

Abstract
We recently reported that the antineoplastic thiodioxopiperazine natural product chaetocin potently induces cellular oxidative stress, thus selectively killing cancer cells. In pursuit of underlying molecular mechanisms, we now report that chaetocin is a competitive and selective substrate for the oxidative stress mitigation enzyme thioredoxin reductase-1 (TrxR1) with lower Km than the TrxR1 native substrate thioredoxin (Trx; chaetocin Km = 4.6 ± 0.6 μM, Trx Km = 104.7 ± 26 μM), thereby attenuating reduction of the critical downstream ROS remediation substrate Trx at achieved intracellular concentrations. Consistent with a role for TrxR1 targeting in the anticancer effects of chaetocin, overexpression of the TrxR1 downstream effector Trx in HeLa cells conferred resistance to chaetocin-induced, but not to doxorubicin-induced, cytotoxicity. As the TrxR/Trx pathway is of central importance in limiting cellular reactive oxygen species (ROS)—and as chaetocin exerts its selective anticancer effects via ROS imposition—the inhibition of TrxR1 by chaetocin has potential to explain its selective anticancer effects. These observations have important implications not just with regard to the mechanism of action and clinical development of chaetocin and related thiodioxopiperazines, but also with regard to the utility of molecular targets within the thioredoxin reductase/thioredoxin pathway in the development of novel candidate antineoplastic agents. Antioxid. Redox Signal. 11, 1097–1106.
Introduction
Thioredoxin reductase-1 (TrxR1) is a 55 kD per subunit homodimeric protein belonging to a family of glutathione reductase-like flavoenzymes. TrxR1 catalyzes the NADPH-dependent reduction of thioredoxin and other substrate disulfide bonds via its selenocysteine/FAD active site. Mammalian TrxR1 consequently participates in diverse metabolic reactions involving oxidation–reduction cycles and is widely believed to be central to intracellular ROS mitigation (1, 4, 26).
The TrxR1/thioredoxin (Trx) pathway may provide plausible molecular targets for cancer therapies for several reasons. First, TrxR1 and/or Trx are known to be upregulated in a variety of human cancers, including lung, colorectal, cervical, hepatic, and pancreatic (5, 20), and Trx overexpression has been linked to aggressive tumor growth and poorer prognosis (11, 19). Second, TrxR1 enhances tumor proliferation via its regulatory effects on the G1 checkpoint during cell cycle progression (22). Third, TrxR1 invokes a pro-survival signaling cascade (22, 25). Further, cells overexpressing TrxR1 are more resistant to some anticancer agents (13). Moreover, upregulated TrxR1/Trx pathway activity may in part account for how cancer cells have adapted to their generally higher basal levels of cellular oxidative stress (18). Therefore, despite providing a potential survival advantage to cancer cells, upregulated TrxR1/Trx pathway activity may also be required for cancer survival in light of increased ROS stress inherent in some cancer cells. In this fashion, the TrxR1/Trx pathway may contain therapeutically useful antineoplastic molecular targets.
Small molecules such as lipid hydroperoxides, selenite, and dehydroascorbate, as well as proteins such as protein disulfide isomerase or glutathione peroxidase along with Trx, are all known substrates of TrxR1, demonstrating its low substrate specificity. There are several known inhibitors of TrxR1 including auranofin (17), cisplatin (2), lipoic acid (3), motexafin gadolinium (7), myricetin and quercetin (15), and 1-methyl-1-propyl-2-imidazolyl disulfide (IV-2, 22). Of these, motexafin gadolinium (7) and IV-2 (22) have anticancer effects putatively attributed to TrxR1 inhibition and are undergoing development as candidate cancer therapeutics.
We recently reported that chaetocin (Fig. 1A), a small molecule thiodioxopiperazine natural product produced by Chaetomium species fungi currently in development as a candidate antimyeloma therapeutic, has potent and selective in vitro and in vivo anticancer activity conveyed by its ability to impose increased levels of cellular oxidative stress (10). Up to this point, however, the mechanism(s) underlying ROS induction and the selective anticancer effects of chaetocin have remained undefined. We now report that chaetocin is a competitive substrate for, and inhibitor of, the central oxidative stress remediation enzyme thioredoxin reductase-1 capable of depletion of levels of the reduced downstream TrxR1 effector thioredoxin. Importantly, transient overexpression of the downstream TrxR1 substrate and effector Trx rescues cancer cells from chaetocin-induced, but not from doxorubicin-induced cell death, thereby providing for the first time evidence of a potential causal linkage between enzyme inactivation, ROS induction, and the antineoplastic activity of chaetocin.
FIG. 1.

Chaetocin inhibits thioredoxin reductase activity. (A) Chemical structure of chaetocin. (B) Thioredoxin reductase activity (assessed by following DTNB reduction) is inhibited by chaetocin in a dose-dependent fashion. (C) Time versus absorbance data showing inhibition of DTNB reduction by chaetocin at various concentrations and times. Data shown in (B) represent the means of triplicate data points, error bars, one standard deviation, *p < 0.05, **p < 0.01, and ***p < 0.001 different from diluent control values; while data shown in (c) are representative of a minimum of three separate experiments.
