

Abstract
Gangliosides have attracted much attention due to their important biological properties. Herein, we report the first chemoenzymatic syntheses of two globo series of ganglioside oligosaccharides, Globo-H 1 and stage-specific embryonic antigen-4 (SSEA-4) 2. The common precursor SSEA-3 pentasaccharide for these two compounds was assembled rapidly using the pre-activation based one-pot glycosylation method. The stereoselectivity in forming the 1,2-cis linkage in SSEA-3 was attributed to a steric buttressing effect of the donor rather than electronic properties of the glycosyl donors. SSEA-3 was then successfully fucosylated by the fucosyltransferase WbsJ and sialylated by sialyltransferases CST-I and PmST1 producing Globo-H and SSEA-4 respectively.
Keywords: carbohydrates, chemoenzymatic synthesis, natural products, oligosaccharides, stereoselectivity
Introduction
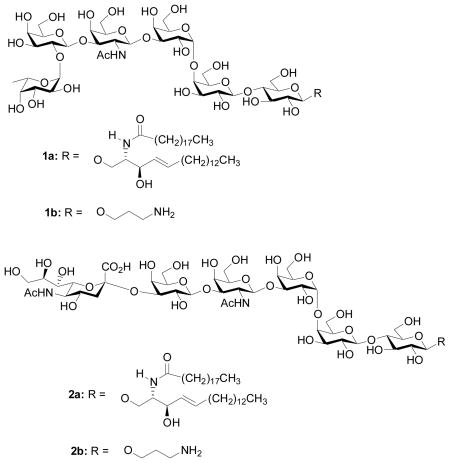
The globo series of gangliosides, including Globo-H 1 and stage-specific embryonic antigen-4 (SSEA-4) 2, possess the β-GalNAc-(1→3)-α-Gal-(1→4)-Gal oligosaccharide moiety.[1,2] This family of glycolipids has attracted much attention due to their roles as tumor-associated antigens[2] and as receptors for bacteria, viruses and toxins, which present excellent targets for novel therapeutics design.[3–5] For example, Globo-H has been found to be over-expressed on a variety of human cancer cells, including breast cancer, prostate cancer, ovarian cancer, and lung carcinomas.[6, 7] Immunotherapy using Globo-H hexasaccharide as a cancer vaccine[8] has received encouraging preliminary results against breast cancer, which is now undergoing phase II clinical trial.[9–11] SSEA-4 is believed to be involved in bacterial and viral infections, serving as receptors for uropathogenic Escherichia coli[12–14] and human parvovirus B19.[15] In addition, SSEA-4 is a marker for human cancer cells[16–19] and the expression level of SSEA-4 shows clear correlation with metastasis potential and malignancy of renal cell carcinoma.[20–23]
Due to their biological importance and the difficulties in accessing these complex oligosaccharides from natural sources, great efforts have been devoted to chemical synthesis.[1,24] Globo-H hexasaccharide has been assembled by a variety of methods including the glycal strategy,[25] the trichloroacetimidate method,[26] two-directional glycosylation,[27] automated solid phase synthesis,[28] the reactivity based one-pot method[29] as well as the pre-activation based one-pot method.[30] With the presence of the sialic acid unit at its non-reducing end, SSEA-4 presents additional synthetic challenges. Although much progress has been made in chemical sialylation,[31,32] it still remains one of the most difficult glycosidic linkages to synthesize. The carboxylic acid moiety in sialic acid also requires additional consideration for protective group compatibility in synthetic design.[33] To date, SSEA-4 has only been assembled twice by the Hasagawa[34] group and the Schmidt/Garegg group.[35] The successful completion of these highly challenging structures serves as highlights of the power of modern carbohydrate synthetic methodologies. However, despite these accomplishments, there is a continual need to further improve synthetic efficiencies of these molecules.
An attractive alternative to chemical synthesis is enzymatic synthesis.[36,37] With their high regio- and stereo-selectivities, glycosyltransferases can facilitate the formation of glycosidic bonds under aqueous conditions without relying heavily on protective groups. However, enzymatic activities can be highly dependent upon structures of both the glycosyl donor and the acceptor. An effective enzymatic synthesis may require the preparation and screening of an array of enzymes, which can be time consuming. Thus, chemoenzymatic synthesis, through the combination of the power of enzymatic synthesis with the flexibility of chemical synthesis, presents a potent approach to access complex oligosaccharides. Several gangliosides such as GM3,[38] GM2,[39] GM1,[39] GD1a,[39] Gb3[40] and SSEA-3[41] have been synthesized chemoenzymatically. Herein, we report our studies on chemoenzymatic syntheses of Globo-H 1b and SSEA-4 2b, both of which contain aminopropyl side chains and can be conjugated to protein carriers for future immunological studies.
Results and Discussion
Retrosynthetically, we envision that both Globo-H and SSEA-4 can be accessed by enzymatic glycosylation of pentasaccharide SSEA-3 3, which is also a tumor associated carbohydrate antigen.[42] The pentasaccharide 3 in turn can be chemically assembled from disaccharide 5, galactoside 6, and lactoside 7 (Figure 1). Based on our previous studies,[30] the presence of electron withdrawing benzoyl moieties on the lactoside acceptor drastically reduced the glycosylation yield. Thus, the electron donating benzyl groups are selected as protective groups masking hydroxyl groups not undergoing glycosylation in order to enhance the nucleophilicity of the acceptors as well as to simplify deprotection procedures. A side effect of using benzyl groups is that building block galactoside 6 possesses high anomeric reactivity (armed)[43] as a glycosyl donor with its multiple electron donating protective groups.[36] Therefore, in chemoselective glycosylation of thiogalactoside 6 by the thioglycoyl donor 5, cautions must be taken to prevent the undesired premature activation of galactoside 6. This can be achieved by pre-activating[44–48] disaccharide 5 first with a stoichiometric promoter, generating a reactive intermediate. Upon complete donor activation, addition of the bifunctional galactoside 6 building block to the reaction mixture will produce the desired trisaccharide. As donor activation and acceptor addition occur at two distinct steps, even though the acceptor 6 has high anomeric reactivity, it will not be activated by the promoter.
Figure 1.

Retrosynthetic analysis.
For the pre-activation based glycosylation approach, we have extensively used the p-TolSCl/AgOTf promoter system,[30,44,49–52] which generates p-TolSOTf in situ. p-TolSOTf is a powerful thiophilic agent, capable of activating thioglycosides even with very low anomeric reactivities.[48,53,54] Moreover, the side product p-tolyl disulfide produced from pre-activation is not electrophilic, thus will not activate the thioglycoside acceptor. Alternatively, reagent combinations such as benzene sulfinyl piperidine/triflic anhydride (Tf2O),[55] S-(4-methoxyphenyl) benzenethiosulfinate/Tf2O,[56] benzene sulfinyl morpholine/Tf2O,[57] and diphenylsulfoxide/Tf2O[58] have also been used as thioglycoside promoters for pre-activation reactions. However, electrophilic side products are typically generated with these promoters, which often require the addition of a quencher such as triethyl phosphite at the end of the reaction to prevent undesired acceptor activation.[45,59] This renders it difficult to carry out multiple sequential glycosylations in the same reaction flask without intermediate purification.
