

Abstract
The homeostatic plasticity hypothesis suggests that neuronal activity scales synaptic strength. This study analyzed effects of activity deprivation on GABAergic synapses in cultured hippocampal neurons using patch clamp electrophysiology to record mIPSCs and immunocytochemistry to visualize presynaptic GAD-65 and the γ2 subunit of the GABAA receptor. When neural activity was blocked for 48 h with tetrodotoxin (TTX, 1 μM), the amplitude of mIPSCs was reduced, corresponding with diminished sizes of GAD-65 puncta and γ2 clusters. Treatment with the NMDA receptor antagonist APV (50 μM) or the AMPA receptor antagonist DNQX (20 μM) mimicked these effects, and co-application of brain-derived neurotrophic factor (BDNF, 100 ng/mL) overcame them. Moreover, when neurons were treated with BDNF alone for 48 h, these effects were reversed via the TrkB receptor. Overall, these results suggest that activity-dependent scaling of inhibitory synaptic strength can be modulated by BDNF/TrkB-mediated signaling.
Keywords: Inhibitory, Homeostatic plasticity, mIPSC, TrkB, NMDA receptor, AMPA receptor
Introduction
The homeostatic plasticity hypothesis predicts that neurons stabilize their firing rates in response to long-term changes in neuronal activity by scaling synaptic strength (Turrigiano, 1999). Turrigiano et al. (1998) first demonstrated this form of homeostatic plasticity at excitatory synapses by showing that their strength was increased by the deprivation of neuronal activity and reduced by the enhancement of neuronal activity, as measured by fluctuations in the amplitude of miniature excitatory postsynaptic currents (mEPSCs). This activity-dependent scaling of excitatory synaptic strength has since been shown to be dependent on postsynaptic depolarization (Leslie et al., 2001) and brain-derived neurotrophic factor (BDNF) (Rutherford et al., 1998), a protein whose expression is induced by neuronal activity (Isackson et al., 1991; Lindholm et al., 1994; Berninger et al., 1995) and that has been implicated in multiple forms of synaptic plasticity (for a review, Poo, 2001).
Neuronal activity can also regulate the strength of inhibitory synapses (Kilman et al., 2002), although the signals that mediate this process are unknown. Similar to excitatory synapses, BDNF is an attractive candidate to scale the strength of inhibitory synapses in response to long-term alterations in neuronal activity. Previous evidence has shown that BDNF modulates the level of functional inhibition in an activity-dependent manner by regulating the number of GABAergic interneurons (Rutherford et al., 1997). Neuronal activity also modulates inhibitory synaptogenesis (Seil and Drake-Baumann, 1994; Marty et al., 2000), and this synapse formation is regulated by BDNF (Seil and Drake-Baumann, 2000). However, the long-term effects of BDNF on the strength of inhibitory synapses are unclear.
This study further explored the mechanisms of the activity-dependent scaling of inhibitory synaptic strength and whether this scaling can be modulated by BDNF. Experiments were performed in cultured hippocampal neurons, which provided the advantages of visibility of and accessibility to individual synapses. Presynaptic and postsynaptic elements were visualized using immunocytochemistry, while synaptic function was measured using whole-cell patch clamp electrophysiology to record miniature inhibitory postsynaptic currents (mIPSCs). We found that 48 h of activity deprivation with tetrodotoxin (TTX) reduced the amplitude of mIPSCs, corresponding with decreased sizes of presynaptic and postsynaptic GABAergic markers. These effects could be mimicked by antagonists of NMDA and AMPA receptors. Moreover, these effects were rescued by co-application of BDNF and reversed by treatment with BDNF alone, suggesting that BDNF scales the strength of inhibitory synapses in an activity-dependent manner.
Results
Activity deprivation reduced the strength of GABAergic synapses
To establish the effects of activity deprivation on GABAergic synaptic transmission in these cultured hippocampal neurons, tetrodotoxin (TTX, 1 μM), a Na+ channel antagonist that blocks action potentials, was applied in the medium of cultured hippocampal neurons from 13 to 15 DIV, a time at which GABAergic synapses have previously been characterized as relatively mature (Swanwick et al., in press). The function of GABAergic synapses was analyzed by whole-cell patch clamp electrophysiology to record miniature inhibitory postsynaptic currents (mIPSCs). In a neuron deprived of activity for 48 h, the strength of GABAergic synaptic transmission was diminished as compared to a control neuron cultured at the same time but not subjected to activity deprivation (Fig. 1). The amplitude of the averaged mIPSC trace recorded from activity-deprived neuron was smaller than that recorded from control neuron (Fig. 1A). The peak of the mIPSC amplitude distribution histogram recorded from an activity-deprived neuron was shifted leftward, towards smaller values compared to the histogram from a control neuron (Fig. 1B). When a group of activity-deprived neurons were compared to control neurons from parallel cultures, the average median amplitude of mIPSCs recorded from activity-deprived neurons was approximately 25% lower than that from control neurons (Table 1).
Fig. 1.

Deprivation of neuronal activity with TTX altered mIPSC kinetics. (A) Average traces of mIPSCs from representative TTX-treated and untreated neurons, (B) distribution histograms of mIPSC amplitude from representative TTX-treated and untreated neurons, (C) 1-min traces from representative TTX-treated and untreated neurons, (D) cumulative probability plots of mIPSC frequency from representative TTX-treated and untreated neurons. Activity blockade reduced mIPSC amplitude but did not affect mIPSC frequency.
Table 1.
Measurements of mIPSC amplitude
| mIPSC amplitude (pA) | ||
|---|---|---|
| TTX | 43.8 ± 1.6 (n = 6) | |
| Control | 58.9 ± 3.6 (n = 7) | *P < 0.005 |
| APV | 49.9 ± 2.7 (n = 14) | |
| Control | 60.1 ± 3.1 (n = 13) | *P < 0.05 |
| DNQX | 47.7 ± 2.7 (n = 14) | |
| Control | 60.1 ± 3.1 (n = 13) | *P < 0.05 |
| TTX + BDNF | 45.5 ± 3.6 (n = 5) | |
| Control | 45.2 ± 3.8 (n = 6) | P = 0.96 |
| BDNF | 62.6 ± 3.7 (n = 9) | |
| Control | 50.1 ± 1.8 (n = 9) | *P < 0.01 |
| BDNF + TrkB-Fc | 46.2 ± 4.3 (n = 5) | |
| Control | 47.7 ± 2.8 (n = 5) | P = 0.8 |
Due to variability between cultures, all treatments performed with parallel controls (n = neurons).
Activity deprivation did not affect the frequency of mIPSCs. One-minute traces from a control and an activity-deprived neuron showed a similar number of events, despite smaller amplitudes of activity-deprived neurons (Fig. 1C). Cumulative probability plots from an activity-deprived neuron and control demonstrated that approximately the same fraction of events from both conditions had similar inter-event intervals (Fig. 1D). These findings were confirmed with groups of activity-deprived and control neurons in which mean mIPSC frequency was similar (0.4 ± 0.1 Hz in 7 activity-deprived neurons and 0.2 ± 0.05 Hz in 6 control neurons, P = 0.2).