Materials and Methods
Reagents
Chaetocin, β-nicotinamide adenine dinucleotide 2′-phosphate reduced tetrasodium salt hydrate (NADPH), rat liver thioredoxin reductase, 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), oxidized glutathione (GSSG), bovine insulin, gliotoxin, thioredoxin reductase assay kit, anti-actin antibody and CelLytic lysis reagent were purchased from Sigma (St. Louis, MO); yeast glutathione reductase and Complete Protease Inhibitor Tablets from Roche (Indianapolis, IN); BCA protein assay from Pierce (Rockford, IL); oxidized E. coli thioredoxin and dithiothreitol (DTT) from Promega (Madison, WI); verruculogen from Calbiochem (San Diego, CA); 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) and Lipofectamine-Plus from Invitrogen (Carlsbad, CA); chetomin from Alexis Biochemicals (San Diego, CA); and anti-thioredoxin antibody from Cell Signaling (Beverly, MA). HeLa cells were obtained from American Type Culture Collection (Manassas, VA) (ATCC).
Thioredoxin reductase activity assay (DTNB method)
Cell-free thioredoxin reductase activity was assayed in 100 mM potassium phosphate (pH 7.0), 10 mM EDTA according to the Sigma kit protocol. Final concentrations were 0.0005 U (Units)/μL of enzyme and 0.24 mM NADPH in the presence of chaetocin as indicated in a 100 μL (microLiter) reaction. The reaction was started by the addition of DTNB (3 mM) and the change in absorbance at 405 nm was monitored in a plate reader. Activity was calculated as the increase in absorbance between 2 and 5 min after DTNB addition.
Glutathione reductase activity assay
Cell-free glutathione reductase activity was assayed in 100 mM potassium phosphate (pH 7.0), 10 mM EDTA. The 200 μL reaction mixture comprised 0.00006 U/μL of enzyme, 0.75 mM DTNB, 0.1 mM NADPH, and varying concentrations of chaetocin as indicated. The reaction was started by addition of oxidized glutathione (1 mM) and was monitored in a plate reader at 405 nm. Activity was calculated as the increase in absorbance between 1 and 3 min after glutathione addition.
Thioredoxin reductase activity assay (gel-based oxidation state of thioredoxin method)
Reduction of thioredoxin by thioredoxin reductase was measured by the decrease in electrophoretic mobility caused by covalent modification of thioredoxin by a thiol-reactive probe (AMS), when the disulfide is reduced. The reaction mix contained 100 mM potassium phosphate (pH 7.0), 10 mM EDTA, 0.24 mM NADPH, chaetocin or other compounds as indicated, 50 μM oxidized thioredoxin, and 0.0002 U/μL thioredoxin reductase (except for the initial rate Ki experiment, which contained 0.00005 U/μL thioredoxin reductase). At the indicated time, a 5 μL sample was removed and immediately added to 5 μL of 30 mM AMS in TE buffer (pH 7.5). The AMS was allowed to react (15 min at 22°C) with reduced thioredoxin sulfhydryl groups (24), then the samples were mixed with nonreducing sample buffer and were electrophoresed on 18% Tris-HCl SDS-PAGE gels. The gels were stained with Coomassie blue, and bands were imaged and quantitated using a Syngene InGenius gel documentation system (Frederick, MD).
Thioredoxin activity assay
The 100 μL reaction contained 100 mM potassium phosphate (pH 7.0), 2 mM EDTA, 0.13 mM bovine insulin, 3.9 μM E. coli thioredoxin, and chaetocin as indicated. The reaction was initiated by addition of 0.33 mM DTT, and turbidity was monitored at 620 nm in a plate reader. The initial linear rate was calculated based on the slope of the line after an increase in absorbance (indicating precipitation of insulin) started to occur (9).
Steady-state kinetics
The 100 μL reaction consisted of 100 mM potassium phosphate (pH 7.0), 10 mM EDTA, 0.0004 U/μL thioredoxin reductase, and chaetocin or other compounds as indicated. The assay was carried out in a 96-well quartz plate, and the oxidation of NADPH was measured as a change in absorption at 340 nm. The initial velocities of the reaction were calculated from the decrease in the absorbance between 0 and 5 min (slope), and on a pathlength of 0.3 cm and NADPH ɛ = 6.22 mmol−1*cm−1*L. The velocity versus concentration data were then analyzed using SigmaPlot Enzyme Kinetics 1.3 program (Systat Software, San Jose, CA).
Synthesis of S-methylchaetocin
Chaetocin was reduced with sodium borohydride and methylated using methyl iodide as previously described (10).
