

Abstract
Modeling the interplay between mutation and selection at the molecular level is key to evolutionary studies. To this end, codon-based evolutionary models have been proposed as pertinent means of studying long-range evolutionary patterns and are widely used. However, these approaches have not yet consolidated results from amino acid level phylogenetic studies showing that selection acting on proteins displays strong site-specific effects, which translate into heterogeneous amino acid propensities across the columns of alignments; related codon-level studies have instead focused on either modeling a single selective context for all codon columns, or a separate selective context for each codon column, with the former strategy deemed too simplistic and the latter deemed overparameterized. Here, we integrate recent developments in nonparametric statistical approaches to propose a probabilistic model that accounts for the heterogeneity of amino acid fitness profiles across the coding positions of a gene. We apply the model to a dozen real protein-coding gene alignments and find it to produce biologically plausible inferences, for instance, as pertaining to site-specific amino acid constraints, as well as distributions of scaled selection coefficients. In their account of mutational features as well as the heterogeneous regimes of selection at the amino acid level, the modeling approaches studied here can form a backdrop for several extensions, accounting for other selective features, for variable population size, or for subtleties of mutational features, all with parameterizations couched within population-genetic theory.
Keywords: codon substitution, Dirichlet process, phylogeny, selection coefficients
Following the seminal works of Muse and Gaut (1) and Goldman and Yang (2), most early applications of codon-based evolutionary models were focused on evaluations of selective effects operating at different positions along a gene or at different time points along the phylogeny (see refs. 3, 4 for reviews). Many of these approaches have modeled selective effects using a parameter representing the nonsynonymous/synonymous rate ratio. However, this may not be ideal, in particular because it amounts to ignoring differences between different pairs of possible amino acid replacements resulting from nonsynonymous point mutations. In recent years, questions regarding selective effects have diversified, such as in the work of Yang and Nielsen (5), who propose a test for selection on codon usage. This test is based on models that invoke a multidimensional specification of scaled selection coefficients, based on either 20 or 61 (under the universal genetic code) scaled fitness parameters—adding 19 or 60 degrees of freedom to the underlying codon substitution model—in contrast with the more conventional use of the single nonsynonymous/synonymous rate ratio parameter, viewing all nonsynonymous events as equivalent (e.g., see ref. 6). By assigning scaled fitness parameters to each of the 20 amino acids, or to the 61 sense codons, Yang and Nielsen obtained scaled selection coefficients associated with each type of possible event from the difference in scaled fitness between the states before and after the event. Moreover, the models have the attractive feature of having direct connections to population genetic theory (7), and Yang and Nielsen have referred to them as mutation-selection substitution models (5).
Beyond addressing specific questions regarding selective regimes, most previous works have given little attention to the level of realism implied by the models used. For instance, most codon substitution models employed to date have the same stationary distribution for all positions. However, if it is not too unreasonable to assume a homogeneous mutation process along the sequences, there are both theoretical and empirical reasons to believe that selection operating at the level of the encoded protein sequences is strongly site specific, thus resulting in a marked differentiation of the stationary distribution across positions (discussed in ref. 8). In practice, of course, codon substitution models were not meant as faithful descriptions of protein-coding gene evolution, but rather as tools for detecting particular selective effects, using appropriate test statistics, which, in the best cases, would be able to offset the model's lack of realism (e.g., ref. 5). However, this might be problematic, as inferences based on too grossly misspecified models can lead to artifactual conclusions. In the mutation-selection framework in particular, this calls for a more elaborate reference model for protein-coding sequence evolution.
Interestingly, the original motivations of Halpern and Bruno (9), over a decade ago, were in fact to account for site specificities of amino acids within the mutation-selection modeling framework. The model used by Halpern and Bruno closely corresponds to a fully site-heterogeneous version of a model described in ref. 5, with a separate set of 20 scaled fitness parameters affiliated to each coding site of the alignment. Although the Halpern and Bruno framework has attractive features, their model has hardly been used since (but see ref. 10). Reasons for this hiatus include the fact that a large number of sequences are required to reliably infer site-specific amino acid fitness parameters and that such site-specific codon substitution models are computationally highly demanding.
Meanwhile, working with models that operate at the amino acid level, several studies have shown that maximally heterogeneous parameterizations may be unreliable, and that a more reasonable balance between this and homogeneous parameterizations is required (e.g., refs. 11 –14). In particular, using mixture models to capture across-site heterogeneities in amino acid propensities in the context of profile HMMs (15), or of protein phylogenomics (11, 16, 17), has generally been found to give an improved model fit over either of these extremes (e.g., refs. 11, 18). Furthermore, algorithmic developments for performing the needed probability calculations [relying on data-augmentation-based Markov chain Monte Carlo (MCMC) sampling] have recently allowed for richer substitution models to become tractable (e.g., refs. 19, 20).