To visualize corresponding synaptic changes that occurred due to activity deprivation, double label immunocytochemistry was performed to analyze synapses between GABAergic interneurons and pyramidal neurons using the 65 kDa isoform of glutamic acid decarboxylase (GAD-65) as a presynaptic marker and the γ2 subunit of the GABAA receptor as a postsynaptic marker (Fig. 2). Similar to the descriptions of GAD-65 immunoreactivity in these cultures provided in detail in the past (Swanwick et al., 2004), large discrete GAD-65 puncta were distributed widely throughout the neuronal processes of control pyramidal neurons (Figs. 2A, C). In an activity-deprived neuron, GAD-65 puncta were also widely distributed throughout neuronal processes, but each puncta appeared smaller than in control neuron (Figs. 2B, D). The size and density of GAD-65 puncta were measured in activity-deprived neurons and in control neurons from parallel cultures. The diameter of GAD-65 puncta in activity-deprived neurons was smaller than that in control neurons (Table 2). However, the number of GAD-65 puncta per 10 μm2 was similar to that in control neurons (Table 3).
Fig. 2.

Deprivation of neuronal activity with TTX reduced the size of presynaptic and postsynaptic GABAergic markers. (A) GAD-65 puncta in untreated neurons, (B) GAD-65 puncta in TTX-treated neurons, (C) magnification of the inset from panel A, (D) magnification of the inset from panel B, (E) γ2 clusters in untreated neurons, (F) γ2 clusters in TTX-treated neurons, (G) magnification of the inset from panel E, (H) magnification of the inset from panel F, (I) merged images of GAD-5 puncta (red) and γ2 clusters (green) from untreated neurons, (J) merged images of GAD-65 puncta (red) and γ2 clusters (green) from TTX-treated neurons. Activity blockade reduced the sizes of GAD-65 puncta and γ2 clusters but did not affect their densities or synaptic localization. Images captured at 60×. Scale bar = 20 μm.
Table 2.
Sizes of GABAergic presynaptic and postsynaptic markers
| GAD-65 | γ2 | |||
|---|---|---|---|---|
| TTX | 4.8 ± 0.3 (n = 30) | 2.0 ± 0.1 (n = 30) | ||
| Control | 7.6 ± 0.5 (n = 31) | *P < 0.0001 | 2.8 ± 0.2 (n = 31) | *P < 0.0005 |
| APV | 2.5 ± 0.2 (n = 46) | 1.8 ± 0.1 (n = 15) | ||
| Control | 5.2 ± 0.2 (n = 44) | *P < 0.001 | 2.4 ± 0.1 (n = 15) | *P < 0.001 |
| DNQX | 4.7 ± 0.1 (n = 45) | 1.8 ± 0.1 (n = 15) | ||
| Control | 5.2 ± 0.2 (n = 44) | *P < 0.05 | 2.4 ± 0.1 (n = 15) | *P < 0.001 |
| TTX + BDNF | 6.0 ± 0.9 (n = 20) | 2.1 ± 0.3 (n = 20) | ||
| Control | 4.9 ± 0.6 (n = 20) | P = 0.3 | 2.2 ± 0.2 (n = 20) | P = 0.8 |
| BDNF | 9.2 ± 0.6 (n = 10) | 1.4 ± 0.07 (n = 20) | ||
| Control | 6.9 ± 0.7 (n = 10) | *P < 0.05 | 1.2 ± 0.04 (n = 30) | *P < 0.05 |
| BDNF + TrkB-Fc | 6.3 ± 0.4 (n = 20) | 1.4 ± 0.07 (n = 13) | ||
| Control | 6.4 ± 0.5 (n = 20) | P = 0.8 | 1.3 ± 0.05 (n = 20) | P = 0.2 |
Due to variability between cultures, all treatments performed with parallel controls. Units = μm diameter (n = neurons).
Table 3.
Densities of GABAergic presynaptic and postsynaptic markers
| GAD-65 | γ2 | |||
|---|---|---|---|---|
| TTX | 2.4 ± 0.4 (n = 16) | 2.2 ± 0.3 (n = 16) | ||
| Control | 2.2 ± 0.3 (n = 15) | P = 0.7 | 2.7 ± 0.4 (n = 15) | P = 0.3 |
| APV | 1.3 ± 0.1 (n = 25) | 2.5 ± 0.2 (n = 15) | ||
| Control | 0.9 ± 0.1 (n = 23) | *P < 0.05 | 2.5 ± 0.3 (n = 15) | P = 0.5 |
| DNQX | 1.4 ± 0.1 (n = 24) | 2.9 ± 0.2 (n = 15) | ||
| Control | 0.9 ± 0.1 (n = 23) | *P < 0.05 | 2.5 ± 0.3 (n = 15) | P = 0.5 |
| BDNF | 1.9 ± 0.2 (n = 18) | 2.8 ± 0.3 (n = 10) | ||
| Control | 2.2 ± 0.2 (n = 20) | P = 0.3 | 3.2 ± 0.4 (n = 10) | P = 0.4 |
Due to variability between cultures, all treatments performed with parallel controls. Units = number per 10 μm2 (n = neurons).
The deprivation of neuronal activity had a similar effect on another presynaptic marker of GABAergic synapses, vesicular inhibitory amino acid transporter (VIAAT). The diameter of VIAAT puncta in activity-deprived neurons (3.3 ± 0.3 μm, n = 12) was smaller than in control neurons (5.3 ± 0.3 μm, n = 12, P < 0.0001). The number of VIAAT puncta per 10 μm2 in activity-deprived neurons was 1.4 ± 0.4 (n = 12), relatively equivalent to 2.2 ± 0.3 puncta per 10 μm2 in control neurons (n = 12, P = 0.2).
GABAA receptors containing the γ2 subunit appeared as clusters that were abundantly expressed throughout the neuronal process of almost all pyramidal neurons in both control neurons (Fig. 2E) and activity-deprived neurons (Fig. 2F). However, in activity-deprived neurons, γ2 clusters were smaller, an effect that was more easily visible at higher magnifications (Figs. 2G, H). In a group of activity-deprived neurons, the diameter of γ2 clusters was shorter than that in parallel control neurons (Table 2). In contrast, the number of γ2 clusters per 10 μm2 in activity-deprived neurons was approximately equivalent to that in control neurons (Table 3).
Activity deprivation did not affect the synaptic localization of γ2 subunit-containing GABAA receptors. In both control neurons (Fig. 2I) and activity-deprived neurons (Fig. 2J), many γ2 clusters (green) were colocalized with GAD-65 puncta (red), but numerous γ2 clusters and GAD-65 puncta were also unmatched. When quantified, the percentage of γ2 clusters colocalized with GAD-65 puncta was 42.6 ± 2.7% in activity-deprived neurons (n = 29) and 39.5 ± 2.1% in control neurons (n = 31, P = 0.4). Previous characterization by this laboratory revealed that approximately 60% of GAD-65 puncta were colocalized with γ2 clusters (Swanwick et al., in press), and these numbers were unchanged in both control and activity-deprived neurons in this study.
Deprivation of glutamate receptor activity mimicked the effects of TTX
We next determined whether reduced activity of glutamate receptors contributes to the reduction in GABAergic synapse strength observed during activity deprivation, as demonstrated by both reduced mIPSC amplitude and diminished sizes of GAD-65 puncta and γ2 clusters. Therefore, GABAergic synapses were analyzed after 48 h of bath application of either the NMDA receptor antagonist APV (50 μM) or the AMPA receptor antagonist DNQX (20 μM) in cultured hippocampal neurons from 13 to 15 DIV (Fig. 3).