Mass spectrometry
Three samples were analyzed: “chaetocin” = 2.04 μg (micrograms) chaetocin in 100 mM potassium phosphate (pH 7.0), 10 mM EDTA; “chaetocin + NADPH” = 2.04 μg chaetocin in 100 mM potassium phosphate (pH 7.0), 10 mM EDTA with 0.24 mM NADPH; and “chaetocin + NADPH + TrxR1” = 2.04 μg chaetocin in 100 mM potassium phosphate (pH 7.0), 10 mM EDTA with 0.24 mM NADPH and 2.04 μg thioredoxin reductase. Samples were allowed to react at 22°C for 15 min and then processed for analysis by desalting and fractionating using C18 ZipTips (Millipore Corporation, Bedford, MA). For the DTT/chaetocin reaction, the reaction proceeded for 20 min and was then spun down to remove precipitated insulin before loading onto ZipTips. ZipTips were conditioned with 60% acetonitrile:39% water:1% acetic acid and equilibrated with 1% acetic acid. Samples were acidified with glacial acetic acid and loaded onto an equilibrated ZipTip and washed with 1% acetic acid. Fractions were step-eluted off the ZipTip using 10 μl of 1% acetic acid in water containing 20, 30, 40, or 60% acetonitrile. Collected fractions were injected by loop injection (2 μL) directly into the mass spectrometer using a mobile phase of 30% acetonitrile:69% water:1% acetic acid at 5 μL/min. Mass spectra were collected on an Agilent Technologies LC/ MSD-TOF mass spectrometer in positive electrospray ionization mode over a m/z range of 400 to 1500. The capillary, fragmentor, skimmer, and OCT RF voltages (3500, 185, 60, 200 volts, respectively) were optimized to enhance signal and minimize instrument fragmentation.
Cell culture, transient transfections, and immunoblotting
HeLa cells were cultured in RPMI 1640 containing L-glutamine and 5% FBS. Cells were passaged twice weekly and maintained in 37°C in an atmosphere containing 95% air-5% CO2 (vol/vol). For transfections, 105 cells per well were plated in 12-well plates and were transfected with 1 μg pcDNA or pcDNA-Trx (8, 12) using standard Lipofectamine-Plus procedures. Transfection efficiency based on cells transfected with GFP was 80%. Twenty four h after transfection the cells were treated with either DMSO, 100 nM (nanomolar) chaetocin or 100 nM doxorubicin for 24 h. The cells were then trypsinized and manually counted in trypan blue to exclude dead cells. The treatments were done in triplicate and the experiment was repeated three times. For immunoblotting (24 h after transfections), cells were trypsinized, washed in cold PBS, and lysed in CelLytic lysis buffer containing protease inhibitors. Protein was analyzed by BCA assay and lysates were electrophoresed on 15% SDS-PAGE gels and transferred to nitrocellulose. Immunoblotting for thioredoxin and actin was then performed.
Statistics
Statistical significance was assessed using pooled estimates of variance and the two-sided T-distribution.
Results
Chaetocin inhibits thioredoxin reductase more potently than glutathione reductase or thioredoxin
Since chaetocin (Fig. 1A) contains two disulfide bonds and is known to induce oxidative stress in cancer cell (10), we hypothesized that chaetocin might interact with oxidative stress-related proteins that rely upon disulfide bond redox cycling for activity. Initial experiments showed that chaetocin inhibited TrxR1-initiated turnover of the synthetic substrate DTNB (Reaction 1) in a cell-free assay in a dose-responsive manner (Figs. 1B and 1C), with an IC50 of about 4 μM.
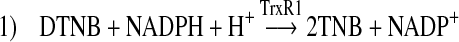 |
The activity of a related family member, glutathione reductase (G.R.; Reaction 2), however, was unaffected by up to 20 μM chaetocin (Fig. 2A).
FIG. 2.

Chaetocin less potently inhibits the activity of glutathione reductase or thioredoxin. Glutathione reductase activity (A) assessed following DTNB reduction, and thioredoxin activity (B) assessed using the insulin precipitation method, are unaffected by chaetocin at concentrations that readily inhibit thioredoxin reductase; however, chaetocin inhibits thioredoxin activity at 20-fold higher concentrations than required to inhibit thioredoxin reductase (note fivefold scale difference between chaetocin concentrations in (A) and (B). Data shown represent single experiments (triplicate data points; error bars, one standard deviation) representative of a minimum of three separate experiments; *p < 0.05, **p < 0.01 different from diluent control.
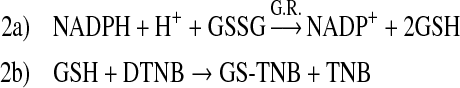 |
We also examined the effects of chaetocin on the reductase activity of Trx, as Trx is a major downstream effector substrate of TrxR1 and because Trx itself is a disulfide-containing reductase. Since we had established that chaetocin inhibits TrxR1 activity, however, we necessarily utilized an activity assay based on insulin reduction (Reaction 3) that did not rely on the coupled TrxR1/Trx reaction.
Additionally, because DTT reduces disulfide bonds and has been previously shown to reduce chaetocin under certain conditions (10), we also examined the reduction state of chaetocin in the presence of DTT by mass spectrometry to establish that, under utilized reaction conditions, DTT itself did not appreciably reduce chaetocin (data not shown). Confident that chaetocin was not reduced by DTT and therefore not inactivated over the time-span of the Trx activity assay, we found that up to 20 μM chaetocin did not appreciably affect Trx activity (Fig. 2B). However, experiments with higher chaetocin concentrations demonstrated Trx activity inhibition with an IC50 of ∼90 μM (Fig. 2B), ∼20 times the chaetocin concentration required to similarly inhibit TrxR1 (Fig. 1B). Collectively, these results indicate that chaetocin inhibits TrxR1 activity with a high degree of selectivity when compared to even closely related reductases, and that chaetocin is therefore not an indiscriminate inhibitor of all disulfide-containing reductases.