Disaccharide donor 5 was prepared from disaccharide 8[30] starting with treatment of sodium hydride and benzyl bromide. Reduction of the azide moiety and subsequent Troc protection produced the disaccharide donor 5 in 81% overall yield for the three steps (Scheme 1). Galactoside 6 and lactoside 7 were prepared according to literature.[30,60]
Scheme 1.

With all necessary building blocks in hand, we performed the assembly of SSEA-3 using the pre-activation based one-pot protocol.[30,44] With future automation in mind, we decided to carry out the one pot synthesis under the reaction condition established previously without optimization. Pre-activation of the disaccharide donor 5 at − 78 °C with p-TolSCl/AgOTf was followed by the addition of the bifunctional building block 6 (Scheme 2a). A sterically hindered base, 2,4,6-tri-tert-butyl-pyrimidine (TTBP)[61] was added with 6 to neutralize triflic acid generated from glycosylation. The reaction temperature was raised to −20 °C to expedite glycosylation, and the acceptor 6 was completely consumed as judged by TLC analysis. The reaction temperature was cooled back down to −78°C, followed by addition of the lactoside acceptor 7 and p-TolSCl/AgOTf. The fully protected SSEA-3 pentasaccharide 4α were obtained in 37% yield from this three component one-pot reaction sequence within six hours, which was fully characterized by 1H-NMR, 13C-NMR, gCOSY, gHMQC and HRMS. In addition, the anomer 4β was also separated in 13% yield. The presence or absence of the base TTBP did not significantly affect either the yield or the stereochemical selectivity of this reaction.[62]
Scheme 2.

Deprotection of the pentasaccharide 4α was performed by first removing the Troc group with 1 M NaOH in THF followed by acetylation. Staudinger reduction of the azide group and subsequent catalytic hydrogenation over Pearlman’s catalyst[30,50,51,63] gave the fully deprotected SSEA-3 3 in 55% overall yield for all the deprotection steps (Scheme 2b).
Formation of the challenging Gal-α-1-4-Gal linkage, stereochemical dependence on donor
We examined next the stereochemical outcome in formation of the α-Gal-(1→4)-Gal linkage, which is a major challenge in syntheses of all globo series of gangliosides, with anomeric mixture of products often obtained.[28,60,64] We have reported that glycosylation of the lactoside acceptor 7 by the galactoside 9[30] gave the Gb3 trisaccharide 10 in a combined 85% yield with an α:βratio of 27:1 (Table 1, entry 1),[30] which compared favorably with literature results on Gb3 synthesis.[28,60,64] The large difference in stereochemical selectivity between Gb3 trisaccharide 10 and SSEA-3 pentasaccharide 4 prompted us to prepare the disaccharide donor 11, the trisaccharide donor 12 and the tetrasaccharide donor 13 (Scheme 3). Pre-activation of donor 14[30] by p-TolSCl/AgOTf followed by addition of the galactoside 6 generated the disaccharide 11 in 70% yield and the glycosylation of 6 by 5 produced the trisaccharide 12 (Scheme 3a,b). The successful glycosylation of armed galactoside 6 by the disarmed donor 14 is a salient feature of the pre-activation method. One pot sequential glycosylation of fucoside 15,30 disaccharide 1630 and galactoside 6 led to the tetrasaccharide donor 13 in 60% overall yield (Scheme 3c).
Table 1.
Evaluation of the formation of the α-Gal-(1→4)-Gal linkage
|
| ||||
|---|---|---|---|---|
| Entry # | Donor | Product (Yield) | α/β | |
| 1 | 9 | 10α(82%) + 10β(3%) | 27 | |
| 2 | 11 | 17α (71%) + 17β (19%) | 3.7 | |
| 3 | 12 | 4α (60%) + 4β (23%) | 2.6 | |
| 4 | 13 | 18α (57%) + 18β (30%) | 1.9 | |
| 5 | 19 | 20α (78%) | α only | |
Scheme 3.

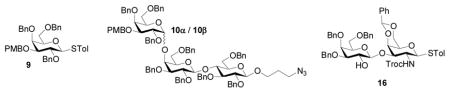
The glycosylation of lactoside 7 by donors 11, 12 and 13 were then performed. The reaction of disaccharide donor 11 with 7 gave tetrasaccharides 17α (71%) and 17β (19%) (17α:17β = 3.7:1, Table 1, entry 2). Trisaccharide 12 glycosylated the lactoside 7 with an α:β ratio of 2.6 (Table 1, entry 3), which was similar to that obtained in one-pot synthesis of SSEA-3 (Scheme 2) indicating that one-pot procedure did not significantly affect the stereoselectivity. A further reduction in α:β selectivity (1.9:1) was observed in glycosylation of 7 by the tetrasaccharide donor 13 (Table 1, entry 4). These results suggested that as the glycosyl donor became larger, the α selectivity became lower although the overall yields were similar. The stereoselectivity change must be reflective of the rate differential of axial versus equatorial approach of the nucleophile onto the reactive intermediate(s),[51, 65] because no anomeric bond isomerizations were observed when the O-glycoside products were re-submitted to the glycosylation reaction condition.
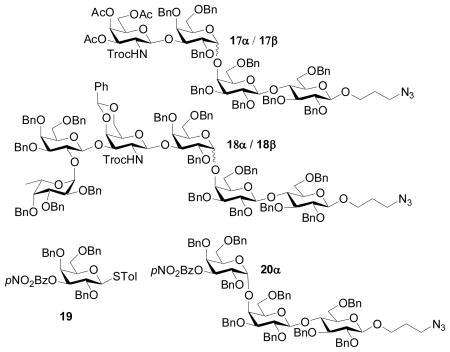
It is possible that the stereochemical divergence in glycosylating the acceptor 7 may be attributed to changes in electronic properties and/or steric encumbrance of the glycosyl donors. The introduction of a single electron withdrawing group on glycosyl donors can greatly influence the stereoselectivity.[66,67] Inductively, the glycosyl units on 3-O of the reducing end galactose in donors 11–13 can be viewed as electron withdrawing groups.[51,68] Therefore we prepared donor 19 bearing a highly electron withdrawing p-nitrobenzoyl (p-NO2Bz) moiety on its 3-O position through nitrobenzoylation of galactoside 6. Glycosylation of the lactoside 7 by the donor 19 produced the trisaccharide 20α in 78% yield with no corresponding β anomer isolated (Table 1, entry 5). This suggests that electronic properties of the donors do not play a major role in determining stereoselectivity in this case.
