. [DOI] [PubMed] [Google Scholar]
- 17.McShea A, Wahl AF, Smith MA. Re-entry into the cell cycle: a mechanism for neurodegeneration in Alzheimer disease. Med Hypotheses. 1999;52:525–527. doi: 10.1054/mehy.1997.0680. [DOI] [PubMed] [Google Scholar]
- 18.Perry G, Nunomura A, Raina AK, Aliev G, Siedlak SL, Harris PL, Casadesus G, Petersen RB, Bligh-Glover W, Balraj E, Petot GJ, Smith MA. A metabolic basis for Alzheimer disease. Neurochem Res. 2003;28:1549–1552. doi: 10.1023/a:1025678510480. [DOI] [PubMed] [Google Scholar]
- 19.Smith MA, Sayre LM, Anderson VE, Harris PL, Beal MF, Kowall N, Perry G. Cytochemical demonstration of oxidative damage in Alzheimer disease by immunochemical enhancement of the carbonyl reaction with 2,4-dinitrophenylhydrazine. J Histochem Cytochem. 1998;46:731–735. doi: 10.1177/002215549804600605. [DOI] [PubMed] [Google Scholar]
- 20.Smith MA, Rudnicka-Nawrot M, Richey PL, Praprotnik D, Mulvihill P, Miller CA, Sayre LM, Perry G. Carbonyl-related posttranslational modification of neurofilament protein in the neurofibrillary pathology of Alzheimer's disease. J Neurochem. 1995;64:2660–2666. doi: 10.1046/j.1471-4159.1995.64062660.x. [DOI] [PubMed] [Google Scholar]
- 21.Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 22.Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castellani RJ, Harris PL, Sayre LM, Fujii J, Taniguchi N, Vitek MP, Founds H, Atwood CS, Perry G, Smith MA. Active glycation in neurofibrillary pathology of Alzheimer disease: N(epsilon)-(carboxymethyl) lysine and hexitol-lysine. Free Radic Biol Med. 2001;31:175–180. doi: 10.1016/s0891-5849(01)00570-6. [DOI] [PubMed] [Google Scholar]
- 24.Mattson MP, Carney JW, Butterfield DA. A tombstone in Alzheimer's? Nature. 1995;373:481. doi: 10.1038/373481a0. [DOI] [PubMed] [Google Scholar]
- 25.Smith MA, Sayre LM, Vitek MP, Monnier VM, Perry G. Early AGEing and Alzheimer's. Nature. 1995;374:316. doi: 10.1038/374316b0. [DOI] [PubMed] [Google Scholar]
- 26.Smith MA. Oxidative stress and iron imbalance in Alzheimer disease: how rust became the fuss! J Alzheimers Dis. 2006;9:305–308. doi: 10.3233/jad-2006-9s334. [DOI] [PubMed] [Google Scholar]
- 27.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 28.Odetti P, Angelini G, Dapino D, Zaccheo D, Garibaldi S, Dagna-Bricarelli F, Piombo G, Perry G, Smith M, Traverso N, Tabaton M. Early glycoxidation damage in brains from Down's syndrome. Biochem Biophys Res Commun. 1998;243:849–851. doi: 10.1006/bbrc.1998.8186. [DOI] [PubMed] [Google Scholar]
- 29.Mecocci P. Oxidative stress in mild cognitive impairment and Alzheimer disease: a continuum. J Alzheimers Dis. 2004;6:159–163. doi: 10.3233/jad-2004-6207. [DOI] [PubMed] [Google Scholar]
- 30.Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 31.Rinaldi P, Polidori MC, Metastasio A, Mariani E, Mattioli P, Cherubini A, Catani M, Cecchetti R, Senin U, Mecocci P. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer's disease. Neurobiol Aging. 2003;24:915–919. doi: 10.1016/s0197-4580(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 32.Migliore L, Fontana I, Trippi F, Colognato R, Coppede F, Tognoni G, Nucciarone B, Siciliano G. Oxidative DNA damage in peripheral leukocytes of mild cognitive impairment and AD patients. Neurobiol Aging. 2005;26:567–573. doi: 10.1016/j.neurobiolaging.