Fig. 3.

Deprivation of NMDA or AMPA receptor activity mimicked the effects of TTX on GABAergic synapses. (A) Average traces of mIPSCs from representative APV-treated and untreated neurons, (B) average traces from representative DNQX-treated and untreated neurons, (C) distribution histograms of mIPSC amplitude from representative APV-treated, DNQX-treated and untreated neurons, (D) GAD-65 puncta in APV-treated, DNQX-treated and untreated neurons, (E) γ2 clusters in APV-treated, DNQX-treated and untreated neurons. Blockade of NMDA or AMPA receptor activity reduced the sizes of GAD-65 puncta and γ2 clusters. Deprivation of NMDA or AMPA receptor activity also increased the density of GAD-65 puncta but did not affect the density of γ2 clusters. Same control neurons used in parallel with both APV- and DNQX-treated cultures. Images captured at 60×. Scale bar = 10 μm.
The amplitude of mIPSCs was suppressed after deprivation of either NMDA receptor or AMPA receptor activity. The amplitude of an averaged mIPSC trace recorded from neurons deprived of either NMDA or AMPA receptor activity was smaller than that from a control neuron (Figs. 3A, B). The peaks of amplitude distribution histograms of mIPSCs recorded from an APV- or DNQX-treated neuron were shifted leftward towards smaller values compared to that from a control neuron (Fig. 3C). When repeated in a group of parallel cultures, the average median amplitude of mIPSCs recorded from control neurons was larger than that recorded from APV-treated neurons and DNQX-treated neurons (Table 1).
Deprivation of NMDA or AMPA receptor activity also resulted in diminished sizes of both GAD-65 puncta (Fig. 3D) and γ2 clusters (Fig. 3E). The diameters of GAD-65 puncta and γ2 clusters in control neurons were longer than in APV-treated neurons and in DNQX-treated neurons (Table 2). In addition, the number of GAD-65 puncta per 10 μm2 was increased after the blockade of NMDA or AMPA receptor activity, although the number of γ2 clusters per 10 μm2 was unaffected (Table 3).
BDNF prevented the effects of activity deprivation
To test whether BDNF regulates the effects of neuronal activity on the strength of GABAergic synapses, BDNF (100 ng/mL) was bath-applied for 48 h to cultured hippocampal neurons from 13 to 15 DIV in which activity was simultaneously blocked with TTX (1 μM) (Fig. 4). BDNF prevented the reduction in mIPSC amplitude observed after activity deprivation. An averaged trace of mIPSCs from an activity-deprived neuron treated with BDNF was approximately equal to that from a control neuron (Fig. 4A). An amplitude histogram recorded from an activity-deprived neuron after BDNF treatment also showed a similar distribution to that recorded from a control neuron, with equivalent peaks (Fig. 4B). Moreover, the average median amplitude of mIPSCs recorded from a group of activity-deprived neurons treated with BDNF was similar to that recorded from parallel control neurons (Table 1).
Fig. 4.

BDNF prevented the effects of activity deprivation on GABAergic synapses. (A) Average traces of mIPSCs from representative TTX + BDNF-treated and untreated neurons, (B) distribution histograms of mIPSC amplitude from representative TTX + BDNF-treated and untreated neurons, (C) GAD-65 puncta from TTX + BDNF-treated neurons and untreated neurons, (D) γ2 clusters from TTX + BDNF-treated neurons and untreated neurons. BDNF prevented the reduction in mIPSC amplitude and sizes of GAD-65 puncta and γ2 clusters observed in activity-deprived neurons. Images captured at 60×. Scale bar = 10 μm.
BDNF also prevented the reductions in sizes of GAD-65 puncta and γ2 clusters observed in activity-deprived neurons. In activity-deprived neurons treated with BDNF, GAD-65 puncta (Fig. 4C) and γ2 clusters (Fig. 4D) were similar in size to control neurons. The diameters of GAD-65 puncta and γ2 clusters in control neurons were approximately equal to those in activity-deprived neurons treated with BDNF (Table 2).
BDNF alone enhanced the strength of GABAergic synapses
To evaluate the effects of BDNF on neurons with intact activity, BDNF (100 ng/mL) was bath-applied for 48 h to cultured hippocampal neurons from 13 to 15 DIV (Fig. 5). BDNF alone enhanced mIPSC amplitude. An averaged trace of mIPSCs recorded from a BDNF-treated neuron was larger than that recorded from a control neuron (Fig. 5A). A distribution histogram of amplitudes from a neuron after BDNF treatment showed that the peak amplitude had shifted rightward towards larger values compared to that from a control neuron (Fig. 5B). In confirmation, the average median amplitude of mIPSCs recorded from BDNF-treated neurons was 20% more than that recorded from control neurons in parallel cultures (Table 1).
Fig. 5.
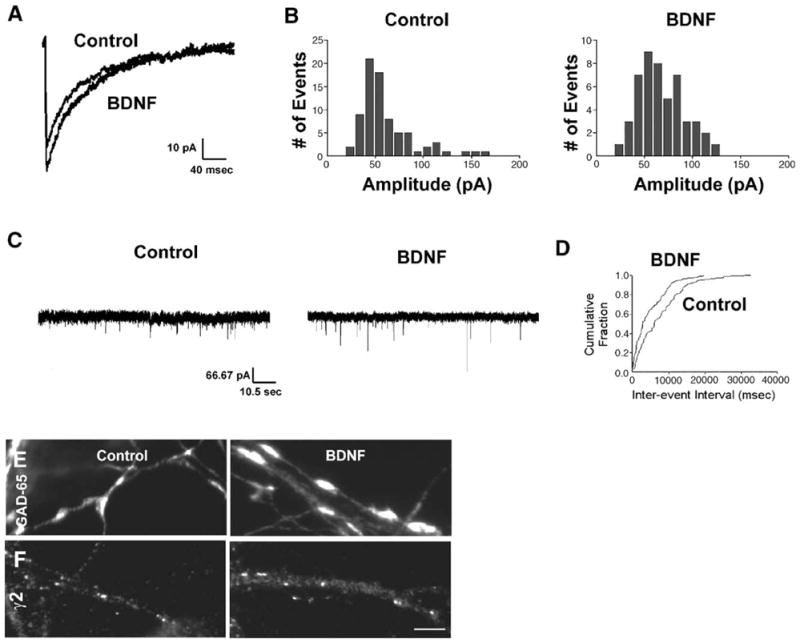
BDNF enhanced GABAergic synapse strength. (A) Average traces of mIPSCs from representative BDNF-treated and untreated neurons, (B) distribution histograms of mIPSC amplitude from representative BDNF-treated and untreated neurons, (C) 1-min traces of mIPSCs from untreated and BDNF-treated neurons, (D) cumulative probability plots of mIPSCs from untreated and BDNF-treated neurons, (E) GAD-65 puncta from BDNF-treated and untreated neurons, (F) γ2 clusters from BDNF-treated and untreated neurons. In neurons with intact activity, BDNF enhanced mIPSC amplitude but did not affect mIPSC frequency. BDNF also increased the sizes of GAD-65 puncta and γ2 clusters but did not affect their densities. Images captured at 60×. Scale bar = 10 μm.
