

Angiogenesis or the growth of new blood vessels from existing host vessels is increasingly being recognized as important in the growth and progression of atherosclerosis, the primary cause of coronary artery and cerebrovascular disease. Neovascularization of atherosclerotic plaques was first noted by Koester in 1876(1) and later Barger et al.(2) proposed that the growth and extension of adventitial blood vessels called vasa vasorum (VV) into the intima occurs as a response to tissue hypoxia which occurs when the intima thickens beyond the diffusion limits of oxygen and nutrients (∼350uM)(3). Indeed, increases in hypoxia inducible factor alpha (HIF-1α), a transcription factor which is upregulated under hypoxic conditions and promotes hypoxia dependent neovascularization, have been found in human atherosclerotic plaques(4).
Because many of the VV that grow into the plaque are immature (i.e. they lack mural cells and competent endothelial cell junctions), they are inherently leaky, permitting inflammatory cell infiltration as well as influx of blood constituents, especially erythrocytes, into the plaque. Most of the intraplaque vessels are endothelialized but only a few possess mural pericytes and smooth muscle cells(5,6). The lack of mural cells and poorly formed endothelial cells junctions contribute to the incompetence of immature intraplaque VV. Erythrocyte membranes, rich in phospholipids and free cholesterol, and free hemoglobin (Hb), a source of oxidative damage and reactive oxygen species (ROS), contribute significantly to necrotic core expansion and inflammatory cell infiltration, two features essential to all high-risk plaques(7).
Neovessels also promote the entry of leukocytes into the plaque by upregulation of adhesion molecules, such as, ICAM-1, VCAM-1, and E-selectin(8). Increased matrix metalloproteinase release from activated macrophages and proteases secreted from mast cells cause further damage to microvessels and facilitate intraplaque hemorrhage(9).
In this issue of iJACC, work by Gossi et al. lends further support to the concept that coronary vasa vasorum (VV) neovascularization plays an important role in plaque progression through promotion of intraplaque hemorrhage(10). Using hearts from 15 patients obtained by autopsy, they demonstrate that VV density was higher in non-stenotic (defined as <50% lumen diameter stenosis) and non-calcified plaques compared to normal segments. As expected, the amount of iron and glycophorin A (an anion exchange protein specific to erythrocytes) was significantly higher in non-stenotic plaques and stenotic plaques as compared to normal and correlated with VV density.
Perhaps more novel and interesting is their finding that in calcified stenotic plaques VV spatial density was lowest yet relatively high amounts of glycophorin A and iron were found in these lesions, suggesting intraplaque hemorrhage is associated with plaque calcification.
These findings confirm earlier work linking hemorrhage to coronary plaque progression. In a relatively large number of human coronary plaques from sudden coronary death victims, Kolodgie et al. demonstrated there was a greater frequency of previous intraplaque hemorrhages in coronary atherosclerotic lesions prone to rupture compared to early lesion morphologies or stable plaques(11). Importantly, the extent of intraplaque hemorrhage corresponded to the size of the necrotic core suggesting that deposition of red cell membranes--rich in free cholesterol and phospholipids--is an important cause of core expansion and plaque progression. While additional information about the type of plaque morphology included in the non-stenotic plaque group would have strengthened the study of Gossi et al. by enhancing our understanding of how neovascularization and hemorrhage contribute to plaque progression, quantitation of VV density using micro-CT images represents significantly novel aspect of this study. Prior quantitation of VV density histologically has also been shown to correlate with plaque progression and reinforces the authors' conclusions(12).
The association of intraplaque hemorrhage with plaque calcification is a more novel aspect of this study. While coronary artery calcification correlates with the severity and extent of coronary disease at autopsy(13), there does not appear to be a linear relationship between coronary calcium and risk for future coronary events(14). Pathologically, ruptured plaques do demonstrate significantly higher calcium than thin cap fibroatheromas (i.e. vulnerable plaques) but the causes for this remain unknown(15). This same relationship holds true for mean number of VV and for hemosiderin-laden macrophages (another sign of previous hemorrhage)(12), so it is tempting to speculate that increases in coronary calcium seen in ruptured plaque may result from hemorrhage. However, neither study demonstrates a cause and effect phenomenon, which is essential to furthering our understanding of the relationship of coronary calcification and plaque stabilization. Instead, it begs the question of how and why heavy calcification occurs in human coronary atherosclerotic disease and whether it is simply a healing response to hemorrhage.