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 33.Schipper HM, Bennett DA, Liberman A, Bienias JL, Schneider JA, Kelly J, Arvanitakis Z. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol Aging. 2006;27:252–261. doi: 10.1016/j.neurobiolaging.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 34.Lovell MA, Markesbery WR. Oxidative damage in mild cognitive impairment and early Alzheimer's disease. J Neurosci Res. 2007;85:3036–3040. doi: 10.1002/jnr.21346. [DOI] [PubMed] [Google Scholar]
- 35.Butterfield DA, Sultana R. Redox proteomics identification of oxidatively modified brain proteins in Alzheimer's disease and mild cognitive impairment: insights into the progression of this dementing disorder. J Alzheimers Dis. 2007;12:61–72. doi: 10.3233/jad-2007-12107. [DOI] [PubMed] [Google Scholar]
- 36.Grant WB. Dietary links to Alzheimer's disease: 1999 update. J Alzheimers Dis. 1999;1:197–201. doi: 10.3233/jad-1999-14-501. [DOI] [PubMed] [Google Scholar]
- 37.Smith MA, Petot GJ, Perry G. Diet and oxidative stress: a novel synthesis of epidemiological data on Alzheimer's disease. J Alzheimers Dis. 1999;1:203–206. doi: 10.3233/jad-1999-14-502. [DOI] [PubMed] [Google Scholar]
- 38.Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sayre LM, Perry G, Harris PL, Liu Y, Schubert KA, Smith MA. In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer's disease: a central role for bound transition metals. J Neurochem. 2000;74:270–279. doi: 10.1046/j.1471-4159.2000.0740270.x. [DOI] [PubMed] [Google Scholar]
- 40.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer's disease senile plaques. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 41.Lavados M, Guillon M, Mujica MC, Rojo LE, Fuentes P, Maccioni RB. Mild cognitive impairment and Alzheimer patients display different levels of redox-active CSF iron. J Alzheimers Dis. 2008;13:225–232. doi: 10.3233/jad-2008-13211. [DOI] [PubMed] [Google Scholar]
- 42.Kala SV, Hasinoff BB, Richardson JS. Brain samples from Alzheimer's patients have elevated levels of loosely bound iron. Int J Neurosci. 1996;86:263–269. doi: 10.3109/00207459608986717. [DOI] [PubMed] [Google Scholar]
- 43.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 44.Goldman WP, Price JL, Storandt M, Grant EA, McKeel DW, Jr, Rubin EH, Morris JC. Absence of cognitive impairment or decline in preclinical Alzheimer's disease. Neurology. 2001;56:361–367. doi: 10.1212/wnl.56.3.361. [DOI] [PubMed] [Google Scholar]
- 45.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 46.Cash AD, Aliev G, Siedlak SL, Nunomura A, Fujioka H, Zhu X, Raina AK, Vinters HV, Tabaton M, Johnson AB, Paula-Barbosa M, Avila J, Jones PK, Castellani RJ, Smith MA, Perry G. Microtubule reduction in Alzheimer's disease and aging is independent of tau filament formation. Am J Pathol. 2003;162:1623–1627. doi: 10.1016/s0002-9440(10)64296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siedlak SL, Casadesus G, Webber KM, Pappolla MA, Atwood CS, Smith MA, Perry G. Chronic antioxidant therapy reduces oxidative stress in a mouse model of Alzheimer's disease. Free Radic Res. 2009;43:156–164. doi: 10.1080/10715760802644694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu X, Lee HG, Perry G, Smith MA. Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta. 2007;1772:494–502. doi: 10.1016/j.bbadis.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 49.Zhu X, Raina AK, Perry G, Smith MA. Alzheimer's disease: the two-hit hypothesis. Lancet Neurol. 2004;3:219–226. doi: 10.1016/S1474-4422(04)00707-0. [DOI] [PubMed] [Google Scholar]