Exogenous BDNF had no significant effect on mIPSC frequency in neurons with preserved activity. One-minute traces from control and BDNF-treated neurons show approximately the same number of events, despite many of the currents from BDNF-treated neurons displaying larger amplitudes (Fig. 5C). Cumulative probability plots show that approximately the same fraction of events had equivalent inter-event intervals in both control neurons and BDNF-treated neurons (Fig. 5D). The mIPSC frequency was 0.1 ± 0.05 Hz in control neurons (n = 9) and 0.2 ± 0.1 in BDNF-treated neurons (n = 9, P = 0.3).
BDNF also enhanced the sizes of GAD-65 puncta and γ2 clusters in neurons with intact activity. Neurons treated with BDNF contained GAD-65 puncta (Fig. 5D) and γ2 clusters (Fig. 5E) that were still widely distributed throughout neuronal processes but were larger in size. The diameters of GAD-65 puncta and γ2 clusters in control neurons were longer than in BDNF-treated neurons (Table 2). In corroboration with the unchanged mIPSC frequency, BDNF had no effect on the numbers of GAD-65 puncta or γ2 clusters per 10 μm2 (Table 3).
To test whether the TrkB receptor mediated these effects of BDNF on GABAergic synapses, TrkB-Fc (2 μg/ml), a receptor body that sequesters ligand for the TrkB receptor, was bath-applied with BDNF to neurons from 13 to 15 DIV (Fig. 6). TrkB-Fc prevented the increase in mIPSC amplitude observed in neurons treated with BDNF alone. An averaged trace of mIPSCs recorded from a BDNF and TrkB-Fc-treated neuron was similar in size to those recorded from control neurons (Fig. 6A). An amplitude distribution histogram of mIPSCs recorded from a BDNF and TrkB-Fc-treated neuron also resembled that recorded from a control neuron (Fig. 6B). In addition, the average median amplitude of mIPSCs recorded from a group of BDNF and TrkB-Fc-treated neurons was approximately equal to that recorded from parallel control neurons (Table 1).
Fig. 6.

The BDNF-induced enhancement of GABAergic synapse strength was mediated by TrkB. (A) Average traces of mIPSCs from representative BDNF + TrkB-Fc-treated and untreated neurons, (B) distribution histograms of mIPSC amplitude from representative BDNF + TrkB-Fc-treated and untreated neurons, (C) GAD-65 puncta from BDNF + TrkB-Fc-treated neurons and untreated neurons, (D) γ2 clusters from BDNF + TrkB-Fc-treated neurons and untreated neurons. BDNF prevented the reduction in mIPSC amplitude and sizes of GAD-65 puncta and γ2 clusters observed in activity-deprived neurons. Images captured at 60×. Scale bar = 10 μm.
TrkB-Fc also prevented the enlargements in sizes of GAD-65 puncta and γ2 clusters observed in neurons treated with BDNF alone. GAD-65 puncta (Fig. 6C) and γ2 (Fig. 6D) clusters in BDNF and TrkB-Fc-treated neurons were similar in size to those in control neurons. The diameters of GAD-65 puncta and γ2 clusters in BDNF + TrkB-Fc-treated neurons were equivalent to those in control neurons (Table 2).
Other effects of activity deprivation on mIPSC kinetics
It is important to note that activity deprivation also affected mIPSC 10–90% rise-time and the rate of mIPSC decay. TTX shortened 10–90% rise-time approximately 33%, from 1.8 ± 0.1 ms in control neurons (n = 7) to 1.2 ± 0.06 ms in activity-deprived neurons (n = 6, P < 0.005). Furthermore, the fast component of decay, τ1, quickened from 59.0 ± 4.7 ms in control neurons (n = 5) to 26.0 ± 2.7 ms in activity-deprived neurons (n = 5, P < 0.0005), and the slow component of decay, τ2, shortened from 143.1 ± 12.8 ms in control neurons (n = 5) to 86.2 ± 5.5 ms in activity-deprived neurons (n = 5, P < 0.005). However, corresponding effects on mIPSC 10–90% rise-time and decay were not observed after the deprivation of glutamate receptors or the addition of BDNF, suggesting that these effects may be regulated by other biological mechanisms.
Discussion
According to the concept of homeostatic plasticity, neurons are regulated to maintain a state of constancy so that fluctuations in neural circuits are prevented from inducing hyper- or hypoactivity. This regulation may occur at multiple levels, including neuronal excitability, synapse formation and synaptic strength (Turrigiano, 1999). The present study shows that neuronal activity scales the strength of GABAergic synapses in cultured hippocampal neurons. The primary conclusions of this study are that (1) neuronal activity scales the strength of GABAergic synapses through coordinated presynaptic and postsynaptic actions, (2) NMDA and AMPA receptors contribute to this activity-dependent scaling of GABAergic synapse strength and (3) BDNF modulates this activity-dependent scaling of GABAergic synapse strength via the TrkB receptor.
Activity-dependent scaling of GABAergic synapse strength
The deprivation of neuronal activity reduced mIPSC amplitude. Alterations in mIPSC amplitude are usually indicative of changes in postsynaptic receptor number, but, in addition to smaller clusters of GABAA receptors, the diminished mIPSC amplitude corresponded with reduced sizes of presynaptic GAD-65 puncta. This suggests that activity blockade may induce a coordinated set of presynaptic and postsynaptic changes at inhibitory synapses. In support, previous evidence demonstrated that variations in both the GABA content of vesicles (Frerking et al., 1995) and the number of postsynaptic GABAA receptors (Nusser et al., 1997, 1998) determine mIPSC amplitude. Moreover, a reduction in both the staining intensity of GAD-65 and synaptic GABAA receptors was previously observed in cultured cortical neurons after activity blockade (Kilman et al., 2002).
Long-term changes in neuronal activity may modulate inter-neurons at multiple levels, including interneuron differentiation and survival. The number of GABAergic interneurons was reduced in the somatosensory and visual cortices after sensory deprivation (Hendry and Jones, 1986, 1988; Benevento et al., 1995). Similarly, the number of GABAergic interneurons was decreased in cortical and hippocampal cultures after activity blockade with tetrodotoxin (Marty et al., 1996; Rutherford et al., 1997).
The present study showed that neuronal activity also affects the strength of synapses projecting from interneurons to pyramidal neurons. This occurred by regulating the size of presynaptic GABAergic components, including GAD-65 and VIAAT. Because GAD-65 is the enzyme responsible for the synthesis of GABA, this suggests that activity blockade reduced the amount of presynaptic GABA. In addition, the diminished size of VIAAT puncta implies that less GABA is transported into presynaptic vesicles after the deprivation of activity. Together, these effects strongly suggest that neuronal activity regulates the strength of GABAergic synapses by modulating the amount of GABA released from the presynaptic terminal. In support, previous studies have reported that activity deprivation regulates neurotransmitter release at excitatory synapses by modulating the sizes of active zones and the number of docked vesicles per active zone (Turrigiano and Nelson, 2004). No corresponding alteration in mIPSC frequency was observed in the present study; however, it is important to note that, although RMS noise was low (approximately 5 pA), changes in mIPSC frequency after activity deprivation may not have been detected to the reduction in mIPSC amplitude.
