

Abstract
Reduced expression of the CDK inhibitor p27Kip1 (p27) in human lung cancer correlates with tumour aggressiveness and poor prognosis. However, the regulation of p27 expression and the role of p27 during lung cancer are poorly understood. Urethane-induced lung tumours in mice frequently harbor mutations in the Kras oncogene and here we use this model to address the regulation of p27 during tumorigenesis. The Ras effector Akt is known to regulate p27 mRNA abundance by phosphorylating and inactivating the FOXO transcription factors. Phosphorylated Akt and FOXO proteins were both increased in lung tumours, correlating with a reduction in p27 mRNA transcript. Akt also directly phosphorylates p27 and regulates its nuclear/cytoplasmic localization. Tumours showed a reduced nuclear/cytoplasmic ratio of p27 protein, together with an increase in phosphorylated Thr198 p27 in the cytoplasmic pool. Treatment of lung tumour-bearing mice with the phosphoinositol-3 kinase inhibitor LY294002 induced a rapid decrease in phosphorylated Akt and phosphorylated p27, concomitant with an increase in nuclear p27. Germline p27 deficiency accelerated both the growth and malignant progression of urethane-induced lung tumours, and did so in a cell autonomous manner, confirming a causal role of p27 in tumour suppression. These results demonstrate that p27 is a potent barrier to the growth and malignant progression of Kras-initiated lung tumours. Further, the reduction of nuclear p27 in tumours is mediated by oncogene signaling pathways, which can be reversed by pharmacologic agents.
Keywords: Kras, p27, Akt, lung cancer, tumour suppressor
Introduction
One approach to cancer treatment is to enhance tumour suppressor activity in tumours. The effectiveness of this strategy has been recently demonstrated using genetically engineered mouse models, where restoration of expression of a tumour suppressor, in this case p53, led to complete tumour regression (Ventura et al. 2007; Martins et al. 2006; Xue et al. 2007). This approach is currently not feasible in humans, the primary limitation being that most known tumour suppressors are mutated or deleted in tumours. The Cdk inhibitor p27Kip1 (p27) is a notable exception to the classic tumour suppressor gene paradigm in that the CDKN1B gene that encodes p27 is rarely mutated in tumours (Kawamata et al. 1995; Ponce-Castaneda et al. 1995). Instead, the levels of p27 protein are reduced or mislocalized and this correlates with tumour aggressiveness and poor prognosis (Chu et al. 2008). A causal role for p27 in tumour suppression has been shown by the increased susceptibility of p27 deficient mice to tumour development (Nakayama et al. 1996; Fero et al. 1996; Kiyokawa et al. 1996; Fero et al. 1998). The fact that the gene encoding p27 remains intact in most human cancers, provides an opportunity to design therapies that might enhance expression of p27. Key elements to the success of this strategy are to understand the mechanism(s) leading to misexpression of p27 and determine whether manipulating these pathways can enhance p27 expression in tumours.
p27 abundance is regulated by a number of different pathways throughout the cell cycle. The levels of p27 are high in quiescent cells and markedly reduced as cells transit through the cell cycle. In S phase, p27 is phosphorylated at Thr187 by CDK2, targeting p27 for ubiquitination by SKP2 and subsequent degradation by the proteasome (Sheaff et al. 1997). The KPC ubiquitin ligase was recently identified as a second mechanism to target p27 for degradation during G1 (Kamura et al. 2004). Microinjection studies showed that active Ras can reduce p27 by three pathways: MAPK in G1, PI3K in G1 and S, and via increased SKP2 in S and G2 (Sa and Stacey 2004). Colorectal cancer cell lines exhibit proteasome-dependent degradation of p27 (Loda et al. 1997), and many tumours express high levels of its ubiquitin ligase, SKP2, suggesting that the levels of p27 in tumours are regulated by proteasomal degradation (Bloom and Pagano 2003). Recent studies revealed that nonreceptor tyrosine kinases, such as Src, phosphorylate p27, consequently decreasing its stability (Chu et al. 2007; Grimmler et al. 2007), which may be yet another mechanism to reduce p27 abundance in tumours.
In addition to decreasing overall p27 levels, exclusion of p27 from the nucleus and retention in the cytoplasm can effectively reduce its CDK inhibitory activity. Cancers of the breast, thyroid, esophagus, and colon show specific reduction in nuclear p27, suggesting p27 mislocalization may be important for tumour progression (Blain and Massague 2002). At least two pathways have been proposed for nuclear export of p27 during G1 and S phase; association with Jab1, a component of the COP9-signalosome complex (Tomoda et al. 1999), or direct binding to the transporter CRM1 via the nuclear export signal of p27 (Connor et al. 2003). Nuclear export is regulated by the phosphorylation of p27 at specific residues. Ser10 phosphorylation of p27 by human kinase-interacting stathmin (hKIS or KIST) increases the binding of p27 to the nuclear exporter CRM1 (Boehm et al. 2002). Experiments utilizing knock-in mice carrying a Ser to Ala mutation at position 10 of p27 showed reduced levels of p27 in cultured MEFs (Kotake et al. 2005) and impaired nuclear export of p27 (Besson et al. 2006). Phosphorylation of Thr198, by either Akt or RSK1/2, results in binding to the protein 14-3-3 and retention in the cytoplasm (Fujita et al. 2003). This increases the stability of p27 by preventing proteasomal turnover (Kossatz et al. 2006). Cytoplasmic retention of p27 in human breast cancer cells was shown to be regulated by phosphorylation of Thr157 by the Akt kinase (Shin et al. 2005; Viglietto et al. 2002; Liang et al. 2002). However, Thr157 is not present in rodent p27, so this pathway is not evolutionarily conserved.
p27 is also regulated at the transcriptional levels. In quiescent cells, FOXO proteins are located within the nucleus where they bind to the p27 promoter and initiate transcription (Dijkers et al. 2000). In response to growth factors or oncogenic signals, FOXO proteins are phosphorylated by Akt, resulting in cytoplasmic sequestration, decreased p27 transcription, and cell proliferation (Medema et al. 2000). As Akt is active in many tumours, this may represent an additional mechanism to reduce p27 during tumour growth.