While not specifically addressed in this study, another important and related question involves understanding how and why hemorrhage occurs in human coronary atherosclerotic lesions. Recently, Sluimer et al. demonstrated abnormalities such as incomplete endothelial junctions and basement membrane detachment in human intraplaque endothelial cells(6). In addition, there was monocyte and mast cell accumulation in these areas, substantiating the concept that intraplaque microvessels are abnormal and thus provide entry points for erythrocyte and other blood cell components into the plaque. However, no change in mural cell coverage was observed from early, advanced, and ruptured coronary plaques, suggesting lack of mural cell coverage alone may not be responsible for intraplaque hemorrhage. This paradigm that pathological angiogenesis within plaques leads to hemorrhage would be strengthed by in vivo imaging data.
Further work needs to be done to understand precisely how and which molecules regulate the key pro and anti-angiogenic factors in the VV within the plaques that show hemorrhage. The mechanisms responsible for the abnormal endothelial cells morphology seen in intraplaque vessels undoubtedly involves VEGF. Angiogenesis depends both on endothelial cells invasion and proliferation as well as pericyte coverage of vascular sprouts, processes which are coordinated by VEGF and platelet derived growth factor (PDGF)(16). VEGF disrupts endothelial cell junctions and disrupts vascular smooth muscle cell function by inhibiting PDGF induced pericyte coverage of nascent vascular sprouts(17,18). Placental like growth factor (PlGF) also stimulates vascular permeability and its expression has been associated with plaque vulnerability(19). The Tie receptors, Tie1 and Tie2, and two ligands for Tie2, Ang1 and Ang2 are also critical for vessel formation and maturation. Sources of Ang1 and Ang2 are mural cells and ECs, respectively. Ang1 is known to stabilize nascent vessels and make them leak-resistant, presumably by facilitating communication between ECs and mural cells. However, the mechanism of vessel maturation by Ang1 is far from clear. The role of Ang2 appears to be contextual. In the absence of VEGF, Ang2 acts as an antagonist of Ang1 and destabilizes vessels, ultimately leading to vessel regression. In the presence of VEGF, Ang2 facilitates vascular sprouting. A high Ang2/Ang1 ratio has been found in vulnerable neovascularized plaques(20). Other molecules and growth factors have also been shown to play a role in vessel maturation.
Another avenue of interest raised by the present study is whether or not it might be possible to prevent, destroy or “normalize” neovessels within plaques. Here there are many parallels to other fields of medicine especially oncology. Given the dependence of tumor growth and metastasis on blood vessels, inhibition of new vessel formation or destruction of existing vessels of tumors has been one of the most exciting areas of cancer research for the past decade. Most of the clinical trials to date have tested agents that neutralize VEGF or inhibit its signaling.
We have recently proposed --and demonstrated both preclinically and clinically - that antiangiogenic agents can prune and “normalize” the abnormal vasculature of tumors (Figure 1)(21). Based on similarities to the structure and function of tumor vessels, could the judicious application of antiangiogenic agents prune and normalize immature intraplaque VV, thereby preventing intraplaque hemorrhage?
Figure 1. Normalization hypothesis.