Here, we integrate recent developments from previous works to explore a nonparametric approach previously used for amino acid level modeling (11, 16) within the mutation-selection codon substitution modeling framework. The basic biological assumptions underlying our model are the same as those in ref. 9: (i) most coding positions are under a regime of purifying selection at the amino acid level (where it is likely that only a few amino acids have comparatively higher fitness values than all other amino acids); (ii) this regime of purifying selection at a coding site is constant over time; and (iii) although selective effects are likely to implicate interdependencies between different coding sites (21), these shall be ignored; in other words, the focus is limited to capturing the marginal site specificities induced by purifying selection, based on site-heterogeneous modeling of amino acid fitness. We illustrate features of the model in a qualitative-to-quantitative presentation, first showing that the integration of modeling approaches from refs. 5, 9, 11, 16, 17 leads to biologically plausible inferences, in particular regarding site-specific amino acid constraints and distributions of scaled selection coefficients. Using a posterior predictive simulation diagnostic, we further show that the modeling approaches capture global features of selection, and, using Bayes factors, that they lead to a significant improvement in statistical fit. We propose the framework as a reference mutation-selection model, and discuss further developments and applications that it could enable, contributing to a merger of phylogenetics and population genetics.
Results and Discussion
Mutation-Selection Modeling with the Dirichlet Process.
We build our model on a basic mutational specification, corresponding to a general time-reversible (GTR) model at the nucleotide level (22). The GTR model is given by two sets of parameters: six parameters (five effective degrees of freedom) governing the exchangeability of each (unordered) pair of nucleotides, as well as four global nucleotide propensity parameters (three effective degrees of freedom, for a total of 5 + 3 = 8 parameters). For modeling selective effects, we incorporated the approach of an “infinite” mixture of amino acid profiles described in refs. 11, 16 into the mutation-selection codon substitution framework inspired by refs. 5, 9. This part of the model is implemented using Markov chain Monte Carlo methods to update configurations of a Dirichlet process (e.g., see ref. 23). A particular configuration of the Dirichlet process is given by a set of amino acid propensity vectors, with each vector having one or more sites affiliated to it. The entries of each vector consist of positive values (summing to 1), and in our parameterization the difference of the logarithm of two entries corresponds to a scaled selection coefficient. This coefficient in turn defines a fixation factor for nonsynonymous events, derived from population-genetic principles (e.g., see refs. 5, 9 for details regarding the specification of a codon substitution matrix in the mutation-selection framework, and also see Materials and Methods). Note that given a particular configuration of the Dirichlet process, two or more sites may have the same fixation factors, if these sites are affiliated to the same amino acid propensity vector; however, through the posterior averaging implemented by the MCMC—updating configurations of the Dirichlet process—each site has its own posterior distribution of amino acid propensity parameters. As for the parameters governing the Dirichlet process itself, often called hyperparameters, these include a “granularity” parameter (controlling the mixture granularity, or “clumpiness”), a vector parameter acting as a base distribution of the amino acid propensity mixture components, and a parameter controlling the dispersal of mixture components around the base distribution (11, 16). We combine MCMC methods for the Dirichlet process with MCMC update operators on mutational parameters, branch lengths, hyperparameters (although using a fixed tree topology), embedded within a data-augmentation-based system (20, 24), giving us the means to sample from the overall joint posterior distribution. Altogether, because the framework enables a model that is derived from the Muse and Gaut approach, and incorporates the mutation-selection framework using the Dirichlet process, we refer to it as the MG-MutSelDP model.
Posterior Distributions.
We report the posterior distributions of all parameters governing our model, obtained under a dozen real data sets, in SI Text and focus here on a few key features. First, because the Dirichlet process approach is aimed at capturing selection, the usual interpretation of the nucleotide propensity parameters must be revised. Under the traditional nucleotide-level models, such as the GTR model, these parameters reflect the long-term proportions of nucleotides that are expected from running the evolutionary model. Indeed, a common practice aimed at simplifying practical problems has consisted in fixing these parameters to the observed nucleotide frequencies. In the present mutation-selection framework, however, one must view the nucleotide propensity parameters as reflecting the long-term proportions of nucleotides expected in the absence of selection. In other words, we do not expect their posterior mean values to resemble the global empirical frequencies of the alignment, because these have presumably been dictated by a tradeoff between mutational and selective effects. Third-position nucleotide frequencies are those least subjected to the effects of selection at the amino acid level, and thus a prediction is that the posterior distributions of nucleotide propensity parameters under our model should tend to be located near these values. Indeed we found that the posterior mean values of nucleotide propensity parameters follow third-position nucleotide frequency values closely. In Fig. 1, we pooled the posterior mean nucleotide propensity values obtained under a dozen data sets and plotted these against the empirical third-position frequencies of each respective data set. The strong correlation provides a first simple validation that our model is able to tease out mutational and selective factors in a biologically meaningful way. This result is in contrast with previous works exploring the mutation-selection framework, in which it was noted that the selective parameterization had little effect on nucleotide propensity parameters (e.g., refs. 5, 20, 21). This contrast is likely due to the very different approach of the present work for modeling selective effects. Moreover, the result suggests that short-cut schemes of fixing nucleotide propensity parameters to the global empirical nucleotide frequencies (as done in the original Halpern-Bruno model) should be avoided.