Lung cancer is among the leading causes of death worldwide and non-small cell lung cancer (NSCLC) accounts for ~87% of these cases (Singhal et al. 2005). Reduced expression of p27 is seen in ~30% of NSCLC cases, and five independent studies have shown a significant correlation of reduced p27 with disease recurrence or death (Chu et al. 2008). Mutations in Kras and EGFR are frequently seen in lung cancer (Ding et al. 2008) and signaling from both can downregulate p27 in cell lines (Sa and Stacey 2004; Busse et al. 2000). Inui et al. reported that p27 mRNA levels were not reduced in lung tumors, although these were not stratified by Ras mutational status or Akt activity (Inui et al. 2003). Catzavelos et al. reported that Ras mutation and p27 expression seem to occur independently in adenocarcinoma of the lung (Catzavelos et al. 1999). Thus, the mechanism(s) contributing to altered regulation of p27 during lung tumourigenesis are unclear.
Urethane treatment of mice induces alveolar/bronchiolar adenomas and adenocarcinomas, which closely resemble human NSCLC adenocarcinoma in morphologic and molecular characteristics (Malkinson 2001). Here we use this model to examine the role of p27 in lung tumour suppression and the mechanisms leading to its misregulation.
Results
p27-deficient mice are susceptible to non-small cell lung cancer
To establish an autochthonous tumour model to examine the role of p27 in lung cancer, cohorts of p27+/+, +/−, and −/− littermates were injected with urethane. The lung tumour incidence for all three genotypes was 100% at both 30 and 50 weeks. At 30 weeks, both tumour multiplicity and tumour size were significantly greater in p27−/− and +/− mice compared to wildtype littermates (Figure 1A). The average number of tumours greater than 1mm in diameter was 1.4 ± 1.87, 4.2 ± 3.5, and 7.0 ± 4.5 for p27+/+, +/− and −/− mice, respectively (p het-wt = 0.026; p null-wt = 0.0012; two-tailed t test). Only four p27−/− mice survived beyond 40 weeks of age (median age 46 weeks), due to complications arising from excess lung tumour burden and pituitary tumours, and data from these four mice are included in the 50 week graph (Figure 1B). Tumour burden was greater at 50 versus 30 weeks for all genotypes. Again, tumour multiplicity and size were both increased in p27 deficient mice compared to wildtype littermates (Figure 1B, 1D). The average number of tumours per mouse at 50 weeks of age in p27 intact mice was similar to the number in p27−/− mice at 30 weeks of age (Figure 1C), suggesting that the primary effect of p27 is to inhibit tumour growth as opposed to tumour initiation.
Figure 1. p27 is a lung tumour suppressor.

Histogram comparing the number and sizes of urethane-induced lung tumours in p27+/+, +/−, and −/− mice at (A) 30 weeks and (B) 50 weeks post-treatment. Only four p27 null mice survived to a median age of 46 weeks and the tumour counts from these mice are graphed in (B). (C) Table showing mean number of tumours per mouse according to genotype and age. (D) Photograph of lungs from p27+/+ and p27−/− mice showing increased tumour burden in p27−/− lungs. (E) Loss of heterozygosity analysis of DNA from tumours from p27+/− mice, n= 24 tumours examined in total. DNA from p27+/+ and −/− tumours are shown as controls. wt= wild type p27 allele. mu= knockout p27 allele. (F) Histogram showing mean number of adenocarcinomas per mouse at 30 and 50 weeks of age. Insufficient numbers of p27−/− mice survived to 50 weeks, so these are not shown. Shown are p values from unpaired t-test for comparisons between genotypes. (G) Photomicrographs of lung sections with an alveolar/bronchiolar adenoma from a p27+/+ mouse (left) and an adenocarcinoma from a p27−/− mouse (right). Note the discrete boundary between adenoma and normal lung (clear arrow) and monomorphic, orderly tumour cells (clear arrowhead), compared to invasive tumour cells of the adenocarcinoma into lung parenchyma and local airway (black arrows) and pleomorphic appearance of tumour cells (black arrowheads).
Since the tumour burden of p27+/− mice was increased relative to p27+/+ mice, we examined the p27 locus for loss of heterozygosity. Analysis of tumours from p27+/− mice showed retention of the wildtype p27 allele in all 24 tumours examined (Figure 1E), consistent with previous findings that p27 is haploinsufficient for tumour suppression (Fero et al. 1998).
Histological evaluation revealed that the majority of tumours were alveolar/bronchiolar adenomas that displayed papillary, solid, or mixed morphologies. In p27 intact mice, adenocarcinomas were not seen until 50 weeks of age, while both p27 null and heterozygous mice developed an average of one malignant tumour per mouse by 30 weeks of age (Figure 1F). These tumours where characterized by marked cellular atypia and local invasion indicating that p27 deficiency increased both tumour growth and malignant tumour progression.
Tumour suppression by p27 is cell autonomous
Detection of mutations in cancer genes in tumours is evidence for a cell autonomous mechanism of action. However, mutations in the CDKN1B gene encoding p27 are rarely observed in tumours. Forced expression of p27 can inhibit tumour cell proliferation, and mice lacking p27 are tumour prone. However, this evidence is insufficient to prove that p27 suppresses tumours through a cell autonomous mechanism and other approaches are required to establish this conclusively. We used a neonatal lung tissue transplantation model to address this. Lung tissue from neonatal p27 deficient or wildtype mice was transplanted to the ear of adult wildtype recipients. After engraftment, mice were treated with urethane and transplant size was measured up to 30 weeks. Tumours developed from ~50% of transplants from p27−/− (10/19) or p27+/− mice (19/39), compared to only 25% (5/20) from p27+/+ mice (Figure 2A). Tumours were larger from both p27−/− and p27+/− transplants (Figure 2B). Histologic evaluation indicated that the majority of tumours were bronchioalveolar adenomas and carcinomas. Thus, cells lacking p27 are more susceptible to tumourigenic outgrowth than are p27 intact cells, indicating a cell autonomous mechanism of tumour suppression by p27.
Figure 2. Tumour suppression by p27 is cell autonomous.

Lung tissue from p27+/+, +/−, or −/− mice was transplanted to the ears of wild type recipients. After engraftment, mice were then treated with urethane and transplant size was measured up to 30 weeks (p27+/+ n=14, p27+/− n=23, p27−/− n=14). (A) Bar graph shows percent transplants that developed tumours. (B) Size of transplanted tumours at 30 weeks. Each symbol represents one transplant.
Mutation of Kras in urethane-induced lung tumours
Sequence analysis of lung tumour DNA revealed that 80% of urethane-induced tumours had a CAA to CTA transversion mutation at codon 61 of the Kras gene, resulting in a Q61L amino acid change (Figure 3A). The frequency of this mutation was similar in tumours from A/J, B6, and B6/129 strains. The frequencies of Kras mutations were also similar in tumours from p27+/+, +/− and −/− mice (Figure 3B).