Deprivation of neuronal activity also reduced the size of clustered GABAA receptors in pyramidal neurons. One possibility is that GABAA receptor trafficking is responsive to long-term changes in neuronal activity. Rapid subcellular redistribution of GABAA receptors has been demonstrated to underlie multiple forms of GABAergic synaptic plasticity (Kittler and Moss, 2003; Goodkin et al., 2005). Activity blockade reduced mIPSC amplitude in cultured cortical neurons by reducing the accumulation of GABAA receptors at synapses (Kilman et al., 2002). In addition, alterations in neuronal activity influence the aggregation of AMPA receptors into clusters at excitatory synapses (O’Brien et al., 1998). However, the 48 h time frame suggests that protein synthesis may also be involved in mediating the reduction of GABAergic synapse strength. Alterations in the number of GABAA receptors are likely to be part of global homeostatic plasticity (Mody, 2005).
NMDA and AMPA receptor activity scales GABAergic synaptic strength
The present study demonstrated that the deprivation of NMDA or AMPA receptor activity mimicked the reduction in GABAergic synaptic strength observed after general activity blockade. Moreover, this decrease in strength was mediated by the same coordinated actions at presynaptic and postsynaptic terminals. However, this does not necessarily imply a direct link between glutamate receptor activity and GABAergic synapse strength because blocking glutamate receptor activity has numerous effects on neurons. For instance, preventing the function of NMDA and AMPA receptors also lowers the overall firing rate of neurons from approximately 1 Hz to 0.033 Hz (H.P. Goodkin, unpublished observations), and spike activity may actually be the critical component of scaling GABAergic synapse strength.
Other studies have suggested that glutamate receptor activity may cause various homeostatic alterations in GABAergic synaptic transmission besides GABAergic synapse strength. An AMPA receptor antagonist induced homeostatic plasticity in spinal cultures by slowing GABAergic events (Galante et al., 2001). Furthermore, NMDA receptor activity has also been reported to increase IPSC decay during postnatal development of the superior colliculus (Henneberger et al., 2005). Moreover, chronic treatment with an ionotropic glutamate antagonist significantly increased paired-pulse depression in cultured hippocampal neurons, although it did not affect the amplitude of evoked IPSCs or the reversal potential of IPSCs (Ivanova et al., 2003).
In addition, presynaptic actions of NMDA on interneurons have previously been demonstrated in rat cerebellar slices (Glitsch and Marty, 1999) and cultured cerebellar neurons (Fiszman et al., 2005), where chronic exposure to NMDA increased spontaneous transmitter release at GABAergic synapses by enhancing the size of GABAergic boutons along the axons of developing GABAergic interneurons. Similar presynaptic effects were reported for AMPA on interneurons in stellate cells from cerebellar slices (Bureau and Mulle, 1998).
Interestingly, whereas the size of GAD-65 puncta diminished after deprivation of NMDA or AMPA receptor activity, the number of GAD-65 puncta increased. This was a surprising result, particularly because a similar effect was not observed after blocking action potentials. It is therefore probably due to a neuronal effect of glutamate receptor activity other than firing rate. It is possible that the increased number of smaller GAD-65 puncta represents the dismantling of presynaptic terminals. However, it could also be a consequence of axonal sprouting of interneurons. Future experiments should investigate the mechanism underlying this phenomenon.
BDNF modulates the activity-dependent scaling of GABAergic synapse strength
Activity-dependent release of BDNF from pyramidal neurons has been demonstrated to scale the strength of excitatory synapses (Rutherford et al., 1998). In addition, BDNF release has been shown to modulate network homeostatic plasticity by regulating the strength of synapses between pairs of pyramidal neurons or between pyramidal neurons and interneurons (Rutherford et al., 1997; Rutherford et al., 1998; Turrigiano and Nelson, 2004). The present study demonstrates a novel role for BDNF in the activity-dependent scaling of inhibitory synapses through coordinated actions at presynaptic and postsynaptic terminals.
BDNF prevented the reduction in size of GAD-65 puncta in activity-deprived neurons. Moreover, BDNF alone enhanced the size of GAD-65 puncta in neurons with normal levels of activity. A similar result has been reported previously in hippocampal cultures prepared from P0 rat pups (McLean Bolton et al., 2000). This suggests that BDNF may strengthen presynaptic terminals by increasing the expression of GAD-65, an effect that might lead to the increased synthesis of GABA. If the synapse is not saturated, increased GABA release might then augment mIPSC amplitude. Previous evidence shows that BDNF can induce presynaptic neurotransmitter release. BDNF-coated beads triggered localized neurotransmitter secretion where they contacted developing spinal cord axons (Zhang and Poo, 2002). Presynaptic N- and P/Q-type Ca2+ channel signaling may mediate this enhanced neurotransmitter release as BDNF induced the synthesis of these channels in cultured hippocampal neurons, thereby enhancing evoked inhibitory currents and paired-pulse depression (Baldelli et al., 2002). Moreover, BDNF facilitated the probability of release of GABA but not of glutamate in hippocampal neurons (Yamada et al., 2002; Ohba et al., 2005).
BDNF did not affect mIPSC frequency, implying that the number of functional synaptic contacts projecting from interneurons to pyramidal neurons remained unchanged. In contrast, previous studies have suggested that BDNF enhances inhibitory synaptic function by increasing the frequency of GABAergic synaptic currents (McLean Bolton et al., 2000). However, those experiments were performed on hippocampal neurons cultured from P0 pups, not E18 embryos, and they were grown for 3 days before recording, unlike 14 days as in this study. Those neurons were also exposed to BDNF for 4–7 days, raising the possibility that mIPSC frequency would have increased in the present study if BDNF treatment had lasted for longer durations. Alternatively, emerging new currents may have been too small to detect above baseline noise. However, baseline noise was minimal and BDNF enhanced the size of mIPSCs, making this scenario unlikely. Another possibility is that mIPSC frequency in the present cultured hippocampal neurons was too variable to detect significant changes. However, the density of GAD-65 puncta also remained unaffected by BDNF, supporting the constant rate of mIPSC frequency.
In activity-deprived neurons, BDNF prevented the reduction in size of GABAA receptor clusters. Moreover, in neurons with preserved activity, BDNF enhanced the size of GABAA receptor clusters. In a similar experimental preparation, Elmariah et al. (2004) reported that exogenous BDNF increased the number and synaptic localization of GABAA receptor clusters without affecting their size. However, several key differences exist between our studies. Most importantly, they were examining the effects of BDNF on earlier developmental timepoints (8–10 DIV). We have previously performed extensive characterization of GABAergic synaptogenesis in cultured hippocampal neurons (Swanwick et al., in press) and found that 8 DIV is the time when the first formation of functional GABAergic synapses can be observed. These GABAergic synapses then undergo a period of enormous transformation, including increased number, size and synaptic localization of GABAA receptors, until they become stable at 14 DIV and are then relatively mature. Because early GABAergic synapses are more plastic, it is likely that BDNF treatment from 8 to 10 DIV was able to induce increases in GABAA receptor number and synaptic localization, whereas BDNF treatment from 14 to 16 DIV was not. Furthermore, whereas Elmariah and colleagues used an antibody against the β2/3 subunits as a marker for GABAA receptors, our laboratory used an antibody against the γ2 subunit. Differences may exist between the two markers in response to BDNF, especially because the γ2 subunit is thought to be targeted to synapses (Essrich et al., 1998).