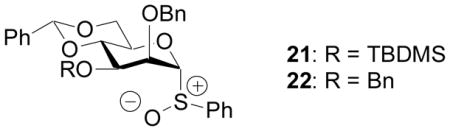
Crich and coworkers have reported that the presence of tertbutyl-dimethyl silyl (TBDMS) group on 3-O of the mannosyl donor 21 led to significant decrease in β mannoside formation compared to the corresponding reaction with the benzyl bearing donor 22.[69] This was rationalized by a steric buttressing effect as the bulky 3-O TBDMS moiety pushes the axial 2-O benzyl group towards the β face of the anomeric center, 1,2-cis (β mannosyl) glycosylation is hindered. Similar phenomena have been observed in modular synthesis of alternating β-(1→3)-β-(1→4) mannans,[70] where increased steric size on 3-O position of the donor in the form of another glycosyl unit reduced the amount of β mannoside product drastically. It is possible in our studies, the glycosyl units on the 3-O of reducing end galactose of donors 11, 12 and 13 present a steric buttressing effect, pushing the equatorial 2-O benzyl towards the anomeric center, thus reducing the amount of 1,2-cis (α galactosyl) products. As the size of the donor increases from monosaccharide 9 to tetrasaccharide 13, the effect of the steric buttressing effect becomes greater resulting in lower α selectivity.
Enzymatic synthesis of Globo-H 1a and MSGG 2a
Fucosylation of the SSEA-3 pentasaccharide 3 was carried out using WbsJ[71] with GDP-fucose. Despite the presence of three galactosyl units in SSEA-3, the 2-OH of the non-reducing terminal galactose was selectively fucosylated leading to Globo-H 1a in 71% yield (Scheme 4a), the identity of which was confirmed by NMR comparison with the Globo-H 1a assembled via chemical synthesis.[30]
Scheme 4.

Sialylation of pentasaccharide 3 was performed with the Campylobacter jejuni Cst-I sialyltransferase (construct CST-06), which cleanly transformed SSEA-3 into SSEA-4 2b with 1.5 eq of CMP-Neu5Ac (Scheme 4a). The conversion was quantitative based on TLC but the isolated yield was 34%. Presumably, some product was lost during the workup and purification processes. SSEA-3 can also be sialylated by a multifunctional Pasteurella multocida sialyltransferase PmST1[72] to form SSEA-4 2b in over 90% yield based on TLC analysis. In addition, we found that SSEA-3 is a substrate for a recombinant Photobacterium damselaα2,6-sialyltransferase Pd2,6ST[73] with a yield of 40% by TLC. These results indicate that the large size of the pentasaccharide acceptor 3 does not present an obstacle to these enzymatic glycosylation reactions.
As we have obtained the tetrasaccharide 17α, we investigated whether its fully deprotected form can also serve as an enzyme substrate. 17α was deprotected in the same manner as described for the pentasaccharide 3, producing the tetrasaccharide 23 in 50% yield (Scheme 4b). Tetrasaccharide 23 can serve as a substrate to galactosyl transferase LgtD,[41,74] providing an alternative access to SSEA-3.
In conclusion, we report the first chemoenzymatic syntheses of two tumor-associated carbohydrate antigens, Globo-H hexasaccharide 1b and SSEA-4 hexasaccharide 2b. The common precursor SSEA-3 pentasaccharide for these two compounds was assembled rapidly using the pre-activation based one-pot method. Enzymatic glycosylations of SSEA-3 by fucosyltransferase WbsJ and sialyltransferases CST-I or PmST1 led to Globo-H and SSEA-4 respectively. Interestingly, we observed that the stereoselectivities in formation of the challenging α-Gal-(1→4)-Gal linkage decreased when larger oligosaccharide donors were used. This was rationalized by a steric buttressing effect due to the glycosyl unit(s) linked to 3-O of the reducing end galactose in the donor.
Experimental Section
Characterization of anomeric stereochemistry
The stereochemistries of the newly formed glycosidic linkages in oligosaccharides are determined by 3JH1.H2 through 1H-NMRand/or 1JC1,H1 through gHMQC 2-D NMR (without 1H decoupling). Smaller coupling constants of 3JH1.H2 (around 3 Hz) indicate 1,2-cis α linkages and larger coupling constants 3JH1.H2 (7.2 Hz or larger) indicate 1,2-trans β linkages. This can be further confirmed by 1JC1,H1 (~170 Hz) for α linkages and 1JC1,H1 (~160 Hz) for β linkages.[75]
General procedure for single step pre-activation based glycosylation
A solution of donor (0.060 mmol) and freshly activated molecular sieve MS 4 Å (200 mg) in CH2Cl2 (2 mL) was stirred at room temperature for 30 minutes, and cooled to −78 °C, which was followed by addition of AgOTf (47 mg, 0.18 mmol) dissolved in Et2O (1 mL) without touching the wall of the flask. After 5 minutes, orange colored p-TolSCl (9.5 μL, 0.060 mmol) was added through a microsyringe. Since the reaction temperature was lower than the freezing point of p-TolSCl, p-TolSCl was added directly into the reaction mixture to prevent it from freezing on the flask wall. The characteristic yellow color of p-TolSCl in the reaction solution dissipated rapidly within a few seconds indicating depletion of p-TolSCl. After the donor was completely consumed according to TLC analysis (about 5 minutes at −78 °C), a solution of acceptor (0.060 mmol) in CH2Cl2 (0.2 mL) was slowly added dropwise via a syringe. The reaction mixture was warmed to − 20 °C under stirring in 2 hours. Then the mixture was diluted with CH2Cl2 (20 mL) and filtered over Celite. The Celite was further washed with CH2Cl2 until no organic compounds were observed in the filtrate by TLC. All CH2Cl2 solutions were combined and washed twice with saturated aqueous solution of NaHCO3 (20 mL) and twice with water (10 mL). The organic layer was collected and dried over Na2SO4. After removal of the solvent, the desired disaccharide was purified from the reaction mixture via silica gel flash chromatography.
p-Tolyl 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→3)-4,6-benzylidene-2-(2,2,2-tri-chloroethoxycarbonylamino)-2-deoxy-1-thio-β-D-galactopyranoside (5)
Galactoside 8[30] (0.4 g, 0.48 mmol) was dissolved in DMF (10 mL) and the solution was cooled to 0 °C. NaH (0.029 g, 60% NaH in mineral oil, 0.72 mmol) was added in portions, followed by addition of benzyl bromide (0.086 mL, 0.72 mmol). The mixture was stirred at room temperature under N2 for 2 h and then diluted with EtOAc (50 mL). The mixture was washed with saturated NaHCO3, water and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded p-tolyl 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→3)-2-azido-4,6-benzylidene-2-deoxy-1-thio-β-D-galactopyranoside S1 as white solid (0.44 g, quantitative); S1 (0.44 g, 0.48 mmol), 1,3-propanedithiol (0.48 mL, 4.8 mmol) and Et3N (0.67 mL, 4.8 mmol) were dissolved in a mixture of CH2Cl2/MeOH (5 mL each). The mixture was heated at reflux overnight under N2 and then concentrated. The resulting residue was diluted with CH2Cl2 (60 mL) and then washed with saturated aqueous solution of NaHCO3 and water, dried over Na2SO4, filtered and concentrated. The resulting residue was purified by quickly passing through a short silica gel column (20:1, CH2Cl2–MeOH) and the obtained solid and solid NaHCO3 (0.078 g, 0.93 mmol) were put into THF (5 mL) and then TrocCl (0.075 mL, 0.56 mmol) was added. The mixture was stirred at room temperature under N2 for 4 hours and filtrated. The filtrate was concentrated and then diluted with CH2Cl2 (50 mL). The mixture was washed with water and brine, dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded compound 5 as white solid (0.42 g, 81% for two steps).