Figure 3. K-ras mutations are frequent in lung tumours.

(A) The chromatograms show the Kras wildtype sequence (left) and an CAA>CTA mutation in codon 61 (right) from a tumour. (B) Table shows frequency of K-ras mutations in tumours from different genetic backgrounds and p27 status.
Nuclear exclusion of p27 protein in lung tumours
Since p27 deficient mice showed increased susceptibility to urethane-induced lung tumours, we examined the status of p27 protein in lung tumours from p27 intact mice. Staining of tissue sections with a monoclonal anti- p27 antibody revealed prominent nuclear p27 staining in normal lung parenchymal cells but reduced nuclear p27 in tumour cells (Figure 4A). Western blot analysis of nuclear and cytoplasmic fractions prepared from frozen tissues revealed that tumours exhibit reduced nuclear p27 while retaining the cytoplasmic pool (Figure 4B). Quantitation with densitometry confirmed the ratio of nuclear to cytoplasmic p27 was markedly reduced in tumours compared to normal lung (Figure 4C). Therefore, both mutation of Kras and aberrant subcellular localization of p27 protein are cardinal features of this tumour model.
Figure 4. Nuclear p27 is reduced in lung tumours.

(A) Photomicrograph of a lung adenoma section from a wild type mouse stained with a p27 antibody. Note strong nuclear staining in normal lung parenchymal cells and reduced nuclear staining in tumour cells. (B) Western blot of extracts from lung tumours and normal lung from wild type mice probed with p27 antibody. n: nuclear extracts, c: cytoplasmic extracts. p27 antibody specificity is indicated by tissue from p27 null mouse (−/−) as a negative control. (C) Quantitation of nuclear (n) to cytoplasmic (c) p27 protein expression from Western blots of normal and tumour lysates. Numbers show median values of n/c ratios.
As both Ras mutation and altered p27 expression are consistently observed together, we asked whether oncogenic signals emanating from mutant Ras protein actively drive p27 mislocalization. We compared the phosphorylation status of specific residues of p27 (Ser10, Thr187, and Thr197) shown to be involved in regulation of cellular localization, as well as upstream candidate regulators between normal lung and tumour lysates.
Ras signaling is expected to induce cyclin D1, activate Cdk2 and drive cell cycle progression. Cdk2, in turn, is known to phosphorylate residue Thr187, targeting p27 for Jab1-mediated export and Skp2 degradation (Sheaff et al. 1997). We found an increase in both cyclin D1 and Cdk2 kinase activity in tumour extracts, consistent with the enhanced proliferation of lung tumour cells (Figure 5A). We also observed an increase in Jab1 and Thr187 phospho- p27 in cytoplasmic fractions of lung tumours (Figure 5B), leading to the conclusion that Cdk2-dependent phospho-Thr187 p27 is exported and accumulates in the cytoplasm. The cytoplasmic levels of two ubiquitin ligases for p27, Skp2 and Kpc, were similar between tumour and normal tissue, suggesting that proteasome degradation via upregulation of Skp2 or Kpc may not be the major pathway to regulate p27 abundance in this tumour model.
Figure 5. Posttranslational regulation of p27 in lung tumours.

(A) Increased Cdk2 activity in lung tumours from wild type mice versus normal lung using histone H1 as a substrate. Western blot of whole cell lysates shows Cdk2 protein levels unchanged and an increase in cyclin D1 in lung tumours compared to normal lung. (B) Western blot of total p27, phospho- T187 p27, Jab1, KPC, and Skp2 in tumours and normal lung, with lamin B1 and β-tubulin as nuclear and cytoplasmic fractionation controls. Note increase in p-T187 p27 in cytoplasmic fractions compared to normal lung. n: nuclear extracts, c: cytoplasmic fractions. (C) Western blot of p-T198 p27, p-S10 p27, and 14-3-3-θ. Note increased p-T198 p27 in cytoplasmic tumour fractions.
Ras also signals through PI3K to activate Akt, which in turn can phosphorylate p27 at multiple sites including Ser10, Thr157 (in human but not in mouse), and Thr198 (Thr197 in the mouse), each of which has been shown to promote nuclear to cytoplasmic shuttling (Susaki and Nakayama 2007). Phosphorylated Ser10 p27 was found in nuclear and cytoplasmic fractions and did not appear to differ between normal and tumour extracts (Figure 5C). However, as total nuclear p27 is markedly reduced in nuclear fractions from tumours, the proportion of p27 that is phosphorylated within tumour nuclear pools could be increased.
Levels of phospho-Thr198 p27 were markedly increased in lung tumours, notably in the cytoplasmic fraction (Figure 5C). The levels of 14-3-3, which binds to cytoplasmic p27 phosphorylated at Thr198 (Fujita et al. 2003) were also modestly elevated, suggesting a plausible scenario of Akt dependent phosphorylation of Thr198 p27 and cytoplasmic retention of p27 by 14-3-3. These data together indicate p27 is phosphorylated at multiple sites in lung tumours, one or more of which could signal nuclear export and cytoplasmic retention.
Reduction of p27 transcript in lung tumours
While most studies have focused on post translational regulation of p27 abundance, transcriptional control is also documented (Philipp-Staheli et al. 2004). Quantitative real time PCR revealed a five to ten fold reduction in p27 mRNA transcript in 13 out of 13 lung tumours examined, as compared to normal lung tissue (Figure 6A). Signaling from Ras has been linked to p27 transcription via Akt dependent phosphorylation of Forkhead transcription factors (Medema et al. 2000). Lung tumour extracts showed an increase in active phospho-Akt (Figure 5C), as well as phosphorylated cytoplasmic FOXO1 and FOXO3a. Phosphorylated FOXO4a, by contrast, shows equivalent expression in tumour and normal lung (Figure 6B). These data are consistent with the idea that oncogenic Kras signaling through PI3K activates Akt, which phosphorylates FOXO transcription factors, resulting in their retention in the cytoplasm, thereby reducing p27 transcription.
Figure 6. Reduced p27 transcript and increased p-Akt and p-FOXO in lung tumours.

(A) Bar graph of p27 mRNA expression levels derived from real-time PCR using total RNA isolated from lung tumours and normal lung (NL) from wild type mice. (B) Western blot shows expression levels of phosphorylated forkhead family members, p-FOXO1, p-FOXO3a, and p-FOXO4a in lung tumour extracts compared to normal lung. n: nuclear extracts, c: cytoplasmic fractions.