An increase in the size of GABAA receptor clusters suggests that BDNF may strengthen GABAergic synaptic transmission by promoting the trafficking of GABAA receptors to synapses. Alternatively, BDNF may increase the synthesis of presynaptic GABAergic components and GABAA receptor subunits. It is important to note that BDNF has vastly different effects on GABAA receptor trafficking over shorter time courses. Acutely, BDNF instead reduces the strength of GABAergic synapses by suppressing the amplitude of inhibitory synaptic currents (Tanaka et al., 1997; Frerking et al., 1998; Brunig et al., 2001; Cheng and Yeh, 2003). Recent experiments from this laboratory also showed that this suppression is mediated by activation of the TrkB receptor leading to an increase in the internalization of GABAA receptors (C.C. Swanwick, J.L. Yeh, and J. Kapur, unpublished observations).
Although BDNF rescued the effects of activity deprivation on GABAergic synapses, a direct link between neuronal activity and BDNF was not provided here. However, previous evidence demonstrates a clear link between them. BDNF expression is induced by neuronal activity (Lindholm et al., 1994; Berninger et al., 1995), and seizure activity causes a rapid increase in BDNF mRNA levels in the hippocampus and cortex (Isackson et al., 1991). BDNF is also released in an activity-dependent manner (Wetmore et al., 1994; Goodman et al., 1996). In addition, neural activity affects TrkB-mediated signaling (Poo, 2001), in part by elevating TrkB receptors on the cell surface (Du et al., 2000). Finally, BDNF provokes spontaneous seizures and enhances seizure propagation in animal models (Binder, 2004).
In conclusion, this study demonstrated homeostatic plasticity of GABAergic synapse strength in response to neuronal activity. It is likely that activity of NMDA and AMPA receptors contributes to these activity-dependent alterations. Moreover, this plasticity can be regulated by BDNF-mediated signaling.
Experimental methods
Cell culture
Neuronal hippocampal/glial co-cultures were prepared from 18-day embryonic rats as in Goslin et al. (1998). Briefly, neurons were isolated by trypsin treatment and triturated and plated on poly-L-lysine-coated glass coverslips in minimum essential medium (MEM) with 15% horse serum at a density of 10,000–100,000 cells/35 mm2. After attachment of cells, the coverslips were transferred, and the neurons were grown over a glial cell monolayer in serum-free MEM with N2 supplements. The population of neurons in culture consists primarily of pyramidal neurons with approximately 7% of the cells being GABAergic (Benson et al., 1994). The neurons were used for immunocytochemistry or electrophysiology at 15 days after plating. Experimental neuron cultures were treated daily at 13 and 14 days after plating before being used. Due to variability in cultured neurons, all treatments were performed with parallel controls.
Immunocytochemistry
Coverslips of neurons were fixed with 4% paraformaldehyde (PFA)/4% sucrose in phosphate-buffered saline (PBS, pH 7.1) for 20 min at room temperature, permeabilized with 0.10% Triton X-100 in PBS for 15 min at room temperature, blocked with 4% normal goat serum for 15 min at room temperature and incubated with primary antibodies overnight at 4°C. Coverslips were washed with 1×PBS the following day and then incubated overnight at 4°C with a second primary antibody. On the final day, the coverslips were washed with 1×PBS and then incubated with appropriate fluorochrome-conjugated secondary antibodies for 45 min on a shaker at room temperature in darkness. Coverslips were mounted in Gel/Mount (Biomeda Corp, Foster City, CA), the edge of each coverslip was sealed with clear nail polish, and slides were stored at −20°C.
Primary antibodies included a mouse monoclonal antibody against GAD-6 clone to recognize GAD-65 (1 μg/mL, Chemicon, Temecula, CA), a rabbit antibody against the γ2 subunit of the GABAA receptor (2 μg/mL, gift, Werner Sieghart, Medical University of Vienna, Austria) and a guinea pig antibody against the vesicular inhibitory amino acid transporter (VIAAT) (1:2000, Chemicon, Temecula, CA). Secondary antibodies included Alexa 488 and Alexa 594 fluorochromes conjugated with goat anti-mouse or goat anti-rabbit IgG (4 μg/mL, Molecular Probes, Eugene, OR). All primary and secondary antibodies were diluted in 1×PBS containing 2% normal goat serum.
Image acquisition and analysis
Fluorescent images of cells were captured on a CoolSNAPcf™ CCD camera (Roper Scientific Photometrics, NJ) mounted on an Eclipse TE200 fluorescent microscope (Nikon, Japan) driven by Metamorph imaging software (Universal Imaging Corporation, Downington, PA). High resolution digital images of each fluorochrome were acquired using a 60×/1.4 NA lens. Brightness and contrast of fluorescent images were adjusted using Metamorph software so that only punctate fluorescence, but no weak diffuse background labeling, was visible.
Definition of puncta/clusters
Clusters were analyzed after a single neuron was centered in the visual field captured at 60×. For cluster density, size and colocalization, thresholds were set to detect punctate fluorescence that was two times higher than diffuse background labeling. Aggregations of ≥2 pixels were selected as clusters, which corresponded to ≥0.15 μm diameter at 60× magnification, as determined by 1 μm diameter fluorescent microspheres. Images were also visually inspected to eliminate the soma, fused puncta or obvious debris from being selected for analysis. Nonspecific labeling in controls lacking primary antibody could appear as granular clusters <7 pixels (<0.54 μm diameter), so, if average cluster size was <7 pixels, cluster number was recorded as 0. Due to fluorescent intensity of the soma, only clusters on processes were quantitated for all neurons.
Analysis of colocalization
A binary image was created from each thresholded image. Binary images were then added together to display overlapping puncta. Number of colocalized puncta or clusters was then measured. Data of number, size and colocalization of puncta or clusters were analyzed using GraphPad Prism 4.0 (San Diego, CA). All values are reported as mean ± SEM. Values compared between two experimental groups were evaluated using a two-tailed Student’s t test with a significance level of P < 0.05. n = neurons.
Photomicrograph production
Images were then saved as 8-bit TIFF files and opened in Adobe Photoshop 6.0 (San Jose, CA), where overall brightness was increased for final production.
mIPSC recording
Synaptic currents mediated by the GABAA receptor were recorded using the whole-cell patch clamp method as described in the past (Hamill et al., 1981; Mangan and Kapur, 2004). Membrane properties of these neurons at 14 days in vitro (DIV) were characterized previously (Mangan and Kapur, 2004). Neurons were studied on the stage of an inverted microscope at room temperature. Thick-walled (1.5 mm outer diameter, 0.86 mm inner diameter) borosilicate patch electrodes (World Precision Instruments, Sarasota, FL) were pulled on a P-97 Flaming-Brown horizontal puller (Sutter Instruments, CA) using a 2-stage pull to a final resistance of 2–5 MΩ.