3-Azidopropyl 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→3)-4,6-benzylidene-2-(2,2,2-tri-chloroethoxycarbonylamino)-2-deoxy-β-D-galactopyranosyl-(1→3)-2,4,6-tri-O-benzyl-α-D-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (4α) and 3-azidopropyl 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→3)-4,6-benzylidene-2-(2,2,2-tri-chloroethoxycarbonylamino)-2-deoxy-β-D-galactopyranosyl-(1→3)-2,4,6-tri-O-benzyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (4β)
After donor 5 (50 mg, 46.7 μmol) and activated molecular sieve MS-4 Å (500 mg) were stirred for 30 minutes at room temperature in CH2Cl2 (3 mL), the solution was cooled to − 78 °C, followed by addition of AgOTf (36 mg, 140 μmol) in Et2O (1.5 mL). The mixture was stirred for 5 minutes at − 78 °C and then p-TolSCl (7.4 μL, 46.7 μmol) was added into the solution. (See the general procedure for single step pre-activation based glycosylation for precautions) The mixture was vigorously stirred for 10 minutes, followed by addition of a solution of acceptor 6 (22.1 mg, 39.7 μmol) and TTBP (6.9 mg, 46.7 μmol) in CH2Cl2 (1 mL). The reaction mixture was stirred for 2 hours from − 78 to − 20 °C and then the mixture was cooled down to − 78 °C, followed by sequential additions of AgOTf (12 mg, 46.7 μmol) in Et2O (1 mL), acceptor 7 (22.5 mg, 23.3 μmol) and TTBP (6.9 mg, 46.7 μmol) in CH2Cl2 (1 mL). The mixture was stirred for 5 minutes at − 78 °C and then p-TolSCl (6.3 μL, 39.7 μmol) was added into the solution. The reaction mixture was stirred for 3 hours from − 78 to 10 °C and then was quenched with Et3N (40 μL), concentrated under vacuum to dryness. The resulting residue was diluted with CH2Cl2 (30 mL), followed by filtration. The organic phase was washed with saturated aqueous NaHCO3, H2O and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded 20 mg of 4α (37%) and 7.2 mg of 4β (13%) respectively as colorless gel.
Compounds 4α and 4β were also synthesized from donor 12 and acceptor 7 in 60% and 23% yield respectively as colorless gel following the general procedure of single step glycosylation.
3-Aminopropyl β-D-galactopyranosyl-(1→3)-2-acetamido-2-deoxy-β-D-galactopyranosyl-(1→3)-α-D-galactopyranosyl-(1→4)-β-D-galactopyranosyl-(1→4)-β-D-glucopyranoside (3)
The mixture of compound 4α (60 mg, 25.5 μmol), 1 M NaOH (4 mL, 0.4 mmol) and THF (4 mL) was stirred at 50 °C overnight and then concentrated to dryness. The resulting residue was diluted with CH2Cl2 (30 mL) and the organic phase was washed by H2O and then dried over Na2SO4, filtered and concentrated to dryness. The resulting residue was dissolved in methanol (2 mL) and acetic anhydride (24 μL, 0.25 mmol) was added dropwise and the mixture was stirred at room temperature under N2 overnight. The reaction was quenched by adding a few drops of H2O and then diluted with CH2Cl2 (30 mL). The organic phase was washed with saturated aqueous solution of NaHCO3, H2O and then dried over Na2SO4, filtered and concentrated to dryness. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded the N-acetylation product as white solid. The mixture of the N-acetylation product, 1 M of PMe3 in THF (0.5 mL, 0.5 mmol), 0.1 M NaOH (0.5 mL, 0.05 mmol) and THF (4 mL) was stirred at 60 °C under N2 overnight. The mixture was concentrated and the resulting residue was diluted with CH2Cl2 (30 mL). The organic phase was washed with H2O and then dried over Na2SO4, filtered and concentrated to dryness. The resulting residue was purified by quickly passing through a short silica gel column (10:1, CH2Cl2–MeOH). The mixture of the obtained solid and Pd(OH)2 in MeOH/H2O/HOAc (3 mL/1 mL/1 mL) was stirred under H2 at room temperature overnight and then filtered. The filtrate was concentrated to dryness under vacuum and then was co-evaporated with H2O (10 mL) three times to remove the HOAc. The aqueous phase was further washed with CH2Cl2 (5 mL × 3) and EtOAc (5 mL×3)and then the aqueous phase was dried under vacuum to afford compound 3 (acetate salt) as white solid (13 mg, 55% for four steps).
p-Tolyl 3,4,6-tri-O-acetyl-2-(2,2,2-tri-chloroethoxycarbonylamino)-2-deoxy-β-D-galactopyranosyl-(1→3)-2,4,6-tri-O-benzyl-1-thio-β-D-galactopyranoside (11)
Compound 11 was synthesized from donor 14 and acceptor 6 in 70% yield following the general procedure of glycosylation.
p-Tolyl 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→3)-4,6-benzylidene-2-(2,2,2-tri-chloroethoxycarbonylamino)-2-deoxy-β-D-galactopyranosyl-(1→3)-2,4,6-tri-O-benzyl-1-thio-β-D-galactopyranoside (12)
Compound 12 was synthesized from donor 5 and acceptor 6 in 71% yield following the general procedure of single step glycosylation.