- 50.Moreira PI, Harris PL, Zhu X, Santos MS, Oliveira CR, Smith MA, Perry G. Lipoic acid and N-acetyl cysteine decrease mitochondrial-related oxidative stress in Alzheimer disease patient fibroblasts. J Alzheimers Dis. 2007;12:195–206. doi: 10.3233/jad-2007-12210. [DOI] [PubMed] [Google Scholar]
- 51.Mecocci P, Cherubini A, Polidori MC, Cecchetti R, Chionne F, Senin U. Oxidative stress and lymphocytes in Alzheimer disease. Arch Gerontol Geriatr. 1998;26(Suppl 1):313–316. [Google Scholar]
- 52.Premkumar DR, Smith MA, Richey PL, Petersen RB, Castellani R, Kutty RK, Wiggert B, Perry G, Kalaria RN. Induction of heme oxygenase-1 mRNA and protein in neocortex and cerebral vessels in Alzheimer's disease. J Neurochem. 1995;65:1399–1402. doi: 10.1046/j.1471-4159.1995.65031399.x. [DOI] [PubMed] [Google Scholar]
- 53.Smith MA, Kutty RK, Richey PL, Yan SD, Stern D, Chader GJ, Wiggert B, Petersen RB, Perry G. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer's disease. Am J Pathol. 1994;145:42–47. [PMC free article] [PubMed] [Google Scholar]
- 54.Schipper HM, Cisse S, Stopa EG. Expression of heme oxygenase-1 in the senescent and Alzheimer-diseased brain. Ann Neurol. 1995;37:758–768. doi: 10.1002/ana.410370609. [DOI] [PubMed] [Google Scholar]
- 55.Connor JR, Snyder BS, Beard JL, Fine RE, Mufson EJ. Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer's disease. J Neurosci Res. 1992;31:327–335. doi: 10.1002/jnr.490310214. [DOI] [PubMed] [Google Scholar]
- 56.Smith MA, Wehr K, Harris PL, Siedlak SL, Connor JR, Perry G. Abnormal localization of iron regulatory protein in Alzheimer's disease. Brain Res. 1998;788:232–236. doi: 10.1016/s0006-8993(98)00002-x. [DOI] [PubMed] [Google Scholar]
- 57.Perry G, Nunomura A, Hirai K, Takeda A, Aliev G, Smith MA. Oxidative damage in Alzheimer's disease: the metabolic dimension. Int J Dev Neurosci. 2000;18:417–421. doi: 10.1016/s0736-5748(00)00006-x. [DOI] [PubMed] [Google Scholar]
- 58.Blass JP, Sheu KF, Piacentini S, Sorbi S. Inherent abnormalities in oxidative metabolism in Alzheimer's disease: interaction with vascular abnormalities. Ann N Y Acad Sci. 1997;826:382–385. doi: 10.1111/j.1749-6632.1997.tb48488.x. [DOI] [PubMed] [Google Scholar]
- 59.Martins IJ, Hone E, Foster JK, Sunram-Lea SI, Gnjec A, Fuller SJ, Nolan D, Gandy SE, Martins RN. Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer's disease and cardiovascular disease. Mol Psychiatry. 2006;11:721–736. doi: 10.1038/sj.mp.4001854. [DOI] [PubMed] [Google Scholar]
- 60.Russell RL, Siedlak SL, Raina AK, Bautista JM, Smith MA, Perry G. Increased neuronal glucose-6-phosphate dehydrogenase and sulfhydryl levels indicate reductive compensation to oxidative stress in Alzheimer disease. Arch Biochem Biophys. 1999;370:236–239. doi: 10.1006/abbi.1999.1404. [DOI] [PubMed] [Google Scholar]