Patch electrodes were filled with internal recording solution containing (in mM): CsCl 153.3, MgCl2 1.0, EGTA 5.0 and HEPES 10.0, with a pH of 7.40 and osmolarity of 290–300 mosM. CsCl was used to block potassium currents. MgATP (4 mM) was included in the intracellular solution before recording. The external recording medium contained (in mM): NaCl 146.0, KCl 2.5, MgCl2 3.0, CaCl2 2.0, Glucose 10.0 and HEPES 10.0, with a pH of 7.4 and osmolarity of 315–330 mosM. Internal and external solutions contained equimolar concentrations of chloride ions, and no GABAA-receptor-mediated inhibitory postsynaptic currents (IPSCs) were evident at a clamped membrane potential of 0 mV. Glutamate-receptor-mediated synaptic currents were blocked using 50 μM D(−)-2-amino-5-phosphono-valeric acid (D-APV) and 20 μM 6,7-dinitroquinoxaline-2,3-dione (DNQX) in the external solution. Action potentials were blocked using 1 μM tetrodotoxin (TTX) in the external solution. Bath application of the GABAA receptor antagonist bicuculline (5 μM) eliminated all currents observed, verifying that recorded currents were GABAergic mIPSCs.
Recordings were made at a holding potential of −60 mV. Currents were recorded with an Axopatch 200 A amplifier and low-pass-filtered at 3 kHz with an 8-pole Bessel filter prior to digitization, storage and display using the patch clamp technique (Hamill et al., 1981). Currents were recorded using Axoscope software (Axon Instruments, CA) digitized at 400 Hz. Series resistance and capacitance were compensated for each neuron. A recording was rejected if the series resistance after compensation was ≥20 MΩ. The resting membrane potential of all recorded neurons was approximately −60 mV. The input resistance of all recorded neurons ranged was approximately 150 MΩ. Neurons were recorded for 10 min.
Electrophysiological analysis
MiniAnalysis software (Synaptosoft, Decatur, GA) was used to analyze mIPSC frequency, amplitude, 10–90% rise-time and decay. Detection thresholds were set at three times the root mean square of baseline noise (typically approximately 5 pA). The mIPSC frequency is described by a mean value. The mIPSC amplitude and 10–90% rise-time are described by median values because their values were not normally distributed due to the loss of smaller currents in the level of background noise. The median mIPSC amplitude and 10–90% rise-time from each neuron were then averaged, and this mean of medians is reported for both amplitude and 10–90% rise-time. Decay was analyzed by fitting with biexponential curves and accepting the fit when R2 > 0.70. Decays were analyzed until 20 values were obtained for each neuron. Because the range of fitted mIPSC decays is also not normally distributed, the mean of median values of the decay of each neuron is also reported. When applicable, standard error of the mean (SEM) is reported. Comparisons between two experimental groups were performed using a two-tailed Student’s t test with a significance level of P < 0.05. n = neurons.
Acknowledgments
This research was supported by NIH-NINDS grants F31 NS 43831, RO1 NS 040337 and RO1 NS 44370. We thank Ms. Ashley Renick, Ms. Bahar Alawi and Dr. Sandra Medina-Ramos for preparing and maintaining the hippocampal cultures.
References
- Baldelli P, Novara M, Carabelli V, Hernandez-Guijo JM, Carbone E. BDNF up-regulates evoked GABAergic transmission in developing hippocampus by potentiating presynaptic N- and P/Q-type Ca2+ channels signaling. Eur J Neurosci. 2002;16:2297–2310. doi: 10.1046/j.1460-9568.2002.02313.x. [DOI] [PubMed] [Google Scholar]
- Benevento LA, Bakkum BW, Cohen RS. γ-Aminobutyric acid and somatostatin immunoreactivity in the visual cortex of normal and dark-reared rats. Brain Res. 1995;689:172–182. doi: 10.1016/0006-8993(95)00553-3. [DOI] [PubMed] [Google Scholar]
- Benson DL, Watkins FH, Steward O, Banker G. Characterization of GABAergic neurons in hippocampal cell cultures. J Neurocytol. 1994;23:279–295. doi: 10.1007/BF01188497. [DOI] [PubMed] [Google Scholar]
- Berninger B, Marty S, Zafra F, da Penha BM, Thoenen H, Lindholm D. GABAergic stimulation switches from enhancing to repressing BDNF expression in rat hippocampal neurons during maturation in vitro. Development. 1995;121:2327–2335. doi: 10.1242/dev.121.8.2327. [DOI] [PubMed] [Google Scholar]
- Binder DK. The role of BDNF in epilepsy and other diseases of the mature nervous system. Adv Exp Med Biol. 2004;548:34–56. doi: 10.1007/978-1-4757-6376-8_3. [DOI] [PubMed] [Google Scholar]
- Brunig I, Penschuck S, Berninger B, Benson J, Fritschy JM. BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABAA receptor surface expression. Eur J Neurosci. 2001;13:1320–1328. doi: 10.1046/j.0953-816x.2001.01506.x. [DOI] [PubMed] [Google Scholar]
- Bureau I, Mulle C. Potentiation of GABAergic synaptic transmission by AMPA receptors in mouse cerebellar stellate cells: changes during development. J Physiol. 1998;509 (Pt 3):817–831. doi: 10.1111/j.1469-7793.1998.817bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Yeh HH. Brain-derived neurotrophic factor attenuates mouse cerebellar granule cell GABAA receptor-mediated responses via postsynaptic mechanisms. J Physiol. 2003;548:711–721. doi: 10.1113/jphysiol.2002.037846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Feng L, Yang F, Lu B. Activity and Ca2+-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. J Cell Biol. 2000;150:1423–1434. doi: 10.1083/jcb.150.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmariah SB, Crumling MA, Parsons TD, Balice-Gordon RJ. Postsynaptic TrkB-mediated signaling modulates excitatory and inhibitory neurotransmitter receptor clustering at hippocampal synapses. J Neurosci. 2004;24:2380–2393. doi: 10.1523/JNEUROSCI.4112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essrich C, Lorez M, Benson JA, Fritschy JM, Luscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the γ2 subunit and gephyrin. Nat Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- Fiszman ML, Barberis A, Lu C, Fu Z, Erdelyi F, Szabo G, Vicini S. NMDA receptors increase the size of GABAergic terminals and enhance GABA release. J Neurosci. 2005;25:2024–2031. doi: 10.1523/JNEUROSCI.4980-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M, Borges S, Wilson M. Variation in GABA mini amplitude is the consequence of variation in transmitter concentration. Neuron. 1995;15:885–895. doi: 10.1016/0896-6273(95)90179-5. [DOI] [PubMed] [Google Scholar]
- Frerking M, Malenka RC, Nicoll RA. Brain-derived neurotrophic factor (BDNF) modulates inhibitory, but not excitatory, transmission in the CA1 region of the hippocampus. J Neurophysiol. 1998;80:3383–3386. doi: 10.1152/jn.1998.80.6.3383. [DOI] [PubMed] [Google Scholar]
- Galante M, Avossa D, Rosato-Siri M, Ballerini L. Homeostatic plasticity induced by chronic block of AMPA/kainate receptors modulates the generation of rhythmic bursting in rat spinal cord organotypic cultures. Eur J Neurosci. 2001;14:903–917. doi: 10.1046/j.0953-816x.2001.01710.x. [DOI] [PubMed] [Google Scholar]