Fig. 1.

Comparison of nucleotide propensity parameters with third-position empirical nucleotide frequencies.
We next highlight how the modeling approach allows us to explore site-heterogeneous selective constraints at the amino acid level. As a simple graphical display that illustrates how the model distinguishes between amino acids, we calculate for each site the average amino acid propensity vector over our sample from the posterior distribution (using the vector affiliated to the site of interest for each draw), and display it as a logo with the height of single letter abbreviations being proportional to the mean propensity parameter for that amino acid (logos are sorted to display the most predominant amino acid at the top of the profile, and in decreasing order going down the profile).
Using two data sets (Globin 17–144 and Rhod 38–300, see SI Text) for illustrative purposes, Fig. 2 contrasts the site-specific amino acid frequencies observed in the true alignments, in the Top row of each panel, with the site-specific posterior mean amino acid propensity vectors obtained under the MG-MutSelDP model in the Second row of each panel, for the first 30 positions. We first observe that the posterior mean amino acid vectors are indeed quite heterogeneous across sites. Moreover, the site-specific posterior mean amino acid propensity vectors are generally suggestive of the true site-specific amino acid frequencies for the Globin 17–144 data set, and the resemblance is graphically more obvious for the Rhod 38–300 data set. We view these results as further validating the modeling approach, in producing qualitatively sensible site-specific inferences. It should be noted that, to date, nearly all of the codon substitution models used have a unique stationary distribution at all sites, implying that the same long-term trend is assumed over the entire gene (i.e., the most probable gene under such models will consist of a repetitive sequence of a single codon). In the present case, the amino acid propensity parameters are involved in the stationary distribution of the Markov process, such that each site will have its own effective limiting distribution (i.e., in this case, the most probable gene will tend to resemble real genes).
Fig. 2.

Site-specific amino acid frequencies and inferred amino acid propensity vectors. (A) For the GLOBIN 17–144 data set, followed by RHOD 38–300 in B. In each panel, the Top Row is a representation of the site-specific amino acid frequencies (with frequencies of amino acids proportional to height of the single letter abbreviation), obtained by translating the true alignment; the Second Row shows the site-specific amino acid propensity parameters (the log of which corresponds to their scaled fitness) obtained under the MG-MutSelDP model.
Distributions of Selection Coefficients.
To summarize site-specific results across all sites into a more quantitative illustration of the properties of the model, we focus on the distributions of scaled selection coefficients. More specifically, we used equations 5 and 6 in ref. 5 (also see Materials and Methods) to calculate, for each site, the proportion of mutations going from codon a to codon b among all possible mutations at stationarity under the Markov process, and the proportion of these falling within particular ranges of selection coefficient values of interest. The proportion of mutations from a to b among all substitutions—i.e., among the mutations that have passed the filtering of selection (5)—can also be calculated as explained in ref. 5, hence allowing us to contrast the distributions of selection coefficients between these two sets of mutation categories. These calculations require parameter values. Here, we simply use the componentwise posterior mean, for global parameters, and site-specific posterior mean amino acid propensity vectors (always based on the affiliation configuration of the Dirichlet process draws) and further average the site-specific results across all sites.
Fig. 3 shows the distributions obtained using our proposed MG-MutSelDP model for the two example data sets. In each panel, the dashed-line histogram reports the proportion of mutations having the scaled selection coefficient displayed in the abscissa. In each case, we first note that most of this distribution is located in the negative region: for the Globin 17–144 data set (Fig. 3A), 87.0% of proposed mutations are either deleterious (i.e., in bins centered on any S < 0, at 61.3%) or neutral (i.e., in the bin centered on S = 0, at 25.7%); and for the Rhod 38–300 data set (Fig. 3B), 92.9% of proposed mutations are either deleterious (bin centers with S < 0 at 67.5%) or neutral (in bin centered on S = 0 at 25.4%). Each panel also shows a solid-line histogram, reporting the proportion of mutations among all substitutions that have the coefficient displayed in the abscissa. These distributions are symmetrical (owing to the time-reversible nature of the models) and the bin centered on S = 0 now attains 37.7% for the Globin 17–144 data set and 50.9% for the Rhod 38–300 data set.
Fig. 3.

Distributions of scaled selection coefficients (S) at equilibrium of the Markov process under the MG-MutSelDP model applied to the Globin 17–144 data set in A and Rhod 38–300 in B. The dashed-line histogram displays the proportion of mutations among all possible mutations having the coefficient value in the abscissa, whereas the solid-line histogram displays the proportion of mutations among all substitutions having the coefficient value in the abscissa.
The distributions of scaled selection coefficients have intuitive interpretations in the mutation-selection modeling framework. There is a long history of empirical observations showing that most possible mutations are detrimental in terms of fitness, and this is represented in the predominantly negative location of the dashed-line histograms. Moreover, the distributions of selection coefficients among substitutions (solid-line histograms) are representative of the model design of capturing the balance between mutation and selection (9), in being centered about 0, and show that the highest proportion of substitutions are those that are neutral (in the bin centered on S = 0) with the remaining being nearly neutral (mildly positive or mildly negative), and more tightly distributed around the bin centered on S = 0. We note that these latter results, which are a basic consequence of the construction of our model, are in accordance with the theoretical developments of Sella and Hirsh (25), who find that the distributions of selection coefficients of mutations that reach fixation should be symmetrical around 0.