- 61.Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, Thibodeau SN, Osborne D. Preclinical evidence of Alzheimer's disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 1996;334:752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- 63.Matthews PM. Molecular imaging of brain amyloid in mild cognitive impairment and Alzheimer's disease. Nat Clin Pract Neurol. 2007;3:366–367. doi: 10.1038/ncpneuro0520. [DOI] [PubMed] [Google Scholar]
- 64.Small GW. Neuroimaging and genetic assessment for early diagnosis of Alzheimer's disease. J Clin Psychiatry. 1996;57 Suppl 14:9–13. [PubMed] [Google Scholar]
- 65.Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sayre LM, Smith MA, Perry G. Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr Med Chem. 2001;8:721–738. doi: 10.2174/0929867013372922. [DOI] [PubMed] [Google Scholar]
- 67.Perry G, Nunomura A, Hirai K, Zhu X, Perez M, Avila J, Castellani RJ, Atwood CS, Aliev G, Sayre LM, Takeda A, Smith MA. Is oxidative damage the fundamental pathogenic mechanism of Alzheimer's and other neurodegenerative diseases? Free Radic Biol Med. 2002;33:1475–1479. doi: 10.1016/s0891-5849(02)01113-9. [DOI] [PubMed] [Google Scholar]
- 68.Yan SD, Yan SF, Chen X, Fu J, Chen M, Kuppusamy P, Smith MA, Perry G, Godman GC, Nawroth P, et al. Non-enzymatically glycated tau in Alzheimer's disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide. Nat Med. 1995;1:693–699. doi: 10.1038/nm0795-693. [DOI] [PubMed] [Google Scholar]
- 69.Castellani RJ, Lee HG, Perry G, Smith MA. Antioxidant protection and neurodegenerative disease: the role of amyloid-beta and tau. Am J Alzheimers Dis Other Demen. 2006;21:126–130. doi: 10.1177/153331750602100213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guglielmotto M, Aragno M, Autelli R, Giliberto L, Novo E, Colombatto S, Danni O, Parola M, Smith MA, Perry G, Tamagno E, Tabaton M. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1alpha. J Neurochem. 2009;108:1045–1056. doi: 10.1111/j.1471-4159.2008.05858.x. [DOI] [PubMed] [Google Scholar]
- 71.Tamagno E, Bardini P, Obbili A, Vitali A, Borghi R, Zaccheo D, Pronzato MA, Danni O, Smith MA, Perry G, Tabaton M. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis. 2002;10:279–288. doi: 10.1006/nbdi.2002.0515. [DOI] [PubMed] [Google Scholar]
- 72.Tamagno E, Guglielmotto M, Aragno M, Borghi R, Autelli R, Giliberto L, Muraca G, Danni O, Zhu X, Smith MA, Perry G, Jo DG, Mattson MP, Tabaton M. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J Neurochem. 2008;104:683–695. doi: 10.1111/j.1471-4159.2007.05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rottkamp CA, Raina AK, Zhu X, Gaier E, Bush AI, Atwood CS, Chevion M, Perry G, Smith MA. Redox-active iron mediates amyloid-beta toxicity. Free Radic Biol Med. 2001;30:447–450. doi: 10.1016/s0891-5849(00)00494-9. [DOI] [PubMed] [Google Scholar]
- 74.Atwood CS, Robinson SR, Smith MA. Amyloid-beta: redox-metal chelator and antioxidant. J Alzheimers Dis. 2002;4:203–214. doi: 10.3233/jad-2002-4310. [DOI] [PubMed] [Google Scholar]
- 75.Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, Atwood CS, Huang X, Farrag YW, Perry G, Bush AI. Evidence that the beta-amyloid plaques of Alzheimer's disease represent the redox-silencing and entombment of abeta by zinc. J Biol Chem. 2000;275:19439–19442. doi: 10.1074/jbc.C000165200. [DOI] [PubMed] [Google Scholar]