Chaetocin and related thiodioxopiperazines inhibit the reduction of thioredoxin by thioredoxin reductase
As the small molecule DTNB utilized in the previous assay is not the native substrate for thioredoxin reductase, we sought to establish whether chaetocin might also impair the ability of TrxR1 to reduce its native substrate, thioredoxin (Reaction 4). We therefore developed a novel gel-based kinetics assay to resolve the oxidized and reduced forms of Trx by rapid covalent modification of the free sulfhydryl groups of Trx with AMS. Using this method, chaetocin was indeed also found to inhibit the ability of TrxR1 to reduce Trx (Fig. 3A) in a dose- and time-dependent fashion. In addition, the structurally related thiodioxopiperazines gliotoxin and chetomin also inhibited the activity of thioredoxin reductase, albeit somewhat less effectively than chaetocin (Fig. 3B). Not surprisingly, intact disulfide bonds of tested thiodioxopiperazines were found to be critical to this inhibition, based on the inability of S-methyl chaetocin or the reduced thiodioxopiperazine verruculogen to recapitulate this effect (Fig. 3B). The IC50 for chaetocin in this reaction was found to be 4 μM under conditions optimized to be within the initial linear portion of the reaction (Fig. 3C).
FIG. 3.

Chaetocin and other intact thiodioxopiperazines inhibit the ability of thioredoxin reductase to reduce its native substrate thioredoxin. (A) Chaetocin inhibits the reduction of thioredoxin by thioredoxin reductase in a dose-dependent fashion. (B) Chaetocin, chetomin, and gliotoxin each inhibit the reduction of thioredoxin by thioredoxin reductase; however, the related compounds S-methyl chaetocin and verruculogen, lacking bridged disulfide bonds, do not. (C) In a representative initial rate experiment, the IC50 of chaetocin inhibiting redox cycling of thioredoxin by thioredoxin reductase is 4 μM. This IC50 value was attained based upon quantitation of the percent control fraction reduced thioredoxin [(reduced/reduced+oxidized)treated/(reduced/reduced+oxidized)diluent] assessed at 5 min reaction times via densitometry of resulting thioredoxin gel bands, as shown in the inset. Data shown are representative of a minimum of three separate experiments.
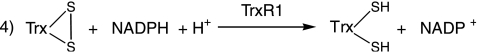 |
As achieved intracellular chaetocin concentrations are as high as 1,000-fold greater than applied extracellular concentrations (10), cytotoxic intracellular chaetocin levels easily reach 25–100 times its IC50 for this reaction (100–400 μM; 10), underscoring the potential biological relevance of TrxR1 inhibition by chaetocin to its anticancer effects.
Chaetocin and related thiodioxopiperazines serve as substrates for thioredoxin reductase
We next hypothesized that chaetocin, which itself contains two disulfides, might inhibit thioredoxin reductase by serving as a competitive substrate for TrxR1. Consistent with this possibility, NADPH is indeed oxidized over time when chaetocin is substituted for Trx in Reaction 4, with a Km for chaetocin of 4.6 ± 0.6 μM, indicative of substrate functionality (Fig. 4A). Interestingly, the Km for Trx in this same assay is, almost 25 times higher than that of chaetocin (Table 1), indicating that chaetocin effectively serves as a more efficient substrate for TrxR1 than its native substrate Trx. Presented data are therefore consistent with the hypothesis that the ability of chaetocin to serve as a TrxR1 inhibitor (Fig. 1) most likely relates to its function as a competitive TrxR1 substrate. Interestingly, the related thiodioxopiperazines gliotoxin and chetomin are also TrxR1 substrates (Table 1), albeit chaetocin is the highest affinity TrxR1 substrate of the series.
FIG. 4.

Chaetocin is a substrate for thioredoxin reductase. (A) Oxidation of NADPH by thioredoxin reductase in response to various chaetocin concentrations in the absence of other thioredoxin reductase substrates, indicating that chaetocin itself has thioredoxin reductase substrate functionality. Data shown are representative of a minimum of three separate experiments. (B) Mass spectrometry results demonstrating that chaetocin disulfide bonds are reduced in the presence of thioredoxin reductase and NADPH, but not NADPH alone. Note the addition of two (m/z 699) and four (m/z 701) Daltons to chaetocin (m/z 697), indicative of the addition of two or four protons respectively, and reflective of reduction of one or both chaetocin disulfide bonds.
Table 1.
Thioredoxin Reductase Steady-State Kinetics Data for Thioredoxin and Other Tested Substrates
| Compound | Km, μM | Model | n (cooperativity) | Velocity |
|---|---|---|---|---|
| chaetocin | 4.6 ± 0.6 | Hill | 4.8 | 8.62e-8 ± 1e-8 |
| gliotoxin | 16.9 ± 5.0 | Hill | 2.7 | 4.07e-7 ± 9e-8 |
| chetomin | 16.1 ± 5.4 | MM | na | 3.97e-7 ± 3e-8 |
| thioredoxin | 104.7 ± 26 | Hill | 1.4 | 1.17e-6 ± 3e-7 |
Km and velocity measurements represent means ± 1 standard deviation obtained from four independent experiments. MM, Michaelis-Menten.