Model Assessment and Ranking.
We performed a simple posterior predictive diagnostic showing that the MG-MutSelDP model captures selective features, reported in SI Text, but otherwise sought to rank the heterogeneous modeling ideas described here with respect to other well-known modeling approaches. We used the thermodynamic integration methods described in ref. 26 to compute Bayes factors. However, as currently implemented, these are computationally intensive MCMC procedures. In particular, they currently do not allow us to exploit the data-augmentation sampling systems that make the Dirichlet process modeling studied here tractable. Hence, to keep the thermodynamic calculations manageable, we only ranked three empirical modeling approaches, with respect to the MG model and only performed this analysis on the smallest of the our data sets (Globin 17–144). For these empirical versions, the mixture model is predefined, in terms of the number of components and in terms of the profiles of each component, which are fixed to those inferred in ref.12; only the weights of the components are free parameters, endowed with a flat Dirichlet prior. Depending on the number of components, we refer to these models as MG-MutSelC20, MG-MutSelC40, and MG-MutSelC60.
The empirical versions considerably reduce the MCMC computational demands, while capturing the essence of the heterogeneous modeling ideas. However, because the 20, 40, or 60 profiles were obtained from a framework operating at the amino acid level only (i.e., that does not attempt to tease out mutational and selective effects), the Bayes factors associated with their use in the mutation-selection framework are likely to be lower than they might be with empirical mixtures obtained by incorporating the nucleotide (codon)-level data within the training methods described in ref. 12. Despite this weakness of the plug-in we attempt here, the natural log Bayes factors are ∼236, 255, and 269 for the MG-MutSelC20, MG-MutSelC40, and MG-MutSelC60 models, respectively (computed against the MG model). These are much greater than the natural log Bayes factor in favor of the MG-NS model, evaluated in ref. 24 at ∼92, or even the MG-NSDP model (which applies a Dirichlet process to the unidimensional nonsynonymous rate factor (27), at ∼195. These results suggest that the approaches studied here could be addressing one of the most important points of articulation in codon substitution model design, although further comparisons with other approaches (e.g., refs. 28 –31) should eventually be performed.
Conclusions
The MG-MutSelDP model we report here rests on three basic biological assumptions previously mentioned: the assumption of a site-heterogeneous background of purifying selection at the amino acid level, of constancy of selective regimes, and of independence between codon sites (or a complete lack of within-gene and general epistasis). Whereas the first assumption appears justified, there have been several reports of violations of the second and third assumptions (e.g., refs. 32, 33, 34 ). Nonetheless, we believe the approach could serve as a basic reference model with direct connections to population-genetic theory (7), while constituting a stepping stone to a more in-depth modeling of protein-coding sequence evolution. We have shown several lines of results that support this viewpoint. First, it appears successful in teasing out mutational tendencies from selective effects, as reflected by the similitude of the posterior distributions of nucleotide propensity parameters and the empirical third-position nucleotide frequencies (Fig. 1). Second, the inferred site-specific selective constraints at the amino acid level appear qualitatively reasonable when contrasted with observed amino acid frequencies (Fig. 2). Third, the model produces biologically sensible distributions of selection coefficients for all possible mutations (predominantly negative values), and theoretically coherent distributions when considering mutations that pass through the filtering of selection (Fig. 3). Finally, the model performs well in posterior predictive checks and markedly outperforms other models as measured by Bayes factors.
Despite the theoretically attractive aspects of Halpern and Bruno's mutation-selection framework, proposed over a decade ago, the approach has received little attention in practice, mainly due to computational difficulties; the Halpern and Bruno model involves as many different codon matrices as there are sites, and even models with a single matrix have generally been considered computationally too expensive, motivating other approaches that circumvent their use (e.g., ref. 35). However, with recent developments in data-augmentation-based MCMC sampling methods, as well as the Dirichlet process modeling approaches, these types of heterogeneous modeling ideas are becoming tractable (at least for performing inferences on the basis of posterior distributions). In our implementation of the MG-MutSelDP model, we observed the data-augmentation-based sampler to be roughly two orders of magnitude faster than the traditional pruning-based sampler, which implies that the latter would require years of CPU instead of weeks in some cases we study here. Although the empirical mixture approaches can provide less taxing models, the Bayes factors reported above (computed using pruning-based sampling) still required over 2 months of CPU time. It is thus of interest to advance further computational methods, both to ameliorate our current data-augmentation-based sampler and to bridge this type of MCMC sampling with our thermodynamic integration methods.