3-Azidopropyl 2,3,4-tri-O-benzyl-α-L-fucopyranosyl-(1→2)-3,4,6-tri-O-benzyl-β-D-galactopyranosyl-(1→3)-4,6-benzylidene-2-(2,2,2-tri-chloroethoxycarbonylamino)-2-deoxy-β-D-galactopyranosyl-(1→3)-2,4,6-tri-O-benzyl-1-thio-β-D-galactopyranoside (13)
Compound 13 was synthesized by a three component one-pot synthesis procedure. After the donor p-tolyl 2,3,4-tri-O-benzyl-1-thio-β-L-fucopyranoside 15 [30] (50 mg, 92.47 μmol) and activated molecular sieve MS-4 Å (500 mg) were stirred for 30 minutes at room temperature in Et2O (4 mL), the solution was cooled to − 78 °C, followed by addition of AgOTf (72 mg, 277.4 μmol) in Et2O (1.5 mL). The mixture was stirred for 5 minutes at − 78 °C and then p-TolSCl (14.7 μL, 92.47 μmol) was added into the solution. (See the general procedure for single step pre-activation based glycosylation for precautions) The mixture was vigorously stirred for 10 minutes, followed by addition of a solution of acceptor p-tolyl 3,4,6-tri-O-benzyl-β-D-galactopyranosyl-(1→3)-4,6-benzylidene-2-(2,2,2-trichloroethoxycarbonylamino)-2-deoxy-1-thio-β-D-galactopyranoside 16[30] (77.1 mg, 78.60 μmol) and TTBP (23 mg, 92.47 μmol) in CH2Cl2 (1 mL). The reaction mixture was stirred for 2 hours from − 78 to − 20 °C and then the mixture was cooled down to − 78 °C, followed by addition of AgOTf (24 mg, 92.47 μmol) in Et2O (1 mL). The mixture was stirred for 10 minutes at − 78 °C and then p-TolSCl (12.5 μL, 78.60 μmol) was added into the solution. After stirred for 5 minutes, a solution of acceptor 6 (30.9 mg, 55.48 μmol) and TTBP (23 mg, 92.47 μmol) in CH2Cl2 (1 mL) was added slowly along the flask wall into the mixture and the reaction mixture was stirred for 2 h from − 78 to − 20 °C and then reaction was quenched with Et3N (40 μL), concentrated under vacuum to dryness. The resulting residue was diluted with CH2Cl2 (30 mL), followed by filtration. The organic phase was washed with saturated aqueous NaHCO3, H2O and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded 13 as colorless gel (60.6 mg, 60 %).
3-Azidopropyl 3,4,6-tri-O-acetyl-2-(2,2,2-tri-chloroethoxycarbonylamino)-2-deoxy-β-D-galactopyranosyl-(1→3)-2,4,6-tri-O-benzyl-α-D-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-D-galactopyranosyl-(1→3)-2,4,6-tri-O-benzyl-β-D-glucopyranoside (17α) and 3-azidopropyl 3,4,6-tri-O-acetyl-2-(2,2,2-tri-chloroethoxycarbonylamino)-2-deoxy-β-D-galactopyranosyl-(1→3)-2,4,6-tri-O-benzyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-D-galactopyranosyl-(1→3)-2,4,6-tri-O-benzyl-β-D-glucopyranoside (17β)
Compound 17α and 17β were synthesized from donor 11 and acceptor 7 in 71% and 19% yield respectively as colorless gel following the general procedure of single step glycosylation.
3-Azidopropyl 2,3,4-tri-O-benzyl-α-L-fucopyranosyl-(1→2)-3,4,6-tri-O-benzyl-β-D-galactopyranosyl-(1→3)-4,6-benzylidene-2-(2,2,2-tri-chloroethoxycarbonylamino)-2-deoxy-β-D-galactopyranosyl-(1→3)-2,4,6-tri-O-benzyl-α-D-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (18α) and 3-azidopropyl 2,3,4-tri-O-benzyl-α-L-fucopyranosyl-(1→2)-3,4,6-tri-O-benzyl-β-D-galactopyranosyl-(1→3)-4,6-benzylidene-2-(2,2,2-tri-chloroethoxycarbonylamino)-2-deoxy-β-D-galactopyranosyl-(1→3)-2,4,6-tri-O-benzyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (18β)
Compound 18α and 18β were synthesized from donor 13 and acceptor 7 in 57% and 30% yield respectively as colorless gel following the general procedure of single step glycosylation. The identities of which were confirmed by NMR comparison with literature data.[30]
p-Tolyl 2,4,6-tri-O-benzyl-3-O-p-nitrobenzoyl-1-thio-β-D-galactopyranoside (19)
Compound 6 (43.5 mg, 0.078 mmol) and N,N-dimethylamino pyridine (9.5 mg, 0.078 mmol) was dissolved in CH2Cl2 (5 mL), followed by the addition of p-nitrobenzoyl chloride (10.9 μL, 0.09 mmol). The mixture was stirred overnight at room temperature and then diluted with CH2Cl2 (20 mL), washed with saturated aqueous NaHCO3 and H2O and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (4:1 Hexanes–EtOAc) afforded 19 as gel-like solid (50.6 mg, 92%).
3-Azidopropyl 2,4,6-tri-O-benzyl-3-O-p-nitrobenzoyl-α-D-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (20α)
After donor 19 (30 mg, 42 μmol), acceptor 7 (37 mg, 38 μmol) and activated molecular sieve MS-4 Å (200 mg) were stirred for 30 min at room temperature in a mixture solvent of Et2O (1 mL) and CH2Cl2 (2 mL), the mixture was cooled to − 78 °C, followed by addition of AgOTf (33 mg, 0.127 mmol) in Et2O (1 mL). The mixture was vigorously stirred for 10 min and then p-TolSCl (6.74 μL, 42 μmol) was added and the reaction mixture was stirred for 2 h from − 78 to − 20 °C. The reaction was quenched by Et3N and then concentrated under vacuum to dryness. The resulting residue was diluted with CH2Cl2 (10 mL), followed by filtration. The organic phase was washed with saturated aqueous NaHCO3 and H2O and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (3:1 Hexanes–EtOAc) afforded 20α as gel-like solid (46.2 mg, 78%).
3-Aminopropyl α-L-fucopyranosyl-(1→2)-β-D-galactopyranosyl-(1→3)-2-acetamido-2-deoxy-β-D-galactopyranosyl-(1→3)-α-D-galactopyranosyl-(1→4)-β-D-galactopyranosyl-(1→4)-β-D-glucopyranoside (1b)
To a mixture of 10 mM SSEA-3 pentasaccharide 3, 15 mM GDP-fucose in 20 mM Tris-HCl buffer (pH 7.5) was added α1,2 fucosyltransferase WbsJ (7 mU). The mixture was incubated at room temperature for 2 days. The Globo-H hexasaccharide 1b was purified by BioGel P-2 gel filteration column (BioRad) followed by Dowex 1x8-400 ion exchange resin (Chloride form) with water as the mobile phase. The fractions containing the desired product 1b (71%) was collected and lyophilized. NMR data of 1b were collected, which were identical to those obtained from Globo-H 1b synthesized chemically.[30]
3-Aminopropyl 5-acetamido-3,5-dideoxy-D-glycero-α-D-galacto-non-2-ulopyranosylonic acid-(2→3)-β-D-galactopyranosyl-(1→3)-2-acetamido-2-deoxy-β-D-galactopyranosyl-(1→3)-α-D-galactopyranosyl-(1→4)-β-D-galactopyranosyl-(1→4)-β-D-glucopyranoside (2b)
The Campylobacter jejuni CST-I α-2,3-sialyltransferase (construct CST-06) was expressed as a fusion protein with the E. coli maltose-binding protein and purified.[39] To a mixture of 10 mM MgCl2, 5 mM SSEA-3 pentasaccharide 3, 7.5 mM CMP-Neu5Ac in Hepes buffer (50 mM, pH 7.5) was added CST-Iα-2,3-sialyltransferase (0.32 U/mL). The reaction was incubated at 37°C for 2 h and TLC (BPH:AMW = 1:1, BPH 1:2:1 nbutanol: propanol: 0.1 M HCl; AMW 1:1:1 CH3CN: methanol: water) showed complete consumption of the pentasaccharide 3. Multiple reactions were set to convert all of the pentasaccharide 3 (9 mg, 9.1 μmol). The reaction volume was reduced using a centrifugal concentrator and the material was loaded (in 3 runs) on a 10 mm × 30 cm Superdex Peptide column (GE Healthcare). The column was eluted with an ammonium acetate buffer (pH 7, 20 mM) at 0.5 mL/min. The product was eluted between 7 and 8 mL. The fractions were pooled and lyophilized three times to remove the ammonium acetate. We recovered 4 mg (3.1 μmol) of purified 2b with a yield of 34%. As the conditions for enzymatic synthesis were very mild and previously we did not observe any product decomposition using this enzyme,[39] the low isolated yield was probably due to multiple small scale reactions performed resulting in loss of product during workup and purification.