- Glitsch M, Marty A. Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J Neurosci. 1999;19:511–519. doi: 10.1523/JNEUROSCI.19-02-00511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, Yeh JL, Kapur J. Status epilepticus increases the intracellular accumulation of GABAA receptors. J Neurosci. 2005;25:5511–5520. doi: 10.1523/JNEUROSCI.0900-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996;7:222–238. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- Goslin K, Asmussen H, Banker G. Culturing Nerve Cells. MIT Press; Cambridge, MA: 1998. Rat Hippocampal Neurons in Low-Density Culture; pp. 339–370. [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hendry SH, Jones EG. Reduction in number of immunostained GABAergic neurones in deprived-eye dominance columns of monkey area 17. Nature. 1986;320:750–753. doi: 10.1038/320750a0. [DOI] [PubMed] [Google Scholar]
- Hendry SH, Jones EG. Activity-dependent regulation of GABA expression in the visual cortex of adult monkeys. Neuron. 1988;1:701–712. doi: 10.1016/0896-6273(88)90169-9. [DOI] [PubMed] [Google Scholar]
- Henneberger C, Juttner R, Schmidt SA, Walter J, Meier JC, Rothe T, Grantyn R. GluR- and TrkB-mediated maturation of GABA receptor function during the period of eye opening. Eur J Neurosci. 2005;21:431–440. doi: 10.1111/j.1460-9568.2005.03869.x. [DOI] [PubMed] [Google Scholar]
- Isackson PJ, Huntsman MM, Murray KD, Gall CM. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron. 1991;6:937–948. doi: 10.1016/0896-6273(91)90234-q. [DOI] [PubMed] [Google Scholar]
- Ivanova SY, Storozhuk MV, Melnick IV, Kostyuk PG. Chronic treatment with ionotropic glutamate receptor antagonist kynurenate affects GABAergic synaptic transmission in rat hippocampal cell cultures. Neurosci Lett. 2003;341:61–64. doi: 10.1016/s0304-3940(03)00154-x. [DOI] [PubMed] [Google Scholar]
- Kilman V, van Rossum MC, Turrigiano GG. Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABAA receptors clustered at neocortical synapses. J Neurosci. 2002;22:1328–1337. doi: 10.1523/JNEUROSCI.22-04-01328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Moss SJ. Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr Opin Neurobiol. 2003;13:341–347. doi: 10.1016/s0959-4388(03)00064-3. [DOI] [PubMed] [Google Scholar]
- Leslie KR, Nelson SB, Turrigiano GG. Postsynaptic depolarization scales quantal amplitude in cortical pyramidal neurons. J Neurosci. 2001;21:RC170. doi: 10.1523/JNEUROSCI.21-19-j0005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm D, Castren E, Berzaghi M, Blochl A, Thoenen H. Activity-dependent and hormonal regulation of neurotrophin mRNA levels in the brain—Implications for neuronal plasticity. J Neurobiol. 1994;25:1362–1372. doi: 10.1002/neu.480251105. [DOI] [PubMed] [Google Scholar]
- Mangan PS, Kapur J. Factors underlying bursting behavior in a network of cultured hippocampal neurons exposed to zero magnesium. J Neurophysiol. 2004;91:946–957. doi: 10.1152/jn.00547.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty S, Berninger B, Carroll P, Thoenen H. GABAergic stimulation regulates the phenotype of hippocampal interneurons through the regulation of brain-derived neurotrophic factor. Neuron. 1996;16:565–570. doi: 10.1016/s0896-6273(00)80075-6. [DOI] [PubMed] [Google Scholar]
- Marty S, Wehrle R, Sotelo C. Neuronal activity and brain-derived neurotrophic factor regulate the density of inhibitory synapses in organotypic slice cultures of postnatal hippocampus. J Neurosci. 2000;20:8087–8095. doi: 10.1523/JNEUROSCI.20-21-08087.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean Bolton M, Pittman AJ, Lo DC. Brain-derived neurotrophic factor differentially regulates excitatory and inhibitory synaptic transmission in hippocampal cultures. J Neurosci. 2000;20:3221–3232. doi: 10.1523/JNEUROSCI.20-09-03221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody I. Aspects of the homeostatic plasticity of GABAA receptor-mediated inhibition. J Physiol. 2005;562:37–46. doi: 10.1113/jphysiol.2004.077362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Cull-Candy S, Farrant M. Differences in synaptic GABAA receptor number underlie variation in GABA mini amplitude. Neuron. 1997;19:697–709. doi: 10.1016/s0896-6273(00)80382-7. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Hajos N, Somogyi P, Mody I. Increased number of synaptic GABAA receptors underlies potentiation at hippocampal inhibitory synapses. Nature. 1998;395:172–177. doi: 10.1038/25999. [DOI] [PubMed] [Google Scholar]
- O’Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- Ohba S, Ikeda T, Ikegaya Y, Nishiyama N, Matsuki N, Yamada MK. BDNF locally potentiates GABAergic presynaptic machineries: target-selective circuit inhibition. Cereb Cortex. 2005;15:291–298. doi: 10.1093/cercor/bhh130. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev, Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, DeWan A, Lauer HM, Turrigiano GG. Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. J Neurosci. 1997;17:4527–4535. doi: 10.1523/JNEUROSCI.17-12-04527.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Seil FJ, Drake-Baumann R. Reduced cortical inhibitory synaptogenesis in organotypic cerebellar cultures developing in the absence of neuronal activity. J Comp Neurol. 1994;342:366–377. doi: 10.1002/cne.903420305. [DOI] [PubMed] [Google Scholar]
- Seil FJ, Drake-Baumann R. TrkB receptor ligands promote activity-dependent inhibitory synaptogenesis. J Neurosci. 2000;20:5367–5373. doi: 10.1523/JNEUROSCI.20-14-05367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanwick CC, Harrison MB, Kapur J. Synaptic and extra-synaptic localization of brain-derived neurotrophic factor and the tyrosine kinase B receptor in cultured hippocampal neurons. J Comp Neurol. 2004;478:405–417. doi: 10.1002/cne.20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanwick CC, Murthy NR, Mtchedlishvili Z, Sieghart W, Kapur J. Development of GABAergic synapses in cultured hippocampal neurons. J Comp Neurol. doi: 10.1002/cne.20897. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci. 1999;22:221–227. doi: 10.1016/s0166-2236(98)01341-1. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev, Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Wetmore C, Olson L, Bean AJ. Regulation of brain-derived neurotrophic factor (BDNF) expression and release from hippocampal neurons is mediated by non-NMDA type glutamate receptors. J Neurosci. 1994;14:1688–1700. doi: 10.1523/JNEUROSCI.14-03-01688.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada MK, Nakanishi K, Ohba S, Nakamura T, Ikegaya Y, Nishiyama N, Matsuki N. Brain-derived neurotrophic factor promotes the maturation of GABAergic mechanisms in cultured hippocampal neurons. J Neurosci. 2002;22:7580–7585. doi: 10.1523/JNEUROSCI.22-17-07580.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Poo MM. Localized synaptic potentiation by BDNF requires local protein synthesis in the developing axon. Neuron. 2002;36:675–688. doi: 10.1016/s0896-6273(02)01023-1. [DOI] [PubMed] [Google Scholar]