In summary, p27 expression in lung tumours is altered in at least two ways: increased phosphorylation leading to nuclear exclusion, and reduced transcription. Both mechanisms could be due to Ras signaling through Akt.
Inhibition of PI3K and Akt restores p27 levels in tumours
Since the gene encoding p27 is intact in tumours, we asked whether its expression could be restored by pharmacologically blocking pathways leading to its misregulation. As Akt plays a role in both transcriptional and posttranslational regulation of p27, we used the PI3K inhibitor LY294002 to inhibit this pathway. Treatment of the mouse lung tumour cell line SP10 with LY294002 led to a reduction of phospho-Akt, a reduction in phospho-FOXO, and an increase in both p27 transcript and protein levels (Figure 7A, B). Therefore, inhibition of the PI-3 Kinase pathway can enhance p27 levels in tumour cells by increasing p27 transcription.
Figure 7. Inhibition of the PI3K pathway increases nuclear p27 in tumours.
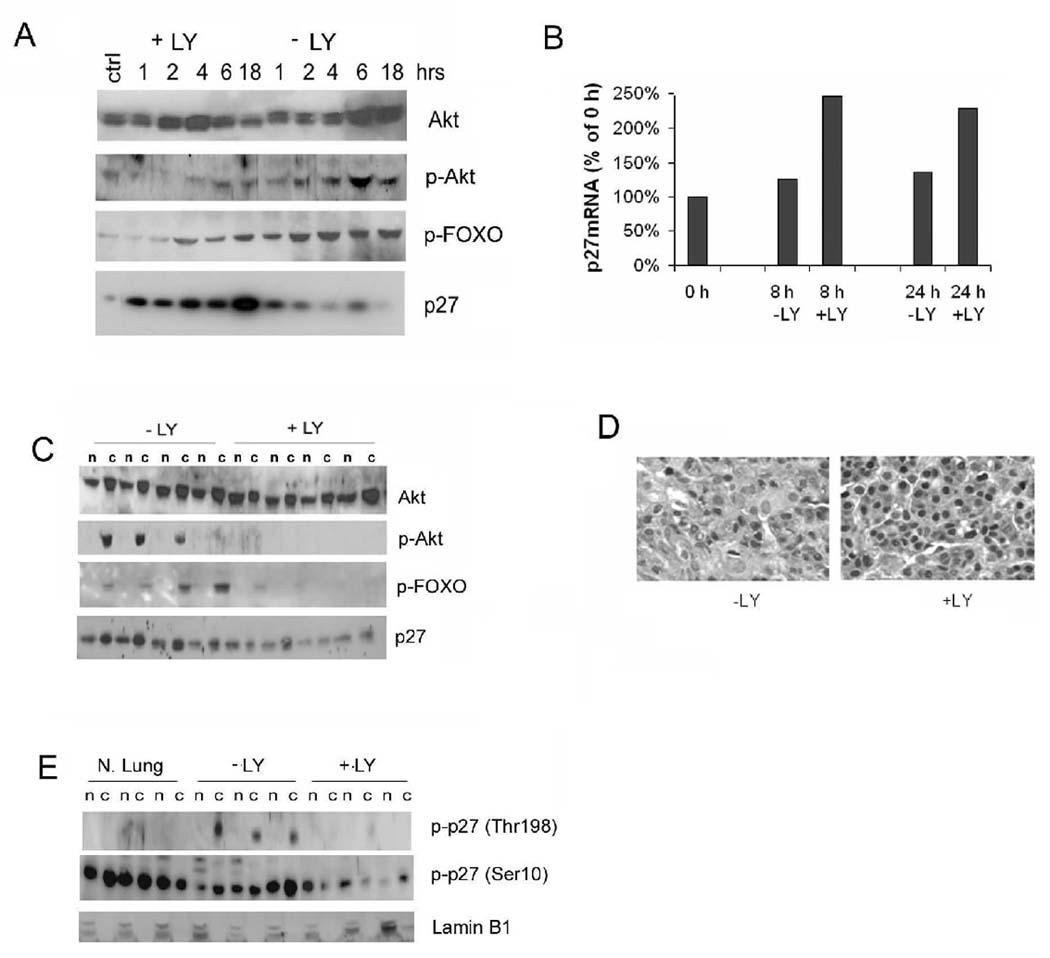
(A) Time course analysis of effects of LY294002 (LY) on SP10 mouse lung tumour cell line. Shown are Western blots of cell lysates probed with antibodies to p-Akt, p-FOXO, and p27 at the indicated times after addition of serum and LY. The control (ctrl) lane is from cells not serum starved. (B) Real-time PCR of p27 mRNA from treated (+LY) and control (−LY) treated SP10 cells 8 and 24 h after start of PI3K inhibition. (C) Western blot of total and p-Akt, p-FOXO, and p27 in lung tumours from wild type mice 5 h after treatment with LY. n: nuclear extracts, c: cytoplasmic fractions. (D) Sections of representative lung tumours immunostained for p27 shows increase in nuclear p27 following LY294002 treatment. (E) Western blot of tumour lysates shows decreased p-T198 p27 and p-S10 p27 in LY294002 treated lung tumours. Lamin B1 is nuclear fraction loading control. N.Lung = normal lung.
We next treated tumour-bearing A/J mice with LY294002 and analyzed tumours for short term changes in Akt signaling and p27 expression. Western blot analysis of tumour lysates showed a marked reduction of phospho-Akt and phosphorylated FOXO in inhibitor treated mice compared to tumours from vehicle treated mice (Figure 7C), while total cellular levels of Akt remained unchanged. This shows that LY294002 treatment effectively inhibited the PI-3K to Akt to FOXO signaling cascade in lung tumours in vivo. Next, we examined p27 expression and phosphorylation. LY294002 treatment reversed the nuclear exclusion of p27 in tumours, as revealed by both Western blots of tumour lysates (Figure 7C) and immunohistochemistry of lung tumour sections (Figure 7D). Relative to untreated tumours, tumours from LY294002 treated mice showed equal nuclear/cytoplasmic distribution of p27 and more prominent nuclear staining for p27. Since LY294002 inhibits Akt, and Akt can phosphorylate p27 directly, we examined the phosphorylation status of p27. Western blotting of lysates from inhibitor treated tumours showed a reduction in both cytoplasmic phospho-Thr198 and phospho-Ser10 p27 (Figure 7E). Together, these findings indicate that aberrant expression of p27 in tumours is an active processes regulated by the PI3K-Akt pathway, and that the mislocalization of p27 in tumours can be reversed through the use of pathway specific inhibitors.