Confirmatory mass spectrometry analyses of chaetocin in the presence and absence of TrxR1 showed the expected parent peak of 697.11 m/z with chaetocin alone or after addition of NADPH (Fig. 4B, upper panels). In the presence of thioredoxin reductase, however, a minor peak at 699.11 m/z (+2 Da; corresponding to the addition of two protons) and a major peak at 701.11 m/z (+4 Da; corresponding to the addition of four protons) were observed (Fig. 4B, lower panel). These results are consistent with the hypothesis that the disulfide bonds of chaetocin are reduced by TrxR1, as expected if chaetocin serves as a substrate for the enzyme. Furthermore, there was no evidence of covalent chaetocin-TrxR1 adduction by mass spectroscopy (data not shown), in contrast to what has been reported in the instance of the interaction of the related thiodioxopiperazine sporidesmin with glutaredoxin (23).
Transient thioredoxin overexpression attenuates chaetocin-induced cell death
The above studies collectively indicate that, although serving as a TrxR1 competitive substrate, chaetocin does not in fact modify TrxR1 itself, but instead attenuates reduction of the downstream TrxR1 effector Trx. Consequently, as we hypothesized that Trx (and not TrxR1) ultimately functions as the primary cellular ROS-scavenger affected by chaetocin, we transiently overexpressed Trx as a means of exploring the potential linkage between the anticancer effects of chaetocin and its ability to inhibit TrxR1. Indeed, Trx overexpression significantly attenuated chaetocin-induced cell death (Fig. 5A), consistent with a linkage between the ability of chaetocin to inhibit the reduction of Trx by TrxR1 and chaetocin-induced cytotoxicity. As a negative control we evaluated the impact of Trx overexpression on doxorubicin-induced cytotoxicity, as doxorubicin-induced cytotoxicity was not attenuated by NAC co-treatment, indicating lesser contributions of ROS to doxorubicin-induced cytotoxicity consistent with induction of cytotoxicity instead via topoisomerase II inhibition. Importantly, parallel experiments demonstrated that Trx overexpression did not rescue cells from doxorubicin-induced cell death (Fig. 5A), indicating that the attenuation of chaetocin-induced cell death in response to transient Trx overexpression was not attributable to indiscriminate induction of pro-survival signaling, but instead to the effects of chaetocin on the TrxR1/Trx pathway.
FIG. 5.

Transient overexpression of the downstream thioredoxin substrate and effector thioredoxin attenuates chaetocin-induced, but not doxorubicin-induced, cytotoxicity; and the reduction of chaetocin and thioredoxin by thioredoxin reductase display sigmoidal kinetics. (A) Transient overexpression of thioredoxin attenuates chaetocin, but not doxorubicin-induced cytotoxicity in HeLa cells. Transfected cells were treated for 24 h; cell viability assessed using a trypan blue exclusion assay. As the primary mechanism of cytotoxicity induction by doxorubicin involves inhibition of topoisomerase II, doxorubicin was utilized as a “negative control” to assure that Trx overexpression would not nonspecifically attenuate drug-induced cytotoxicity known to be conferred by effects on non-Trx/TrxR1 pathways. NS, not statistically significantly different from doxorubicin pc-DNA control. Inset: Immunoblotting showing increased level of thioredoxin in transiently transfected HeLa cells compared to empty-vector transfected cells. (B) and (C) Michaelis-Menton plot fitted with the Hill equation for chaetocin (B) and thioredoxin (C). Note the different concentration and rate scales between the chaetocin and thioredoxin plots. Data shown represent single experiments (triplicate data points; error bars, one standard deviation), representative of a minimum of three separate experiments. Error bars not evident are hidden by data points.
Discussion
This article presents for the first time data potentially linking the effects of chaetocin on a specific molecular target, TrxR1 (Figs. 1–4 and Table 1), to its ability to impose cellular oxidative stress (10) and induce death in cancer cells (Fig. 5A, ref. 10). To our knowledge, this article represents the first report to identify a specific molecular target of chaetocin to be causally linked to its antineoplastic effects. Moreover, not only chaetocin, but also several other structurally related thiodioxopiperazines including gliotoxin and chetomin, competitively inhibit TrxR1, suggesting that thiodioxopiperazines as a class may target TrxR1. Finally, in addition to providing important insights into the cytotoxic molecular targets of chaetocin and related thiodioxopiperazines, the presented work also highlights the potential utility of the TrxR1/Trx pathway as an important source of promising candidate antineoplastic molecular targets.