Enzymatic sialylation of pentasaccharide SSEA-3 using PmST1 and Pd2,6ST
The enzymatic assays were carried out in a total volume of 10 μL in a Tris–HCl buffer (100 mM, pH 8.5) containing CMP–Neu5Ac (20 mM), SSEA-3 3 (10 mM), MgCl2 (20 mM) and the corresponding sialyltransferases (PmST1 or Pd2,6ST). Reactions were allowed to proceed for 60 min at 37 °C. The reactions were then monitored by TLC (BPH: AMW= 1:1) and the products formed were confirmed by MS. ESI-MS for sialylation product catalysed by PmST1: [M+Na]+ C46H78N3Na2O34 calcd 1262.43, obsd 1262.29; ESI-MS for sialylation product catalysed by Pd2,6ST: [M+Na]+ C46H78N3Na2O34 calcd 1262.43, obsd 1262.28.
3-Aminopropyl 2-acetamido-2-deoxy-β-D-galactopyranosyl-(1→3)-α-D-galactopyranosyl-(1→4)-β-D-galactopyranosyl-(1→4)-β-D-glucopyranoside (23)
The mixture of compound 17α (0.050 g, 0.027 mmol), 1 M NaOMe (4.0 mL, 4.0 mmol) and THF (4 mL) was stirred at 50 °C overnight and then concentrated to dryness. The resulting residue was diluted with CH2Cl2 (50 mL) and the organic phase was washed by H2O and then dried over Na2SO4, filtered and concentrated to dryness. The resulting residue was dissolved in methanol (3 mL). Acetic anhydride (1.0 mL) was added dropwise and the mixture was stirred at room temperature under N2 for 6 hours. The reaction was quenched by adding EtOH and then diluted with EtOAc (20 mL). The organic phase was washed with saturated aqueous solution of NaHCO3, H2O and then dried over Na2SO4, filtered and concentrated to dryness. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded the N-acetylation product as white solid. The mixture of the N-acetylation product, 1 M of PMe3 in THF (0.5 mL, 0.5 mmol) and THF (3 mL) was stirred at 50 °C under N2 overnight. The mixture was concentrated and the resulting residue was diluted with CH2Cl2 (50 mL). The organic phase was washed with H2O and then dried over Na2SO4, filtered and concentrated to dryness. The resulting residue was purified by quickly passing through a short silica gel column (10:1, CH2Cl2–MeOH). The mixture of the obtained solid and Pd(OH)2 in MeOH/H2O/HOAc (5 mL, 3:1:1) was stirred under H2 at room temperature overnight and then filtered. The filtrate was concentrated to dryness under vacuum and then was co-evaporated with H2O (10 mL) three times to remove the HOAc. The aqueous phase was further washed with CH2Cl2 (5 mL × 3) and EtOAc (5 mL × 3) and then the aqueous phase was dried under vacuum to afford compound 23 as white solid (10.4 mg, 50% for three steps).
Enzymatic galactosylation of tetrasaccharide 23
To a mixture of 10 mM tetrasaccharide 23, 20 mM UDP-galactose, 1 mM MnCl2, 1 mM dithiothreitol and 1 % BSA in 20 mM Tris-HCl buffer (pH 7.5) was added β1,3 galactosyltransferase (LgtD) (2 mU). The mixture was incubated at room temperature for 2 days. The SSEA-3 penta-saccharide 3 (70%) was purified by BioGel P-2 gel filteration column (BioRad) followed by Dowex 1x8-400 ion exchange resin (chloride form) with water as the mobile phase.
Supplementary Material
Acknowledgments
We are grateful for financial supports from the National Institutes of Health (R01-GM-72667 to XH, R01GM076360 to XC). We would like to thank Dr. Warren Wakarchuk (National Research Council Canada) for helpful suggestions, Dr. Jianjun Li (National Research Council Canada) for mass spectrometry analysis of the sialylated product and Marie-France Karwaski (National Research Council Canada) for technical help.
Footnotes
Dedicated to Prof. Chi-Huey Wong on the occasion of his 60th birthday for his ground breaking contributions to chemistry.
Supporting information Available: Supporting information for this article is available on the WWW under http://asc.wiley-vch.de/home/.