Many other applications can be envisioned for these evolutionary models. For instance, although we have reported distributions of selection coefficients averaged across all sites, the modeling framework allows for site-specific distributions to be inferred as well; as a specific example, from the posterior distribution of parameters under the MG-MutSelDP model obtained from, say, a mammalian alignment (and tree), one could evaluate the selection coefficients associated with all possible point mutations of a given human gene sequence (e.g., see ref. 36). Also, the model could be applied to the problem of phylogenetic inference per se, particularly as algorithmic developments progress; the amino acid level CAT model has been found to be more robust in conditions where homogeneous models are susceptible to the long branch attraction artifact (16), and recent work has shown the presence of phylogenetically relevant synonymous signals even in highly diverged proteins (37). The MG-MutSelDP model accounts for both these recent observations and could therefore be expected to provide a particularly robust framework for phylogenetic inference. Finally, the model could constitute a more relevant background against which to distinguish positive selection. All our experiments show that purifying selection is both widespread and overwhelming in intensity in protein-coding sequences. It seems possible that weak signals of positive selection (possibly sporadic) are overwhelmed by such a strong purifying selection environment. Our posterior predictive experiments (Supporting Information) show that we offset the background of purifying selection in a significant way, but they also reveal the potential presence of weak signals of positive selection. Exploring these directions is likely to be computationally challenging for large data sets, although promising advances continue to be made (e.g., ref. 38).
Among the useful possible extensions to the model, it could allow one to tease out mutational and selective contributions to the compositional variations across proteomes of different species (39). Or, the overall efficacy of selection could be modulated over the phylogeny by having a variable effective population size over the tree, which could also enable a probabilistic reconstruction of ancestral population sizes. A global modeling of selection on codon usage (as in ref. 5) could be incorporated in a straightforward way and could be expanded to incorporate heterogeneities at this level as well. Combinations with context-dependent mutational modeling (e.g., refs. 40, 41) could also be envisaged, as well as accommodating levels of dependence between codons (21). More generally, any aspect could be modeled as lineage dependent, site dependent, gene dependent, dependent on the chromosomal region, etc., thereby providing a foundation for contrasting evolutionary patterns under a broad spectrum of conditions.
Materials and Methods
Models.
We build the MG-MutSelDP model as follows: mutational parameters corresponding to the exchangeability of each (unordered) pair of nucleotides are written as 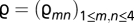 , with the (arbitrary) constraint
, with the (arbitrary) constraint 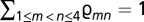 ; mutational parameters governing nucleotide propensities are written as φ = (φn)1 ≤ n ≤ 4, with
; mutational parameters governing nucleotide propensities are written as φ = (φn)1 ≤ n ≤ 4, with 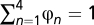 ; a particular configuration of the Dirichlet process is given by a set of K (1 ≤ K ≤ N, where N is the number of codon sites in the alignment) amino acid propensity vectors, written as ψ = (ψ(k)
l)1 ≤ l ≤ 20, 1 ≤ k ≤ K (with
; a particular configuration of the Dirichlet process is given by a set of K (1 ≤ K ≤ N, where N is the number of codon sites in the alignment) amino acid propensity vectors, written as ψ = (ψ(k)
l)1 ≤ l ≤ 20, 1 ≤ k ≤ K (with 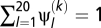 for all k), and an allocation vector z = (zi)1 ≤ i ≤ N. For a particular site i, the entry of the allocation vector, zi, returns an index, in the range 1 to K, corresponding to the particular amino acid propensity vector currently affiliated to site i. With this detailed configuration, for any site i the scaled selection coefficient for an event from codon a to codon b is given by
for all k), and an allocation vector z = (zi)1 ≤ i ≤ N. For a particular site i, the entry of the allocation vector, zi, returns an index, in the range 1 to K, corresponding to the particular amino acid propensity vector currently affiliated to site i. With this detailed configuration, for any site i the scaled selection coefficient for an event from codon a to codon b is given by 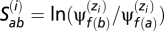 , where f(a) returns an index corresponding to the amino acid encoded by codon a. Note that two or more sites may have the same scaled selection coefficients, if these sites are affiliated to the same amino acid propensity vector; however, through the fluctuations of the MCMC—updating configurations of the Dirichlet process—each site has its own effective distribution. The Markov generator at a particular site i, for a particular configuration of the Dirichlet process, then has three possible off-diagonal entries: (i)
, where f(a) returns an index corresponding to the amino acid encoded by codon a. Note that two or more sites may have the same scaled selection coefficients, if these sites are affiliated to the same amino acid propensity vector; however, through the fluctuations of the MCMC—updating configurations of the Dirichlet process—each site has its own effective distribution. The Markov generator at a particular site i, for a particular configuration of the Dirichlet process, then has three possible off-diagonal entries: (i) 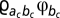 , for the case of a synonymous event from codon a to codon b, where c is 1, 2, or 3, for the first, second, and third within-codon positions, and ac gives the index corresponding to the nucleotide at the c
th position of codon a; (ii)
, for the case of a synonymous event from codon a to codon b, where c is 1, 2, or 3, for the first, second, and third within-codon positions, and ac gives the index corresponding to the nucleotide at the c
th position of codon a; (ii) 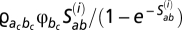 , for the case of a nonsynonymous event from codon a to codon b, where
, for the case of a nonsynonymous event from codon a to codon b, where 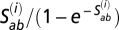 is the fixation factor; and (iii) 0 for cases with codons differing by more than one nucleotide (only point mutations are allowed by the model). As in ref. 16, we draw amino acid propensity vectors from a base prior governed by a 20 dimensional (19 effective degrees of freedom) probability vector, as well as a parameter controlling the dispersion around this vector, and another parameter controlling the granularity of the mixture. The base prior probability vector is endowed with a Dirichlet hyperprior, and unidimensional parameters are endowed with exponential hyperpriors of mean 1. For the MG-MutSelC20, MG-MutSelC40, and MG-MutSelC60 models described in the main text, the only free parameters involved in the mixture are the weights of the components; these weights are implicitly integrated out via an allocation system, in a manner that is equivalent to having a flat Dirichlet prior on them (12). The priors on all other parameters are as described in ref. 24, as are the priors for the MG, MG-NS, and MG-NSDP models (described in ref. 24 and the main text).