Discussion
These studies demonstrate that p27 acts as a barrier to the growth and malignant progression of Kras driven NSCLC. Both homozygous and hemizygous germline deletion of p27 led to an increase in both the size and malignancy of tumours. The observation that the average number of tumours in wild type mice at 50 weeks was similar to the number of tumours in p27 deficient mice at 30 weeks, together with the observation that tumour suppression by p27 is cell autonomous, argues that the primary effect of p27 is to inhibit the clonal expansion of initiated tumour cells. Tumours from all p27 genotypes harbored mutations in the Kras oncogene, establishing that p27 specifically inhibits Kras-driven tumour growth. The ubiquitous reduction of nuclear p27 in tumours further implies that cell cycle inhibition by p27 is a critical barrier to be overcome for such clonal expansion.
p27 and malignant progression
In addition to inhibiting tumour growth, p27 suppressed malignant lung tumour progression. By 30 weeks of age, 67% of p27 deficient mice had malignant disease, averaging one adenocarcinoma per mouse, while no carcinomas were observed in p27 intact mice. It is at present not clear whether this is due to increased tumour growth, leading to an increase in rate of tumour evolution toward malignancy, or is a more direct consequence of p27 functions; e.g. due to loss of differentiation or increased invasion. p27 can induce cell differentiation in vitro (reviewed in (Philipp-Staheli et al. 2001)), and p27 deficiency leads to impaired differentiation of several cell types in vivo (Casaccia-Bonnefil et al. 1997; Nakayama et al. 1996; Zindy et al. 1999; Tong et al. 1998). Although the mechanisms by which p27 can trigger differentiation are poorly understood, p27 induced cell cycle exit appears to be tightly linked to terminal differentiation. p27 also plays a role in cell migration, independent of its Cdk inhibitory function (Besson et al. 2004a). p27 has been shown to inhibit cancer cell invasion (Yuan et al. 2007; Schiappacassi et al. 2008) by binding to microtubule destabilizing protein, stathmin (Baldassarre et al. 2005). On the other hand, a promigratory role for cytoplasmic p27, through interaction with the Rho family of GTPases and the cytoskeleton, has also been described (McAllister et al. 2003; Besson et al. 2004b). Hence, tumours with reduced or mislocalized p27 may have impaired capacity to differentiate and/or enhanced migratory ability thereby increasing their malignant behavior.
Malignant tumour progression due to reduced p27 has been documented in other mouse cancer models (Philipp et al. 1999; Philipp-Staheli et al. 2002; Shaffer et al. 2005), indicating this is a characteristic trait of tumour suppression by p27. In human cancer, including NSCLC, reduced p27 expression correlates with higher tumour grade and stage, features which incorporate differentiation, invasion and metastasis (Chu et al. 2008). Together, this indicates that p27 is a barrier to malignant progression and the misregulation of p27 in tumours is causally linked to tumour aggressiveness.
p27 misregulation in tumours
Having established that p27 inhibits both the growth and malignant progression of NSCLC, we next examined the mechanism by which tumours circumvent p27. Lung tumours showed a consistent reduction in the nuclear pool of p27, while largely retaining cytoplasmic expression, as well as a marked reduction of mRNA transcript. Thus, transcriptional control as well as subcellular localization contribute to a reduction of nuclear p27 in tumours, which can lead to increased CDK activity. Activating mutations in Kras were also cardinal features of this tumour model, suggesting a mechanistic link between Kras signaling and p27 loss. Indeed, an emerging model to explain the basis of cooperation between oncogene activation and tumour suppressor gene loss posits that oncogenes directly signal to tumour suppressor genes. The Ras effector, Akt, has been shown to regulate p27 at multiple levels, so we asked whether p27 misregulation in tumours is a direct consequence of Akt signaling. p27 can be barred from the nucleus by pathways involving phosphorylation at Ser10, Thr157 (human but not mouse), and Thr198, all of which have been reported as Akt targets. We did not observe significant changes in abundance of phospho-Ser10 p27 in lung extracts. However, as the total nuclear p27 levels were decreased in tumours, it is possible that the ratio of phospho-Ser10 p27 to total p27 is increased in these tumours. Akt can also phosphorylate p27 on Thr198 and promote binding to 14-3-3 in the cytoplasm (Fujita et al. 2003; Motti et al. 2005; Motti et al. 2004). Lung tumours showed a marked increase in phospho-Thr198 p27, specifically in the cytoplasmic pool, together with a modest increase in 14-3-3, suggesting phospho-Thr198 may direct nuclear export and increase stability of p27 in the cytoplasm. Indeed, inhibition of Akt in both mouse lung tumour cell lines and in tumour-bearing mice led to a reduction in phospho-Ser10 and phospho-Thr198 p27 and redistribution of p27 to the nucleus.
Akt is also a key player in mediating signaling from Ras through the FOXO transcription factors to regulate p27 transcription (Medema et al. 2000). Increased phosphorylation of FOXO proteins was seen in cytoplasmic fractions from lung tumours, correlating with reduced p27 mRNA levels. LY294002 reduced phosphorylation of both Akt and FOXO and increased p27 transcript and nuclear p27 protein levels. Together these findings demonstrate that Ras signaling through the PI3K/Akt pathway regulates p27 abundance and localization at both the transcriptional and post-translational levels.
In summary, p27 acts as a potent barrier to Kras initiated NSCLC that must be overcome for efficient clonal expansion and malignant progression. However, in contrast to most known tumour suppressors which are inactivated by mutation, p27 reduction is an active and reversible process; in this case, mediated by oncogenic signaling. If a similar mechanism occurs in human lung cancer, this would create a window of opportunity for therapeutic intervention.
Materials and methods
Mice
Experiments were approved by the FHCRC Institutional Animal Care and Use Committee. Two cohorts of 129×C57BL/6J F1 p27−/−, p27+/−, and p27+/+ mice (Fero et al. 1996) as well as C57BL/6J and A/J mice were treated with urethane (1mg/g bodyweight, i.p.) at 12 days of age. Cohorts were sacrificed at 30 or 50 weeks of age. Tumours visible on the surface of all lung lobes were counted and measured using a dissecting microscope. Normal and tumour tissues were either snap frozen in liquid nitrogen for DNA, RNA, and protein extraction or fixed in neutral buffered formalin. Fixed tissue was processed and stained with H&E for histological examination. DNA for genotyping was obtained by digestion of tissue with proteinase K in InstaGene Matrix solution (Bio-Rad, Hercules, CA, USA). Each mouse was genotyped by PCR using primers specific for the wild-type or neo interrupted p27 locus (Fero et al. 1996).