References
- 1.Ishida H, Kiso M. Trends Glycosci Glycotech. 2001;13:57–64. [Google Scholar]
- 2.Hakomori S, Zhang Y. Chem Biol. 1997;4:97–104. doi: 10.1016/s1074-5521(97)90253-2. [DOI] [PubMed] [Google Scholar]
- 3.Wedeking A, van Echten-Deckert G. Curr Org Chem. 2007;11:579–598. [Google Scholar]
- 4.Izumi M, Uzawa H. Trends Glycosci Glycotech. 2005;17:107–119. [Google Scholar]
- 5.Birkle S, Zeng G, Gao L, Yu RK, Aubry J. Biochimie. 2003;85:455–463. doi: 10.1016/s0300-9084(03)00006-3. [DOI] [PubMed] [Google Scholar]
- 6.Zhang S, Zhang HS, Reuter VE, Slovin SF, Scher HI, Livingston PO. Clin Cancer Res. 1998;4:295–302. [PubMed] [Google Scholar]
- 7.Bremer EG, Levery SB, Sonnino S, Ghidoni R, Canevari S, Kannagi R, Hakomori S. J Biol Chem. 1984;259:14773–14777. [PubMed] [Google Scholar]
- 8.Danishefsky SJ, Allen JR. Angew Chem Int Ed. 2000;39:836–863. doi: 10.1002/(sici)1521-3773(20000303)39:5<836::aid-anie836>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 9.Gilewske T, Ragupathi G, Bhuta S, Williams LJ, Musselli C, Zhang XF, Bencsath KP, Panageas KS, Chin J, Hudis CA, Norton L, Houghton AN, Livingston PO, Danishefsky SJ. Proc Natl Acad Sci U S A. 2001;98:3270–3275. doi: 10.1073/pnas.051626298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang ZG, Williams LJ, Zhang XF, Zatorski A, Kudrya-shov V, Ragupathi G, Spassova M, Bornmann W, Slovin SF, Scher HI, Livingston PO, Lloyd KO, Danishefsky SJ. Proc Natl Acad Sci U S A. 2000;97:2719. doi: 10.1073/pnas.97.6.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slovin SF, Ragupathi G, Adluri S, Ungers G, Terry K, Kim S, Spassova M, Bornmann WG, Fazzari M, Dantis L, Olkiewicz K, Lloyd KO, Livingston PO, Danishefsky SJ, Scher HI. Proc Natl Acad Sci U S A. 1999;96:5710. doi: 10.1073/pnas.96.10.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stapleton AE, Stroud MR, Hakomori SI, Stamm WE. Infect Immun. 1998;66:3856–3861. doi: 10.1128/iai.66.8.3856-3861.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stroud MR, Stapleton AE, Levery SB. Biochem. 1998;37:17420–17428. doi: 10.1021/bi9814639. [DOI] [PubMed] [Google Scholar]
- 14.Stapleton A, Nudelman E, Clausen H, Hakomori S, Stamm WE. J Clin Invest. 1992;90:965–972. doi: 10.1172/JCI115973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooling LLW, Koerner TAW, Naides SJ. J Infect Dis. 1995;172:1198–1205. doi: 10.1093/infdis/172.5.1198. [DOI] [PubMed] [Google Scholar]
- 16.Katagiri YU, Ohmi K, Katagiri C, Sekino T, Nakajima H, Ebata T, Kiyokawa N, Fujimoto J. Glycoconjugate J. 2001;18:347–353. doi: 10.1023/a:1013673300717. [DOI] [PubMed] [Google Scholar]
- 17.Wenk J, Andrews PW, Casper J, Hata J, Pera MF, von Keitz A, Damjanov I, Fenderson BA. Int J Cancer. 1994;58:108–115. doi: 10.1002/ijc.2910580118. [DOI] [PubMed] [Google Scholar]
- 18.Krupnick JG, Damjanov I, Damjanov A, Zhu ZM, Fenderson BA. Int J Cancer. 1994;59:692–698. doi: 10.1002/ijc.2910590518. [DOI] [PubMed] [Google Scholar]
- 19.Kannagi R, Levery SB, Ishigami F, Hakomori S, Shevinsky LH, Knowles BB, Solter D. J Biol Chem. 1983;258:8934–8942. [PubMed] [Google Scholar]
- 20.Saito S, Aoki H, Ito A, Ueno S, Wada T, Mitsuzuka K, Satoh M, Arai Y, Miyagi T. J Biol Chem. 2003;278:26474–26479. doi: 10.1074/jbc.M213223200. [DOI] [PubMed] [Google Scholar]
- 21.Steelant WF, Kawakami Y, Ito A, Handa K, Bruyneel EA, Mareel M, Hakomori S. FEBS Lett. 2002;531:93–98. doi: 10.1016/s0014-5793(02)03484-1. [DOI] [PubMed] [Google Scholar]
- 22.Saito S, Orikasa S, Satoh M, Ohyama C, Ito A, Takahashi T. Jpn J Cancer Res. 1997;88:652–659. doi: 10.1111/j.1349-7006.1997.tb00433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saito S, Orikasa S, Ohyama C, Satoh M, Fukushi Y. Int J Cancer. 1991;49:329–334. doi: 10.1002/ijc.2910490303. [DOI] [PubMed] [Google Scholar]
- 24.Kiso M, Ishida H, Hiromne A. In: Carbohydrate-Based Drug Discovery. Wong C-H, editor. Vol. 1. Wiley-VCH; Weinheim: 2003. pp. 37–54. [Google Scholar]
- 25.Allen JR, Allen JG, Zhang XF, Williams LJ, Zatorski A, Ragupathi G, Livingston PO, Danishefsky SJ. Chem Eur J. 2000;6:1366–1375. doi: 10.1002/(sici)1521-3765(20000417)6:8<1366::aid-chem1366>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 26.Lassaletta JM, Schmidt RR. Liebigs Ann Chem. 1996:1417–1423. [Google Scholar]
- 27.Zhu T, Boons GJ. Angew Chem Int Ed. 1999;38:3495–3497. doi: 10.1002/(sici)1521-3773(19991203)38:23<3495::aid-anie3495>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 28.Werz DD, Castagner B, Seeberger PH. J Am Chem Soc. 2007;129:2770–2771. doi: 10.1021/ja069218x. [DOI] [PubMed] [Google Scholar]
- 29.Huang CY, Thayer DA, Chang AY, Best M, Hoffmann L, Head S, Wong CH. Proc Natl Acad Sci U S A. 2006;100:15–20. doi: 10.1073/pnas.0509693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Z, Zhou L, El-boubbou K, Ye X-S, Huang X. J Org Chem. 2007;72:6409–6420. doi: 10.1021/jo070585g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crich D, Wu B. Tetrahedron. 2008;64:2042–2047. doi: 10.1016/j.tet.2007.12.026. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boons GJ, Demchenko A. Chem Rev. 2000;100:4539–4565. doi: 10.1021/cr990313g. [DOI] [PubMed] [Google Scholar]
- 33.Seifert J, Lergenmuller M, Ito Y. Angew Chem Int Ed. 2000;39:531–534. doi: 10.1002/(sici)1521-3773(20000204)39:3<531::aid-anie531>3.3.co;2-6. [DOI] [PubMed] [Google Scholar]
- 34.Ishida H, Miyawaki R, Kiso M, Hasegawa A. J Carbohydr Chem. 1996;15:163–182. doi: 10.1016/0008-6215(96)00016-x. [DOI] [PubMed] [Google Scholar]
- 35.Lassaletta JM, Carlsson K, Garegg PJ, Schmidt RR. J Org Chem. 1996;61:6873–6880. doi: 10.1021/jo9608073. [DOI] [PubMed] [Google Scholar]