is the fixation factor; and (iii) 0 for cases with codons differing by more than one nucleotide (only point mutations are allowed by the model). As in ref. 16, we draw amino acid propensity vectors from a base prior governed by a 20 dimensional (19 effective degrees of freedom) probability vector, as well as a parameter controlling the dispersion around this vector, and another parameter controlling the granularity of the mixture. The base prior probability vector is endowed with a Dirichlet hyperprior, and unidimensional parameters are endowed with exponential hyperpriors of mean 1. For the MG-MutSelC20, MG-MutSelC40, and MG-MutSelC60 models described in the main text, the only free parameters involved in the mixture are the weights of the components; these weights are implicitly integrated out via an allocation system, in a manner that is equivalent to having a flat Dirichlet prior on them (12). The priors on all other parameters are as described in ref. 24, as are the priors for the MG, MG-NS, and MG-NSDP models (described in ref. 24 and the main text).
MCMC Sampling.
We used sampling methodologies described elsewhere (e.g., refs. 19, 20) that consist of drawing data augmentations conditional on parameters (and auxiliary variables) followed by updates on parameters conditional on the data augmentations. We used the approach described in ref. 24 to draw data augmentations, although here the draws are directly available from the posterior distribution (because the models are site independent). Thermodynamic integrations for computing Bayes factors were performed as in ref. 26.
Distributions of Selection Coefficients.
We used the approach described in ref. 5 to produce distributions of selection coefficients. Briefly, the distributions of selection coefficients are those at stationarity under the Markov process; the stationary distribution at a given codon site i under a particular configuration of the Dirichlet process is 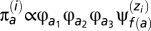 . The proportion of mutations from a to b among all possible mutations at site i, μ(i)
ab is then given by
. The proportion of mutations from a to b among all possible mutations at site i, μ(i)
ab is then given by 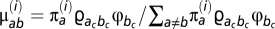 . The collection of all values of μ(i)
ab are then binned to produce a histogram (Fig. 3, dashed line). The proportion of mutations from a to b among all substitutions at site i is calculated in the same way, but including the selective factor in the latter equation, and the collection of values are again binned to produce a histogram (Fig. 3, solid line).
. The collection of all values of μ(i)
ab are then binned to produce a histogram (Fig. 3, dashed line). The proportion of mutations from a to b among all substitutions at site i is calculated in the same way, but including the selective factor in the latter equation, and the collection of values are again binned to produce a histogram (Fig. 3, solid line).
Data.
We used 12 data sets to demonstrate the modeling ideas in practice. These data sets were previously assembled and part of published works (6, 42 –47). The data sets are described in Supporting Information, along with the trees used.
Supplementary Material
Acknowledgments
We thank Stéphane Aris-Brosou for discussions regarding this work, as well as the anonymous reviewers for comments on the manuscript. We also thank the Réseau Québécois de Calcul de Haute Performance for computational resources. N.R. was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), H.P. was supported by the Canadian Research Chair Program, and N.L. and H.P. were supported by NSERC.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0910915107/DCSupplemental.