Kras sequence analysis
Genomic DNA was isolated from lung tumours using the QIAGEN QIAamp DNA mini kit. The Kras locus at codon 61 was amplified using primers 5’-GACTCCTACAGGAAACAAGT-3’ and 5’–GAAGATATTCACCATTATAG-3’ (You et al. 1989) by PCR and sequenced using the Big Dye Sequencing protocol (PE-Biosystems).
Western blot analysis
Nuclear and cytoplasmic protein extracts were prepared as described (Schreiber et al. 1989), with modifications (Philipp-Staheli et al. 2002). Protein concentrations were standardized using the Bradford assay (BioRad), and equal loading was confirmed by Ponceau S staining of PVDF membranes after blotting (Immun-Blot™ PVDF membrane, BioRad). The following antibodies were used for Western blotting: p27 (sc-528), phospho- Thr187 p27 (sc-16324), phospho- Ser10 p27 (sc-12939), cyclinD1 (sc-755), cdk2 (sc-163), Jab1 (sc-9074), 14-3-3 θ (sc-732), β-tubulin (sc-9104) and lamin B1 (sc-20682) were from Santa Cruz Biotechnology (Santa Cruz, CA). p45/Skp2 antibody (ab19877) was purchased from Abcam and the phospho-Thr198 p27 purchased from R&D Systems (AF3994). The anti-KPC1 antibody (H00063891-A01) was obtained from Novus Biologicals. Akt phospho-specific (9271, 9275) and FOXO family (9464, 9466, 9471) antibodies were purchased from Cell Signaling (Beverly, MA).
Histopathology and immunohistochemistry
Lung lobes with embedded tumours were removed and fixed in 10% neutral buffered formalin for 4–6 h, processed and embedded in paraffin. Step sections were made, with every fifth section stained with H&E for the most accurate histologic quantification of tumour multiplicity and malignancy. All slides were reviewed by a pathologist (SK) blind to the genotype. Diagnosis of adenoma was based on discrete, well demarcated foci consisting of a uniform population of epithelial cells with round nuclei and a moderate amount of eosinophilic cytoplasm. Compared with adenomas, adenocarcinomas showed greater cellular atypia, regional variation of growth patterns, increased mitotic activity, and local invasion into adjacent bronchioles or vessels. Immunohistochemistry for p27 was performed as described (Philipp-Staheli et al. 2002) using a mouse monoclonal antibody from Neomarkers (Fremont, CA). Controls included no primary antibody and/or normal rabbit serum, and tissues from p27 null mice.
Histone kinase assay
Protein extracts were prepared as described above and stored in 50% glycerol. 400 µg of protein were incubated on ice in 500 µl RIPA buffer containing Cdk2 antibody. Then 30 µl of washed Protein A-Sepharose beads were added, and the reaction was rotated for 1 h at 4°C. Beads were washed twice in RIPA and once in a buffer containing 25 mM Tris HCl (pH 7.5), 70 mM NaCl, 10mM MgCl2, and 1 mM DTT. The beads were then incubated with Histone H1 as a substrate and 32P-γATP for 30 min at 37°C. The reaction was stopped by the addition of sample buffer, denatured for 5 min at 95°C, and the protein separated on a 12% SDS acrylamide gel run for 45 min at 150 volts. The gel was washed, fixed and exposed to X-ray film to visualize bands.
Real-time PCR assay
The relative levels of p27 mRNA were assessed using RTPCR as described (Philipp-Staheli et al. 2004), using the forward primer 5’-AGGCTGGGTTAGCGGAGC-3’; reverse primer 5’-GAACCGTCTGAAACATTTTCTTCTGT-3’; and probe 5’ FAM-ACCTTGCTGCAGAAGATTCTTCTTCGCAA-TAMRA 3’.
Tumour DNA isolation and LOH analysis
DNA was extracted from frozen tumours using BioRad InstaGene Matrix and p27 PCR conditions are available on request. PCR products were run on a 1% agarose gel stained with ethidium bromide.
Ear engraftment
Lungs were removed from neonatal mice derived from NIH p27+/−×B6/129 p27+/− crosses and cut into 2mm3 sections and kept on ice. Recipient NIH wild type adult mice were anesthetized with avertin. A small pouch was made in both ears using a scalpel and scissors. A 2 mm3 section of lung was inserted, and the pouch sealed with super glue. For a given recipient, transplant of a different p27 genotype in each ear served as a control. Mice were injected with urethane once a week for 6 weeks (0.25 mg/g body weight i.p.). The ear grafts were measured every other week and mice were sacrificed at 30–38 weeks, and tissues processed as described above.
PI3K inhibition
SP10 lung tumour cells (ATCC), derived from a spontaneous lung tumour from an A/J strain mouse, were grown to 70% confluence, serum starved overnight, then serum containing media with 50 µg/ml LY294002 (Sigma) was added. Cells were harvested at 1, 2, 4, 6, and 18 h post-treatment and whole cell lysates were made using buffer containing 150 mM NaCl, 50 mM Tris pH 8, and 1% NP-40. Cells not subjected to serum starvation or inhibitor treatment were used as controls in Western analysis. For in vivo studies, nine month old lung tumour-bearing A/J mice were injected with LY294002 (100 mg/kg body weight, i.p.) or DMSO three times over a six hour period.
Acknowledgments
This work was funded by a NIH T32 CA8046 Interdisciplinary Training Grant in Cancer Research (KSK-S), and an ACS Research Scholar Award and a NIH R01 Research Grant (CJK).