- 36.Koeller KM, Wong CH. Chem Rev. 2000;100:4465–4493. doi: 10.1021/cr990297n. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 37.Wymer N, Toone EJ. Curr Opin Chem Biol. 2000;4:110–119. doi: 10.1016/s1367-5931(99)00061-7. [DOI] [PubMed] [Google Scholar]
- 38.Duclos RI. Carbohydr Res. 2000;328:489–507. doi: 10.1016/s0008-6215(00)00121-x. [DOI] [PubMed] [Google Scholar]
- 39.Blixt O, Vasiliu D, Allin K, Jacobsen N, Warnock D, Razi N, Paulson JC, Bernatchez S, Gilbert M, Wakarchuk W. Carbohydr Res. 2005;340:1963–1972. doi: 10.1016/j.carres.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 40.Kamath VP, Yeske RE, Gregson JM, Ratcliffe RM, Fang YR, Palcic MM. Carbohydr Res. 2004;339:1141–1146. doi: 10.1016/j.carres.2003.12.027. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 41.Randriantsoa M, Drouillard S, Breton C, Samain E. FEBS Lett. 2007;581:2652–2656. doi: 10.1016/j.febslet.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 42.Kannagi R, Cochran NA, Ishigami F, Hakomori S, Andrews PW, Knowles BB, Solter D. EMBO J. 1983;2:2355–2361. doi: 10.1002/j.1460-2075.1983.tb01746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mootoo DR, Konradsson P, Udodong U, Fraser-Reid B. J Am Chem Soc. 1988;110:5583–5584. [Google Scholar]
- 44.Huang X, Huang L, Wang H, Ye XS. Angew Chem Int Ed. 2004;42:5221–5224. doi: 10.1002/anie.200460176. [DOI] [PubMed] [Google Scholar]
- 45.Codee JDC, van den Bos LJ, Litjens REJN, Overkleeft HS, van Boeckel CAA, van Boom JH, van der Marel GA. Tetrahedron. 2004;60:1057–1064. [Google Scholar]
- 46.Yamago S, Yamada T, Maruyama T, Yoshida JI. Angew Chem Int Ed. 2004;43:2145–2148. doi: 10.1002/anie.200353552. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen HM, Poole JL, Gin DY. Angew Chem Int Ed. 2001;40:414–417. doi: 10.1002/1521-3773(20010119)40:2<414::AID-ANIE414>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 48.Crich D, Sun S. Tetrahedron. 1998;54:8321–8348. [Google Scholar]
- 49.Huang L, Huang X. Chem Eur J. 2007;13:529–540. doi: 10.1002/chem.200601090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miermont A, Zeng Y, Jing Y, Ye XS, Huang X. J Org Chem. 2007;72:8958–8961. doi: 10.1021/jo701694k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Teumelsan N, Huang X. J Org Chem. 2007;72:8976–8979. doi: 10.1021/jo7013824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang L, Wang Z, Li X, Ye XS, Huang X. Carbohydr Res. 2006;341:1669–1679. doi: 10.1016/j.carres.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang L, Wang Z, Huang X. Chem Commun. 2004;17:1960–1961. doi: 10.1039/b405886k. [DOI] [PubMed] [Google Scholar]
- 54.Martichonok V, Whitesides GM. J Org Chem. 1996;61:1702–1706. doi: 10.1021/jo951711w. [DOI] [PubMed] [Google Scholar]
- 55.Crich D, Smith M. J Am Chem Soc. 2001;123:9015–9020. doi: 10.1021/ja0111481. [DOI] [PubMed] [Google Scholar]
- 56.Crich D, Smith M. Org Lett. 2000;2:4067–4069. doi: 10.1021/ol006715o. [DOI] [PubMed] [Google Scholar]
- 57.Wang C, Wang H, Huang X, Zhang LH, Ye XS. Synlett. 2006:2846–2850. [Google Scholar]
- 58.Codée JDC, Litjens REJN, den Heeten R, Overkleeft HS, van Boom JH, van der Marel GA. Org Lett. 2003;5:1519–1522. doi: 10.1021/ol034312t. [DOI] [PubMed] [Google Scholar]
- 59.Codée JDC, van den Bos LJ, Litjens REJN, Overkleeft HS, van Boom JH, van der Marel GA. Org Lett. 2003;5:1947–1950. doi: 10.1021/ol034528v. [DOI] [PubMed] [Google Scholar]
- 60.Pornsuriyasak P, Demchenko AV. Carbohydr Res. 2006;341:1458–1466. doi: 10.1016/j.carres.2006.03.041. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 61.Crich D, Smith M, Yao Q, Picione J. Synthesis. 2001:323–326. [Google Scholar]
- 62.Gen Y, Zhang LH, Ye XS. Chem Commun. 2008:597–599. doi: 10.1039/b712591g. [DOI] [PubMed] [Google Scholar]
- 63.Li J, Wang J, Czyryca PG, Chang H, Orsak TW, Evanson R, Chang CWT. Org Lett. 2004;6:1381–1384. doi: 10.1021/ol0497685. [DOI] [PubMed] [Google Scholar]
- 64.Park TY, Kim IJ, Hu S, Bilodeau MT, Randolph JT, Kwon O, Danishefsky SJ. J Am Chem Soc. 1996;118:11488–11500. [Google Scholar]
- 65.Garcia B, Gin DY. J Am Chem Soc. 2000;122:4269–4279. [Google Scholar]
- 66.Abdel-Rahman AA-H, Jonke SJ, El Ashry ESH, Schmidt RR. Angew Chem Int Ed. 2004;43:4389. doi: 10.1002/1521-3773(20020816)41:16<2972::AID-ANIE2972>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 67.Crich D, Picione J. Org Lett. 2003;5:781–784. doi: 10.1021/ol0340890. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Z, Ollman IR, Ye XS, Wischnat R, Baasov T, Wong CH. J Am Chem Soc. 1999;121:734–753. [Google Scholar]
- 69.Crich D, Dudkin V. Tetrahedron Lett. 2000;41:5643–5646. [Google Scholar]
- 70.Crich D, Li W, Li H. J Am Chem Soc. 2004;126:15081–15086. doi: 10.1021/ja0471931. [DOI] [PubMed] [Google Scholar]
- 71.Yi W, Shao J, Zhu L, Li M, Singh M, Lu Y, Lin S, Li H, Ryu K, Shen J, Guo H, Yao Q, Bush CA, Wang PG. J Am Chem Soc. 2005;127:2040–2041. doi: 10.1021/ja045021y. [DOI] [PubMed] [Google Scholar]
- 72.Yu H, Chokhawala H, Karpel H, Yu H, Wu B, Zhang J, Zhang Y, Jia Q, Chen X. J Am Chem Soc. 2005;127:17618–17619. doi: 10.1021/ja0561690. [DOI] [PubMed] [Google Scholar]
- 73.Yu H, Huang S, Chokhawala H, Sun M, Zheng H, Chen X. Angew Chem Int Ed. 2005;45:3938–3944. doi: 10.1002/anie.200600572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Su DM, Eguchi H, Yi W, Li L, Wang PG, Xia C. Org Lett. 2008:10. doi: 10.1021/ol703121h. in press. [DOI] [PubMed] [Google Scholar]
- 75.Bock K, Pedersen C. J Chem Soc Perkin Trans. 1974;2:293–297. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.