References
- 1.Muse SV, Gaut BS. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol Biol Evol. 1994;11:715–724. doi: 10.1093/oxfordjournals.molbev.a040152. [DOI] [PubMed] [Google Scholar]
- 2.Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994;11:725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- 3.Anisimova M, Kosiol C. Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol Biol Evol. 2009;26:255–271. doi: 10.1093/molbev/msn232. [DOI] [PubMed] [Google Scholar]
- 4.Delport W, Scheffler K, Seoighe C. Models of coding sequence evolution. Brief Bioinform. 2009;10:97–109. doi: 10.1093/bib/bbn049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Z, Nielsen R. Mutation-selection models of codon substitution and their use to estimate selective strengths on codon usage. Mol Biol Evol. 2008;25:568–579. doi: 10.1093/molbev/msm284. [DOI] [PubMed] [Google Scholar]
- 6.Yang Z, Nielsen R, Goldman N, Pedersen A-MK. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thorne JL, Choi SC, Yu J, Higgs PG, Kishino H. Population genetics without intraspecific data. Mol Biol Evol. 2007;24:1667–1677. doi: 10.1093/molbev/msm085. [DOI] [PubMed] [Google Scholar]
- 8.Choi SC, Redelings BD, Thorne JL. Basing population genetic inferences and models of molecular evolution upon desired stationary distributions of DNA or protein sequences. Philos Trans R Soc Lond B Biol Sci. 2008;363:3931–3939. doi: 10.1098/rstb.2008.0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halpern AL, Bruno WJ. Evolutionary distances for protein-coding sequences: Modeling site-specific residue frequencies. Mol Biol Evol. 1998;15:910–917. doi: 10.1093/oxfordjournals.molbev.a025995. [DOI] [PubMed] [Google Scholar]
- 10.Holder MT, Zwickl DJ, Dessimoz C. Evaluating the robustness of phylogenetic methods to among-site variability in substitution processes. Philos Trans R Soc B. 2008;363:4013–4021. doi: 10.1098/rstb.2008.0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lartillot N, Philippe H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol. 2004;21:1095–1109. doi: 10.1093/molbev/msh112. [DOI] [PubMed] [Google Scholar]
- 12.Quang S, Gascuel O, Lartillot N. Empirical profile mixture models for phylogenetic reconstruction. Bioinformatics. 2008;24:2317–2323. doi: 10.1093/bioinformatics/btn445. [DOI] [PubMed] [Google Scholar]
- 13.Huelsenbeck JP, Joyce P, Lakner C, Ronquist F. Bayesian analysis of amino acid substitution models. Philos Trans R Soc B. 2008;363:3941–3953. doi: 10.1098/rstb.2008.0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H-C, Li K, Susko E, Roger AJ. A class frequency mixture model that adjusts for site-specific amino acid frequencies and improves inference of protein phylogeny. BMC Evol Biol. 2008;8:33. doi: 10.1186/1471-2148-8-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sjölander K, et al. Dirichlet mixtures: A method for improved detection of weak but significant protein sequence homology. Bioinformatics. 1996;12:327–345. doi: 10.1093/bioinformatics/12.4.327. [DOI] [PubMed] [Google Scholar]
- 16.Lartillot N, Brinkmann H, Philippe H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol Biol. 2007;7(Suppl 1):S4. doi: 10.1186/1471-2148-7-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lartillot N, Lepage T, Blanquart S. PhyloBayes 3: A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 2009;25:2286–2288. doi: 10.1093/bioinformatics/btp368. [DOI] [PubMed] [Google Scholar]
- 18.Lartillot N, Philippe H. Computing Bayes factors using thermodynamic integration. Syst Biol. 2006;55:195–207. doi: 10.1080/10635150500433722. [DOI] [PubMed] [Google Scholar]
- 19.Lartillot N. Conjugate Gibbs sampling for Bayesian phylogenetic models. J Comput Biol. 2006;13:1701–1722. doi: 10.1089/cmb.2006.13.1701. [DOI] [PubMed] [Google Scholar]
- 20.Rodrigue N, Philippe H, Lartillot N. Uniformization for sampling realizations of Markov processes: Applications to Bayesian implementations of codon substitution models. Bioinformatics. 2008;24:56–62. doi: 10.1093/bioinformatics/btm532. [DOI] [PubMed] [Google Scholar]
- 21.Robinson DM, Jones DT, Kishino H, Goldman N, Thorne JL. Protein evolution with dependence among codons due to tertiary structure. Mol Biol Evol. 2003;18:1692–1704. doi: 10.1093/molbev/msg184. [DOI] [PubMed] [Google Scholar]
- 22.Lanave C, Preparata G, Saccone C, Serio G. A new method for calculating evolutionary substitution rates. J Mol Evol. 1984;20:86–93. doi: 10.1007/BF02101990. [DOI] [PubMed] [Google Scholar]
- 23.Neal RM. Markov chain sampling methods for Dirichlet process mixture models. J Comput Graph Stat. 2000;9:249–265. [Google Scholar]
- 24.Rodrigue N, Philippe H, Lartillot N. Assessment and ranking of phylogenetic models of coding sequence evolution with dependence between codons. Mol Biol Evol. 2009;26:1663–1676. doi: 10.1093/molbev/msp078. [DOI] [PubMed] [Google Scholar]