Although we herein present compelling evidence that chaetocin is a competitive substrate and inhibitor of TrxR1, while not as potently inhibiting several related reductases including glutathione reductase and thioredoxin, it is always possible that chaetocin might inhibit other yet untested reductases. The fact that intracellular chaetocin concentrations reach >100 μM (10) clearly lends plausibility to this possibility, and data shown in Fig. 2B indicate that even Trx activity can be partially inhibited at achieved intracellular chaetocin concentrations. Therefore, this issue of whether inhibition of reductases in addition to TrxR1 might contribute to the biological activity of chaetocin remains an open question, with work to be undertaken to determine more comprehensively the effects of chaetocin across a wider spectrum of cellular reductases. Second, we have sought to clarify whether chaetocin might also have effects on other antioxidant systems such as glutathione, catalase, and/or superoxide dismutases (SODs)—finding that although chaetocin does not deplete levels of reduced intracellular glutathione (10), exogenously applied catalase or forced SOD2 overexpression somewhat attenuate chaetocin-induced cytotoxicity (data not shown). Although we as of yet have no evidence that chaetocin directly affects catalase or SOD2 activities, the possibility exists that effects of chaetocin on these other ROS mitigation enzymes might also contribute to its redox effects and investigation is currently underway. Third, it is of importance to note that another group identified chaetocin as an inhibitor of histone methyltransferase (HMT) H3:K9 via a high throughput screening initiative (6), and it is of course therefore possible that the effects of chaetocin on this other target might also contribute to its anticancer activity. This having been said, our experience nonetheless indicates that the cytotoxic effects of chaetocin in both lung cancer and myeloma cell lines is completely abrogated by co-treatment with the cell permeable reduced glutathione precursor N-acetyl cysteine (NAC, 10). These observations are clearly more consistent with the hypothesis that chaetocin-induced cytotoxicity might be conferred via ROS imposition, than with the hypothesis that its cytotoxicity might instead be conferred by HMT H3:K9 inhibition. Nevertheless, attained micromolar chaetocin concentrations might easily allow it to have multiple molecular targets all in concert contributing to its anticancer effects.
While at first glance chaetocin might otherwise appear to be a classical inhibitor of TrxR1 (Figs. 1–3), careful scrutiny indicated instead that chaetocin does not act as pure inhibitor of TrxR1, but rather as a competitive TrxR1 substrate (Fig. 4). Because chaetocin exhibits a significantly lower TrxR1 Km than that of the TrxR1 native substrate Trx (Km for chaetocin 4.6 ± 0.6 vs. 104.7 ± 26 μM for Trx), chaetocin effectively spends more time associated with the enzyme, consequently serving as a noncovalent TrxR1 inhibitor. This is supported by the observation that TrxR1 regains its activity at high chaetocin concentrations at later time points (Fig. 3A), when chaetocin is completely reduced by TrxR1 and therefore no longer capable of TrxR1 inhibition as demonstrated for related compounds lacking intact bridged disulfide bonds (Fig. 3B; S-methyl chaetocin, verruculogen). These data bolster the contention that chaetocin's functionality as a TrxR1 substrate, and not other classical inhibitor qualities such as covalent binding or protein structure rearrangement, ultimately results in the inhibition of TrxR1 by chaetocin. Furthermore, mass spectrometric analyses provide no hint of covalent modification of TrxR1 by chaetocin.
Human TrxR1 contains two redox sites, a Cys59–Cys64 active site pair, and a selenoCys 496′–Cys495′ pair in the C-terminal region that interacts with the active site cysteine pair (26). Glutatione reductase (GR) and Trx, however, each contain only solitary active sites, a cysteine–cysteine pair. Based on the ability of chaetocin to act as an substrate/inhibitor for TrxR1 but not GR or Trx, it is interesting to postulate that chaetocin might primarily interact with the C-terminal selenoCys 496′–Cys495′. Studies with TrxR1 mutants lacking the selenocysteine active site would be required to further examine this possibility. Also, since TrxR1 contains two active sites, it is intriguing that the initial rate (v) versus concentration [S] kinetics plots were best fit by the Hill equation for chaetocin and Trx (Figs. 5B and C). This sigmoidal v by [S] plot often indicates cooperative binding of substrate to the active site. Such behavior is most common for substrates interacting with multimeric enzymes containing several interacting active sites, and has been described for TrxR1 family members (16) but not specifically to the best of our knowledge for TrxR1 itself.
Importantly, by inhibiting TrxR1, chaetocin attenuates otherwise normal levels of TrxR1 redox cycling of its major downstream effector Trx (Fig. 3), thereby apparently compromising cellular ROS mitigation capacity, consequently lending an explanation for the increased cellular ROS previously observed accompanying the treatment of cancer cells with chaetocin (10). Moreover, the observation that chaetocin-induced cytotoxicity is attenuated by Trx overexpression in cancer cells (Fig. 5A) establishes a potential linkage between TrxR1 inhibition by chaetocin and chaetocin-induced ROS and anticancer activity. It is also possible, however, that the observed but less potent inhibition of Trx by chaetocin (Fig. 2B) may augment these upstream effects on TrxR1.
Although increased levels of cellular oxidative stress promulgated by the inhibition of TrxR1 by chaetocin might alone affect cytotoxicity in accord with the rationale articulated in the preceding paragraph, it is also possible that downstream effects on Trx/apoptosis signal regulating kinase-1 (ASK-1) interactions may also be contributory. In particular, oxidation of Trx leads to disruption of Trx/ASK-1 binding and consequent activation of the proapoptotic mitogen activated protein kinase kinase kinase ASK-1 (21)—as reduced Trx appears to be a regulator of ASK-1 levels by promoting ASK-1 ubiquitination and degradation (14). Hence, TrxR1 inhibition by chaetocin might additionally affect cell death via direct effects on apoptotic regulators including ASK-1.