References
- Baldassarre G, Belletti B, Nicoloso MS, Schiappacassi M, Vecchione A, Spessotto P, Morrione A, Canzonieri V, Colombatti A. p27(Kip1)-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell. 2005;7:51–63. doi: 10.1016/j.ccr.2004.11.025. [DOI] [PubMed] [Google Scholar]
- Besson A, Assoian RK, Roberts JM. Regulation of the cytoskeleton: an oncogenic function for CDK inhibitors? Nat Rev Cancer. 2004a;4:948–955. doi: 10.1038/nrc1501. [DOI] [PubMed] [Google Scholar]
- Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. A pathway in quiescent cells that controls p27/Kip1 stability, subcellular localization, and tumor suppression. Genes & Dev. 2006;20:47–64. doi: 10.1101/gad.1384406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004b;18:862–876. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blain SW, Massague J. Breast cancer banishes p27/Kip1 from the nucleus. Nature Medicine. 2002;8:1076–1078. doi: 10.1038/nm1002-1076. [DOI] [PubMed] [Google Scholar]
- Bloom J, Pagano M. Deregulated degradation of the cdk inhibitor p27 and malignant transformation. Semin Cancer Biol. 2003;13:41–47. doi: 10.1016/s1044-579x(02)00098-6. [DOI] [PubMed] [Google Scholar]
- Boehm M, Yoshimoto T, Crook MF, Nallamshetty S, True A, Nabel GJ, Nabel EG. A growth factor-dependent nuclear kinase phosphorylates p27(Kip1) and regulates cell cycle progression. EMBO J. 2002;21:3390–3401. doi: 10.1093/emboj/cdf343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse D, Doughty RS, Ramsey TT, Russell WE, Price JO, Flanagan WM, Shawver LK, Arteaga CL. Reversible G(1) arrest induced by inhibition of the epidermal growth factor receptor tyrosine kinase requires up-regulation of p27(KIP1) independent of MAPK activity. J Biol Chem. 2000;275:6987–6995. doi: 10.1074/jbc.275.10.6987. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Tikoo R, Kiyokawa H, Friedrich V, Jr, Chao MV, Koff A. Oligodendrocyte precursor differentiation is perturbed in the absence of the cyclin-dependent kinase inhibitor p27Kip1. Genes & Dev. 1997;11:2335–2346. doi: 10.1101/gad.11.18.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catzavelos C, Tsao MS, DeBoer G, Bhattacharya N, Shepherd FA, Slingerland JM. Reduced expression of the cell cycle inhibitor p27Kip1 in non-small cell lung carcinoma: a prognostic factor independent of Ras. Cancer Res. 1999;59:684–688. [PubMed] [Google Scholar]
- Chu I, Sun J, Arnaout A, Kahn H, Hanna W, Narod S, Sun P, Tan CK, Hengst L, Slingerland J. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell. 2007;128:281–294. doi: 10.1016/j.cell.2006.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- Connor MK, Kotchetkov R, Cariou S, Resch A, Lupetti R, Beniston RG, Melchior F, Hengst L, Slingerland JM. CRM1/Ran-mediated nuclear export of p27(Kip1) involves a nuclear export signal and links p27 export and proteolysis. Mol Biol Cell. 2003;14:201–213. doi: 10.1091/mbc.E02-06-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkers PF, Medema RH, Pals C, Banerji L, Thomas NS, Lam EW, Burgering BM, Raaijmakers JA, Lammers JW, Koenderman L, Coffer PJ. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1) Mol Cell Biol. 2000;20:9138–9148. doi: 10.1128/mcb.20.24.9138-9148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K, Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR, Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby SD, Sawyer CS, Vickery T, Sander S, Robinson J, Winckler W, Baldwin J, Chirieac LR, Dutt A, Fennell T, Hanna M, Johnson BE, Onofrio RC, Thomas RK, Tonon G, Weir BA, Zhao X, Ziaugra L, Zody MC, Giordano T, Orringer MB, Roth JA, Spitz MR, Wistuba II, Ozenberger B, Good PJ, Chang AC, Beer DG, Watson MA, Ladanyi M, Broderick S, Yoshizawa A, Travis WD, Pao W, Province MA, Weinstock GM, Varmus HE, Gabriel SB, Lander ES, Gibbs RA, Meyerson M, Wilson RK. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396:177–180. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, Kaushansky K, Roberts JM. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27-deficient mice. Cell. 1996;85:733–744. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- Fujita N, Sato S, Tsuruo T. Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2003;278:49254–49260. doi: 10.1074/jbc.M306614200. [DOI] [PubMed] [Google Scholar]
- Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, Jakel H, Kullmann M, Kriwacki RW, Hengst L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128:269–280. doi: 10.1016/j.cell.2006.11.047. [DOI] [PubMed] [Google Scholar]
- Inui N, Kitagawa K, Miwa S, Hattori T, Chida K, Nakamura H, Kitagawa M. High expression of Cks1 in human non-small cell lung carcinomas. Biochem Biophys Res Commun. 2003;303:978–984. doi: 10.1016/s0006-291x(03)00469-8. [DOI] [PubMed] [Google Scholar]
- Kamura T, Hara T, Matsumoto M, Ishida N, Okumura F, Hatakeyama S, Yoshida M, Nakayama K, Nakayama KI. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol. 2004;6:1229–1235. doi: 10.1038/ncb1194. [DOI] [PubMed] [Google Scholar]
- Kawamata N, Morosetti R, Miller CW, Park D, Spirin KS, Nakamaki T, Takeuchi S, Hatta Y, Simpson J, Wilcyznski S. Molecular analysis of the cyclin-dependent kinase inhibitor gene p27/Kip1 in human malignancies. Cancer Res. 1995;55:2266–2269. [PubMed] [Google Scholar]
- Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1) Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- Kossatz U, Vervoorts J, Nickeleit I, Sundberg HA, Arthur JS, Manns MP, Malek NP. C-terminal phosphorylation controls the stability and function of p27kip1. EMBO J. 2006;25:5159–5170. doi: 10.1038/sj.emboj.7601388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotake Y, Nakayama K, Ishida N, Nakayama KI. Role of serine 10 phosphorylation in p27 stabilization revealed by analysis of p27 knock-in mice harboring a serine 10 mutation. J Biol Chem. 2005;280:1095–1102. doi: 10.1074/jbc.M406117200. [DOI] [PubMed] [Google Scholar]
- Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, Franssen E, Slingerland JM. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- Loda M, Cukor B, Tam SW, Lavin P, Fiorentino M, Draetta GF, Jessup JM, Pagano M. Increased proteasome-dependent degradation of the cyclin- dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nature Medicine. 1997;3:231–234. doi: 10.1038/nm0297-231. [DOI] [PubMed] [Google Scholar]