- 25.Sella G, Hirsh AE. The application of statistical physics to evolutionary biology. Proc Natl Acad Sci USA. 2005;102:9541–9546. doi: 10.1073/pnas.0501865102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodrigue N, Lartillot N, Philippe H. Bayesian comparisons of codon substitution models. Genetics. 2008;180:1579–1591. doi: 10.1534/genetics.108.092254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huelsenbeck JP, Jain S, Frost SWD, Pond SL. A Dirichlet process model for detecting positive selection in protein-coding DNA sequences. Proc Natl Acad Sci USA. 2006;103:6263–6268. doi: 10.1073/pnas.0508279103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doron-Faigenboim A, Pupko T. A combined empirical and mechanistic codon model. Mol Biol Evol. 2007;24:388–397. doi: 10.1093/molbev/msl175. [DOI] [PubMed] [Google Scholar]
- 29.Kosiol C, Holmes I, Goldman N. An empirical codon model for protein sequence evolution. Mol Biol Evol. 2007;24:1464–1479. doi: 10.1093/molbev/msm064. [DOI] [PubMed] [Google Scholar]
- 30.Mayrose I, Doron-Faigenboim A, Bacharach E, Pupko T. Towards realistic codon models: Among site variability and dependency of synonymous and non-synonymous rates. Bioinformatics. 2007;23:i319–i327. doi: 10.1093/bioinformatics/btm176. [DOI] [PubMed] [Google Scholar]
- 31.Seo T-K, Kishino H. Statistical comparison of nucleotide, amino acid, and codon substitution models for evolutionary analysis of protein-coding sequences. Syst Biol. 2009;58:199–210. doi: 10.1093/sysbio/syp015. [DOI] [PubMed] [Google Scholar]
- 32.Choi SC, Hobolth A, Robinson DM, Kishino H, Thorne JL. Quantifying the impact of protein tertiary structure on molecular evolution. Mol Biol Evol. 2007;24:1769–1782. doi: 10.1093/molbev/msm097. [DOI] [PubMed] [Google Scholar]
- 33.Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19:908–917. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- 34.Guindon S, Rodrigo AG, Dyer KA, Huelsenbeck JP. Modeling the site-specific variation of selection patterns along lineages. Proc Natl Acad Sci USA. 2004;101:12957–12962. doi: 10.1073/pnas.0402177101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Brien JD, Minin VN, Suchard MA. Learning to count: Robust estimates for labeled distances between molecular sequences. Mol Biol Evol. 2009;26:801–814. doi: 10.1093/molbev/msp003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stone EA, Sidow A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 2005;15:978–986. doi: 10.1101/gr.3804205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seo T-K, Kishino H. Synonymous substitutions substantially improve evolutionary inference from highly diverged proteins. Syst Biol. 2008;57:367–377. doi: 10.1080/10635150802158670. [DOI] [PubMed] [Google Scholar]
- 38.de Koning APJ, Gu W, Pollock DD. Rapid likelihood analysis on large phylogenies using partial sampling of substitution histories. Mol Biol Evol. 2010;27:249–265. doi: 10.1093/molbev/msp228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foster PG, Jermiin LS, Hickey DA. Nucleotide composition bias affects amino acid content in proteins coded by animal mitochondria. J Mol Evol. 1997;44:282–288. doi: 10.1007/pl00006145. [DOI] [PubMed] [Google Scholar]
- 40.Siepel A, Haussler D. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol Biol Evol. 2004;21:468–488. doi: 10.1093/molbev/msh039. [DOI] [PubMed] [Google Scholar]
- 41.Baele G, Van de Peer Y, Vansteelandt S. A model-based approach to study nearest-neighbor influences reveals complex substitution patterns in non-coding sequences. Syst Biol. 2008;57:675–692. doi: 10.1080/10635150802422324. [DOI] [PubMed] [Google Scholar]
- 42.Yokoyama S, Tada T, Zhang H, Britt L. Elucidation of phenotypic adaptations: Molecular analyses of dim-light vision proteins in vertebrates. Proc Natl Acad Sci USA. 2008;105:13480–13485. doi: 10.1073/pnas.0802426105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kullberg MK, Nilsson MA, Arnason U, Harley EH, Janke A. Housekeeping genes for phylogenetic analysis of eutherian relationships. Mol Biol Evol. 2006;23:1493–1503. doi: 10.1093/molbev/msl027. [DOI] [PubMed] [Google Scholar]
- 44.Poux C, Chevret P, Huchon D, de Jong WW, Douzery EJP. Arrival and diversification of caviomorph rodents and platyrrhine primates in South America. Syst Biol. 2006;55:228–244. doi: 10.1080/10635150500481390. [DOI] [PubMed] [Google Scholar]
- 45.Steppan SJ, Adkins RM, Anderson J. Phylogeny and divergence-date estimates of rapid radiations in muroid rodents based on multiple nuclear genes. Syst Biol. 2004;53:533–553. doi: 10.1080/10635150490468701. [DOI] [PubMed] [Google Scholar]
- 46.Li C, Lu G, Ortí G. Optimal data partitioning and a test case for ray-finned fishes (Actinopterygii) based on ten nuclear loci. Syst Biol. 2008;57:519–539. doi: 10.1080/10635150802206883. [DOI] [PubMed] [Google Scholar]
- 47.Friedlander TP, Regier JC, Mitter C, Wagner DL. A nuclear gene for higher level phylogenetics: Phosphoenolpyruvate carboxykinase tracks mesozoic-age divergences within Lepidoptera (Insecta) Mol Biol Evol. 1996;13:594–604. doi: 10.1093/oxfordjournals.molbev.a025619. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.