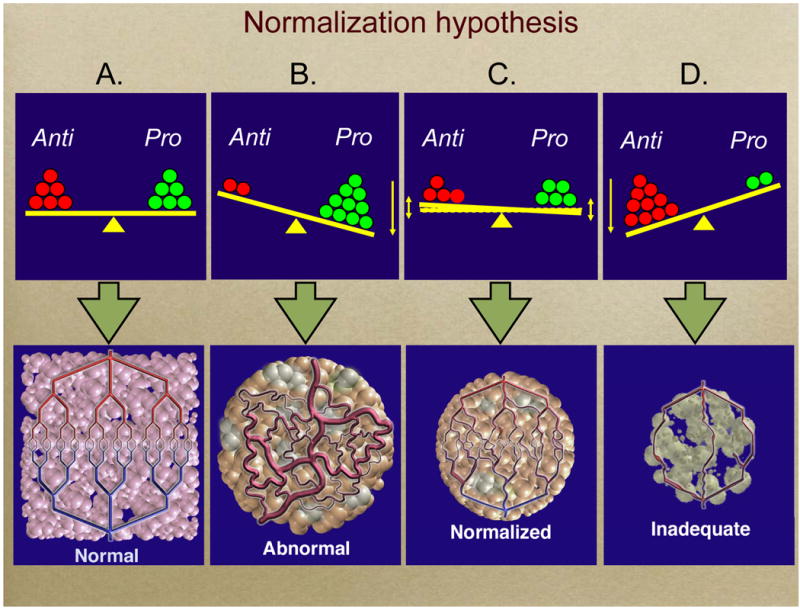
(A) (top and bottom) In normal tissue the effect of pro-angiogenic molecules (e.g., VEGF, bFGF, IL-8) is balanced by that of anti-angiogenic moelcules (e.g., thrombospondin 1, 2). Therefore blood vessels in normal tissues have normal structure and function. (B) When this balance tips in favor of pro-angiogenic molecules, the resulting vessels are abnormal structurally and functionally, often leading to hypoxia. More than 70 diseases are characterized by abnormal vasculature, e.g., cancer, atherosclerosis, and macular degeneration. (C) Our hypothesis is that judicious administration of effective antiangiogenic agents can “normalize” abnormal vessels and make them less leaky and resembling normal vasculature. (D) Higher doses of potent antiangiogenic agents may be able to completely destroy these abnormal vessels, but they may also harm the vasculature of some normal tissues. This “Janus” effect is one reason we propose to use lower doses of antiangiogenic agents rather higher doses that can flat-out inhibit angiogenesis. (From Jain RK (2005) Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy.
The successes of anti-VEGF strategies in cancer biology raise the intriguing question of whether such therapy may be applicable to atherosclerosis. However, this strategy may hold more potential pitfalls than promises given the increases risks of atherothrombotic events seen with systemic antiangiogenic therapies and the reliance of coronary collateral formation on similar angiogenic pathways (Figure 1). Newer agents that might normalize abnormal vasculature without affecting healthy vessels such as Anti-PlGF antibodies are currently being developed and may be a more tenable strategy(22,23).
In conclusion, the work of Gossi et al. lends further insights into the association of neovascularization and hemorrhage with coronary atherosclerosis. Further work needs to be done to understand the causal relationship between hemorrhage and plaque calcification. Until the regulatory molecular mechanisms of intraplaque microvessel formation are better understood we cannot be sure that the two are related or just coincidental bystanders. These and other recent data suggest that strategies to “normalize” intraplaque neovsssels to prevent hemorrhage may be promising new avenues for prevention of coronary events.
Footnotes
Competing interests: Dr. Jain reports receiving consulting fees from Dyax, AstraZeneca, Millennium, and Enlight Biosciences, lecture fees from Pfizer and Roche, and grant support from Dyax and AstraZeneca and owning equity in SynDevRx and Enlight Biosciences. AV Finn declares no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Koester W. Endarteritis amd arteritis. 1876 [Google Scholar]
- 2.Barger AC, Beeuwkes R, 3rd, Lainey LL, Silverman KJ. Hypothesis: vasa vasorum and neovascularization of human coronary arteries. A possible role in the pathophysiology of atherosclerosis. N Engl J Med. 1984;310:175–7. doi: 10.1056/NEJM198401193100307. [DOI] [PubMed] [Google Scholar]
- 3.Geiringer E. Intimal vascularization and atherosclerosis. J Pathol Bacteriol. 1951;63:201–11. doi: 10.1002/path.1700630204. [DOI] [PubMed] [Google Scholar]
- 4.Sluimer JC, Gasc JM, van Wanroij JL, et al. Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J Am Coll Cardiol. 2008;51:1258–65. doi: 10.1016/j.jacc.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 5.Dunmore BJ, McCarthy MJ, Naylor AR, Brindle NP. Carotid plaque instability and ischemic symptoms are linked to immaturity of microvessels within plaques. J Vasc Surg. 2007;45:155–9. doi: 10.1016/j.jvs.2006.08.072. [DOI] [PubMed] [Google Scholar]