- Malkinson AM. Primary lung tumors in mice as an aid for understanding, preventing, and treating human adenocarcinoma of the lung. Lung Cancer. 2001;32:265–279. doi: 10.1016/s0169-5002(00)00232-4. [DOI] [PubMed] [Google Scholar]
- Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–1334. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- McAllister SS, Becker-Hapak M, Pintucci G, Pagano M, Dowdy SF. Novel p27(kip1) C-terminal scatter domain mediates Rac-dependent cell migration independent of cell cycle arrest functions. Molecular & Cellular Biology. 2003;23:216–228. doi: 10.1128/MCB.23.1.216-228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–787. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- Motti ML, Califano D, Troncone G, De MC, Migliaccio I, Palmieri E, Pezzullo L, Palombini L, Fusco A, Viglietto G. Complex regulation of the cyclin-dependent kinase inhibitor p27kip1 in thyroid cancer cells by the PI3K/AKT pathway: regulation of p27kip1 expression and localization. Am J Pathol. 2005;166:737–749. doi: 10.1016/S0002-9440(10)62295-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motti ML, De MC, Califano D, Fusco A, Viglietto G. Akt-dependent T198 phosphorylation of cyclin-dependent kinase inhibitor p27kip1 in breast cancer. Cell Cycle. 2004;3:1074–1080. [PubMed] [Google Scholar]
- Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh D. Mice lacking p27 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- Philipp J, Vo K, Gurley KE, Seidel K, Kemp CJ. Tumor suppression by p27/Kip1 and p21/Cip1 during chemically induced skin carcinogenesis. Oncogene. 1999;18:4689–4698. doi: 10.1038/sj.onc.1202840. [DOI] [PubMed] [Google Scholar]
- Philipp-Staheli J, Kim KH, Liggitt D, Gurley KE, Longton G, Kemp CJ. Distinct roles for p53, p27Kip1, and p21Cip1 during tumor development. Oncogene. 2004;23:905–913. doi: 10.1038/sj.onc.1207220. [DOI] [PubMed] [Google Scholar]
- Philipp-Staheli J, Kim KH, Payne SR, Gurley KE, Liggitt D, Longton G, Kemp CJ. Pathway-specific tumor suppression. Reduction of p27 accelerates gastrointestinal tumorigenesis in Apc mutant mice, but not in Smad3 mutant mice. Cancer Cell. 2002;1:355–368. doi: 10.1016/s1535-6108(02)00054-5. [DOI] [PubMed] [Google Scholar]
- Philipp-Staheli J, Payne SR, Kemp CJ. p27(Kip1): regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp Cell Res. 2001;264:148–168. doi: 10.1006/excr.2000.5143. [DOI] [PubMed] [Google Scholar]
- Ponce-Castaneda MV, Lee MH, Latres E, Polyak K, Lacombe L, Montgomery K, Mathew S, Krauter K, Sheinfeld J, Massague J, et al. p27Kip1: chromosomal mapping to 12p12-12p13.1 and absence of mutations in human tumors. Cancer Res. 1995;55:1211–1214. [PubMed] [Google Scholar]
- Sa G, Stacey DW. P27 expression is regulated by separate signaling pathways, downstream of Ras, in each cell cycle phase. Exp Cell Res. 2004;300:427–439. doi: 10.1016/j.yexcr.2004.07.032. [DOI] [PubMed] [Google Scholar]
- Schiappacassi M, Lovat F, Canzonieri V, Belletti B, Berton S, Di SD, Vecchione A, Colombatti A, Baldassarre G. p27Kip1 expression inhibits glioblastoma growth, invasion, and tumor-induced neoangiogenesis. Mol Cancer Ther. 2008;7:1164–1175. doi: 10.1158/1535-7163.MCT-07-2154. [DOI] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with 'mini-extracts', prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer DR, Viale A, Ishiwata R, Leversha M, Olgac S, Manova K, Satagopan J, Scher H, Koff A. Evidence for a p27 tumor suppressive function independent of its role regulating cell proliferation in the prostate. Proc Natl Acad Sci U S A. 2005;102:210–215. doi: 10.1073/pnas.0407362102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes & Dev. 1997;11:1464–1478. doi: 10.1101/gad.11.11.1464. [DOI] [PubMed] [Google Scholar]
- Shin I, Rotty J, Wu FY, Arteaga CL. Phosphorylation of p27Kip1 at Thr-157 interferes with its association with importin alpha during G1 and prevents nuclear re-entry. J Biol Chem. 2005;280:6055–6063. doi: 10.1074/jbc.M412367200. [DOI] [PubMed] [Google Scholar]
- Singhal S, Vachani A, ntin-Ozerkis D, Kaiser LR, Albelda SM. Prognostic implications of cell cycle, apoptosis, and angiogenesis biomarkers in non-small cell lung cancer: a review. Clin Cancer Res. 2005;11:3974–3986. doi: 10.1158/1078-0432.CCR-04-2661. [DOI] [PubMed] [Google Scholar]
- Susaki E, Nakayama KI. Multiple mechanisms for p27(Kip1) translocation and degradation. Cell Cycle. 2007;6:3015–3020. doi: 10.4161/cc.6.24.5087. [DOI] [PubMed] [Google Scholar]
- Tomoda K, Kubota Y, Kato J. Degradation of the cyclin-dependent-kinase inhibitor p27(Kip1) is instigated by Jab1. Nature. 1999;398:160–165. doi: 10.1038/18230. [DOI] [PubMed] [Google Scholar]
- Tong W, Kiyokawa H, Soos TJ, Park MS, Soares VC, Manova K, Pollard JW, Koff A. The absence of p27Kip1, an inhibitor of G1 cyclin-dependent kinases, uncouples differentiation and growth arrest during the granulosa->luteal transition. Cell Growth & Differentiation. 1998;9:787–794. [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- Viglietto G, Motti ML, Bruni P, Melillo RM, D'Alessio A, Califano D, Vinci F, Chiappetta G, Tsichlis P, Bellacosa A, Fusco A, Santoro M. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002;8:1136–1144. doi: 10.1038/nm762. [DOI] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M, Candrian U, Maronpot RR, Stoner GD, Anderson MW. Activation of the Ki-ras protooncogene in spontaneously occurring and chemically induced lung tumors of the strain A mouse. Proc Natl Acad Sci USA. 1989;86:3070–3074. doi: 10.1073/pnas.86.9.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Qin L, Liu D, Wu RC, Mussi P, Zhou S, Songyang Z, Xu J. Genetic screening reveals an essential role of p27kip1 in restriction of breast cancer progression. Cancer Res. 2007;67:8032–8042. doi: 10.1158/0008-5472.CAN-07-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F, Cunningham JJ, Sherr CJ, Jogal S, Smeyne RJ, Roussel MF. Postnatal neuronal proliferation in mice lacking Ink4d and Kip1 inhibitors of cyclin-dependent kinases. Proc Natl Acad Sci USA. 1999;96:13462–13467. doi: 10.1073/pnas.96.23.13462. [DOI] [PMC free article] [PubMed] [Google Scholar]