It is noteworthy that inherently upregulated Trx in some cancers appears to convey worse prognosis, and also resistance to some conventional chemotherapeutics (11, 19, 13). This may be attributable to what has been referred to as oncogene addiction, whereby tumor cells overexpress a particular gene in order to gain a growth and/or survival advantage, but then go on to become dependent upon the upregulated signaling. TrxR1 overexpression may in part consequently represent an adaptive mechanism facilitating mitigation of otherwise cytotoxic higher basal ROS levels characteristic of many cancers (18), thereby making TrxR1 overexpression required for survival in these cancers. Consequently, agents like chaetocin that specifically target TrxR1 may represent especially attractive target-directed and potentially cancer-selective therapeutics for TrxR1-overexpressing neoplasms. Moreover, it is interesting to speculate that TrxR1 overexpression might be usable as a biomarker to define cancers most likely to respond to chaetocin therapy.
It is also noteworthy that several thiodioxopiperazines (gliotoxin, chetomin) that are structurally related to chaetocin were also found to inhibit TrxR1, albeit somewhat less effectively than chaetocin (Fig. 3, Table 1). This suggests that, as a class, thiodioxopiperazines may generally serve as TrxR1 competitive substrates and inhibitors. Of note, however, is that the structurally-related thiodioxopiperazine sporidesmin has previously been implicated as an inhibitor of glutaredoxin, yet was reported not to significantly inhibit TrxR1 (23). If indeed different thiodioxopiperazines have selectivity in inhibiting distinct cellular reductases, there may be further opportunities to develop members of this class of compounds as inhibitors of other reductases of potential relevance to cancer and other diseases.
In summary, we present herein evidence for the first time that thioredoxin reductase is a molecular target of chaetocin of potential relevance and importance to its previously reported selective anticancer effects. In particular, our data support a model (Fig. 6) whereby chaetocin serves as a potent competitive substrate for the redox cycling enzyme thioredoxin reductase, competing with thioredoxin for reduction by TrxR1, and thereby serving to deplete levels of reduced cellular Trx, a survival-critical ROS remediation substrate and downstream effector of TrxR1. Although chaetocin has recently been shown to affect other molecular targets of potential relevance to cancer pathogenesis (6) and may affect presently unidentified additional redox targets, data presented herein are the first to provide a plausible direct causal linkage between the redox targeting of chaetocin and its selective antineoplastic effects (10). These results highlight the potential of the Trx/TrxR1 pathway as an important source of candidate anticancer molecular targets with relevance not only with regard to the clinical development of chaetocin and related thiodioxopiperazines, but also with regard to advancing the understanding of altered redox signaling pathways in cancer.
FIG. 6.

Model of observed effects of chaetocin on Trx/TrxR1 pathway. Chaetocin competes with thioredoxin as a substrate for thioredoxin reductase, thereby serving to attenuate reduction of the thioredoxin reductase downstream substrate and effector thioredoxin. Figure numbers corresponding to experimental data supporting indicated effects are shown.
Footnotes
Author contributions: Jennifer D. Tibodeau, designed research, performed experiments, wrote and edited manuscript; Linda M. Benson, assisted in experimental design and performed experiments (mass spectrometry), edited manuscript; Crescent R. Isham, contributed to experimental design, performed experiments, and edited manuscript; Whyte G. Owen, assisted in experimental design (enzymology), edited manuscript; Keith C. Bible, designed research, wrote and edited manuscript.
Acknowledgments
The authors would like to thank Jared J. Paul, Ph.D. (UNC-CH) and Joel M. Reid, Ph.D. (Mayo Clinic) for helpful discussions regarding enzyme kinetics experiments. We are also indebted to David Gius (NCI/NIH, Bethesda, MD) for providing the pcDNA-Trx expression plasmid. This work was supported in part by grants from the Multiple Myeloma Research Foundation (Postdoctoral Fellowship; JDT) and from the National Institutes of Health/National Cancer Institute (NIH/NCI; R01 CA97129 and R01 CA 98118, KCB).
Abbreviations
AMS, 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid; DTNB, 5,5′-dithiobis-(2-nitrobenzoic acid); DTT, dithiothreitol; GR, glutathione reductase; GSH, reduced glutathione; GSSG, oxidized glutathione; IV-2, 1-methyl-1-propyl-2-imidazolyl disulfide; NAC, N-acetyl cysteine; ROS, reactive oxygen species; Trx, thioredoxin; TrxR1, thioredoxin reductase 1; V, initial rate.
Disclosure Statement
The authors have no competing financial interest at this time. However, a use patent application for chaetocin is currently under review.