- 76.McKeel DW, Jr, Burns JM, Meuser TM, Morris JC. Neuropathology of preclinical and clinical Alzheimer's disease. In: McKeel DW Jr, Burns JM, Meuser TM, Morris JC, editors. Dementia: an Atlas of Investigation and Diagnosis. Clinical Publishing; Oxford, UK: 2007. pp. 71–94. [Google Scholar]
- 77.Bertoni-Freddari C, Fattoretti P, Solazzi M, Giorgetti B, Di Stefano G, Casoli T, Meier-Ruge W. Neuronal death versus synaptic pathology in Alzheimer's disease. Ann N Y Acad Sci. 2003;1010:635–638. doi: 10.1196/annals.1299.116. [DOI] [PubMed] [Google Scholar]
- 78.Bertoni-Freddari C, Fattoretti P, Paoloni R, Caselli U, Meier-Ruge W. Impaired dynamic morphology of cerebellar mitochondria in physiological aging and Alzheimer's disease. Ann N Y Acad Sci. 1997;826:479–482. doi: 10.1111/j.1749-6632.1997.tb48508.x. [DOI] [PubMed] [Google Scholar]
- 79.Thomann PA, Schlafer C, Seidl U, Santos VD, Essig M, Schroder J. The cerebellum in mild cognitive impairment and Alzheimer's disease - a structural MRI study. J Psychiatr Res. 2008;42:1198–1202. doi: 10.1016/j.jpsychires.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 80.Wang J, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J Neurochem. 2006;96:825–832. doi: 10.1111/j.1471-4159.2005.03615.x. [DOI] [PubMed] [Google Scholar]
- 81.Ghanbari HA, Ghanbari K, Harris PL, Jones PK, Kubat Z, Castellani RJ, Wolozin BL, Smith MA, Perry G. Oxidative damage in cultured human olfactory neurons from Alzheimer's disease patients. Aging Cell. 2004;3:41–44. doi: 10.1111/j.1474-9728.2004.00083.x. [DOI] [PubMed] [Google Scholar]
- 82.Mecocci P, Polidori MC, Cherubini A, Ingegni T, Mattioli P, Catani M, Rinaldi P, Cecchetti R, Stahl W, Senin U, Beal MF. Lymphocyte oxidative DNA damage and plasma antioxidants in Alzheimer disease. Arch Neurol. 2002;59:794–798. doi: 10.1001/archneur.59.5.794. [DOI] [PubMed] [Google Scholar]
- 83.Liu G, Men P, Kudo W, Perry G, Smith MA. Nanoparticle-chelator conjugates as inhibitors of amyloid-beta aggregation and neurotoxicity: A novel therapeutic approach for Alzheimer disease. Neurosci Lett. 2009;455:187–190. doi: 10.1016/j.neulet.2009.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Castellani RJ, Moreira PI, Liu G, Dobson J, Perry G, Smith MA, Zhu X. Iron: the Redox-active center of oxidative stress in Alzheimer disease. Neurochem Res. 2007;32:1640–1645. doi: 10.1007/s11064-007-9360-7. [DOI] [PubMed] [Google Scholar]
- 85.Liu G, Men P, Harris PL, Rolston RK, Perry G, Smith MA. Nanoparticle iron chelators: a new therapeutic approach in Alzheimer disease and other neurologic disorders associated with trace metal imbalance. Neurosci Lett. 2006;406:189–193. doi: 10.1016/j.neulet.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 86.Liu G, Garrett MR, Men P, Zhu X, Perry G, Smith MA. Nanoparticle and other metal chelation therapeutics in Alzheimer disease. Biochim Biophys Acta. 2005;1741:246–252. doi: 10.1016/j.bbadis.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 87.Amit T, Avramovich-Tirosh Y, Youdim MB, Mandel S. Targeting multiple Alzheimer's disease etiologies with multimodal neuroprotective and neurorestorative iron chelators. FASEB J. 2008;22:1296–1305. doi: 10.1096/fj.07-8627rev. [DOI] [PubMed] [Google Scholar]
- 88.Bush AI. Drug development based on the metals hypothesis of Alzheimer's disease. J Alzheimers Dis. 2008;15:223–240. doi: 10.3233/jad-2008-15208. [DOI] [PubMed] [Google Scholar]