- 6.Sluimer JC, Kolodgie FD, Bijnens AP, et al. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. J Am Coll Cardiol. 2009;53:1517–27. doi: 10.1016/j.jacc.2008.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moreno PR, Purushothaman KR, Sirol M, Levy AP, Fuster V. Neovascularization in human atherosclerosis. Circulation. 2006;113:2245–52. doi: 10.1161/CIRCULATIONAHA.105.578955. [DOI] [PubMed] [Google Scholar]
- 8.O'Brien KD, McDonald TO, Chait A, Allen MD, Alpers CE. Neovascular expression of E-selectin, intercellular adhesion molecule-1, and vascular cell adhesion molecule-1 in human atherosclerosis and their relation to intimal leukocyte content. Circulation. 1996;93:672–82. doi: 10.1161/01.cir.93.4.672. [DOI] [PubMed] [Google Scholar]
- 9.Kaartinen M, Penttila A, Kovanen PT. Mast cells accompany microvessels in human coronary atheromas: implications for intimal neovascularization and hemorrhage. Atherosclerosis. 1996;123:123–31. doi: 10.1016/0021-9150(95)05794-3. [DOI] [PubMed] [Google Scholar]
- 10.Gossi M, Versari D, Hildebrandt HA, et al. Journal of the American College of Cardiology Cardiovascular Imaging. 2009. Segmental heterogeneity of vaso vasorum Neovasculaization in human coronary athersclerosis and its role in intraplque hemorrhage in plaque calcification. [Google Scholar]
- 11.Kolodgie FD, Gold HK, Burke AP, et al. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–25. doi: 10.1056/NEJMoa035655. [DOI] [PubMed] [Google Scholar]
- 12.Virmani R, Kolodgie FD, Burke AP, et al. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. 2005;25:2054–61. doi: 10.1161/01.ATV.0000178991.71605.18. [DOI] [PubMed] [Google Scholar]
- 13.Eggen DA, Strong JP, McGill HC., Jr Coronary calcification. Relationship to clinically significant coronary lesions and race, sex, and topographic distribution. Circulation. 1965;32:948–55. doi: 10.1161/01.cir.32.6.948. [DOI] [PubMed] [Google Scholar]
- 14.Raggi P, Callister TQ, Cooil B, et al. Identification of patients at increased risk of first unheralded acute myocardial infarction by electron-beam computed tomography. Circulation. 2000;101:850–5. doi: 10.1161/01.cir.101.8.850. [DOI] [PubMed] [Google Scholar]
- 15.Kolodgie FD, Burke AP, Farb A, et al. The thin-cap fibroatheroma: a type of vulnerable plaque: the major precursor lesion to acute coronary syndromes. Curr Opin Cardiol. 2001;16:285–92. doi: 10.1097/00001573-200109000-00006. [DOI] [PubMed] [Google Scholar]
- 16.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–93. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 17.Suarez S, Ballmer-Hofer K. VEGF transiently disrupts gap junctional communication in endothelial cells. J Cell Sci. 2001;114:1229–35. doi: 10.1242/jcs.114.6.1229. [DOI] [PubMed] [Google Scholar]
- 18.Greenberg JI, Shields DJ, Barillas SG, et al. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature. 2008;456:809–13. doi: 10.1038/nature07424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pilarczyk K, Sattler KJ, Galili O, et al. Placenta growth factor expression in human atherosclerotic carotid plaques is related to plaque destabilization. Atherosclerosis. 2008;196:333–40. doi: 10.1016/j.atherosclerosis.2006.10.038. [DOI] [PubMed] [Google Scholar]
- 20.Post S, Peeters W, Busser E, et al. Balance between angiopoietin-1 and angiopoietin-2 is in favor of angiopoietin-2 in atherosclerotic plaques with high microvessel density. J Vasc Res. 2008;45:244–50. doi: 10.1159/000112939. [DOI] [PubMed] [Google Scholar]
- 21.Jain RK, Finn AV, Kolodgie FD, Gold HK, Virmani R. Antiangiogenic therapy for normalization of atherosclerotic plaque vasculature: a potential strategy for plaque stabilization. Nat Clin Pract Cardiovasc Med. 2007;4:491–502. doi: 10.1038/ncpcardio0979. [DOI] [PubMed] [Google Scholar]
- 22.Fischer C, Jonckx B, Mazzone M, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–75. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 23.Jain RK. A new target for tumor therapy. N Engl J Med. 2009;360:2669–71. doi: 10.1056/NEJMcibr0902054. [DOI] [PMC free article] [PubMed] [Google Scholar]