References
- 1.Arnér ES. Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267:6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 2.Arnér ES. Nakamura H. Sasada T. Yodoi J. Holmgren A. Spyrou G. Analysis of the inhibition of mammalian thioredoxin, thioredoxin reductase, and glutaredoxin by cis-diamminedichloroplatinum (ii) and its major metabolite, the glutathione-platinum complex. Free Rad Biol Med. 2001;31:1170–1178. doi: 10.1016/s0891-5849(01)00698-0. [DOI] [PubMed] [Google Scholar]
- 3.Arnér ES. Nordberg J. Holmgren A. Efficient reduction of lipoamide and lipoic acid by mammalian thioredoxin reductase. Biochem Biophys Res Commun. 1996;225:268–274. doi: 10.1006/bbrc.1996.1165. [DOI] [PubMed] [Google Scholar]
- 4.Becker K. Gromer S. Schirmer RH. Müller S. Thioredoxin reductase as a pathophysiological factor and drug target. Eur J Biochem. 2000;267:6118–6125. doi: 10.1046/j.1432-1327.2000.01703.x. [DOI] [PubMed] [Google Scholar]
- 5.Biaglow JE. Miller RA. The thioredoxin reductase/thioredoxin system: novel redox targets for cancer therapy. Can Biol Ther. 2005;4:6–13. doi: 10.4161/cbt.4.1.1434. [DOI] [PubMed] [Google Scholar]
- 6.Greiner D. Bonaldi T. Eskeland R. Roemer E. Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nat Chem Biol. 2005;1:143–145. doi: 10.1038/nchembio721. [DOI] [PubMed] [Google Scholar]
- 7.Hashemy SI. Ungerstedt JS. Avval FZ. Holmgren A. Motexafin gadolinium, a tumor-selective drug targeting thioredoxin reductase and ribonucleotide reductase. J Biol Chem. 2006;281:10691–10697. doi: 10.1074/jbc.M511373200. [DOI] [PubMed] [Google Scholar]
- 8.Hirota K. Murata M. Sachi Y. Nakamura H. Takeuchi J. Mori K. Yodoi J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. J Biol Chem. 1999;274:27891–27897. doi: 10.1074/jbc.274.39.27891. [DOI] [PubMed] [Google Scholar]
- 9.Holmgren A. Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J Biol Chem. 1979;254:9627–9632. .19. [PubMed] [Google Scholar]
- 10.Isham CR. Tibodeau JD. Jin W. Xu R. Timm MM. Bible KC. Chaetocin: a promising new antimyeloma agent with in vitro and in vivo activity mediated via imposition of oxidative stress. Blood. 2007;109:2579–2587. doi: 10.1182/blood-2006-07-027326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kakolyris S. Giatromanolaki A. Koukourakis M. Powis G. Souglakos J. Sivridis E. Thioredoxin expression is associated with lymph node status and prognosis in early operable non-small cell lung cancer. Clin Can Res. 2001;7:3087–3091. [PubMed] [Google Scholar]
- 12.Karimpour S. Lou J. Linm LL. Rene LM. Lagunas L. Ma X. Karra S. Bradbury CM. Markovina S. Goswami PC. Spitz DR. Hirota K. Kalvakolanu DV. Yodoi J. Gius D. Thioredoxin reductase regulates AP-1 activity as well as thioredoxin nuclear localization via active cysteines in response to ionizing radiation. Oncogene. 2002;21:6317–6327. doi: 10.1038/sj.onc.1205749. [DOI] [PubMed] [Google Scholar]
- 13.Kirkpatrick DL. Kuperus M. Dowdeswell M. Potier N. Donald LJ. Kunkel M. Mechanisms of inhibition of the thioredoxin growth factor system by antitumor 2-imidazolyl disulfides. Biochem Pharmacol. 1998;55:987–994. doi: 10.1016/s0006-2952(97)00597-2. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y. Min W. Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1-mediated apoptosis in a redox activity-independent manner. Circ Res. 2002;90:1259–1266. doi: 10.1161/01.res.0000022160.64355.62. [DOI] [PubMed] [Google Scholar]
- 15.Lu J. Papp LV. Fang J. Rodriguez–Nieto S. Zhivotovsky B. Holmgren A. Inhibition of mammalian thioredoxin reductase by some flavonoids: implications for myricetin and quercetin anticancer activity. Cancer Res. 2006;66:4410–4418. doi: 10.1158/0008-5472.CAN-05-3310. [DOI] [PubMed] [Google Scholar]
- 16.Mukhopadhyay R. Shi J. Rosen BP. Purification and characterization of ACR2p, the Saccharomyces cerevisiae arsenate reductase. J Biol Chem. 2000;275:21149–21157. doi: 10.1074/jbc.M910401199. [DOI] [PubMed] [Google Scholar]
- 17.Omata Y. Folan M. Shaw M. Messer RL. Lockwood PE. Hobbs D. Bouillaguet S. Sano H. Lewis JB. Wataha JC. Sublethal concentrations of diverse gold compounds inhibit mammalian thioredoxin reductase (TrxR1) Tox In Vitro. 2006;20:882–890. doi: 10.1016/j.tiv.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 18.Pelicano H. Carney D. Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Update. 2004;7:97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 19.Raffel J. Bhattacharyya AK. Gallegos A. Cui H. Einspahr JG. Alberts DS. Increased expression of thioredoxin-1 in human colorectal cancer is associated with decreased patient survival. J Lab Clin Med. 2003;42:46–51. doi: 10.1016/S0022-2143(03)00068-4. [DOI] [PubMed] [Google Scholar]
- 20.Rundlöf AK. Arnér E. Regulation of the mammalian selenoprotein thioredoxin reductase 1 in relation to cellular phenotype, growth, and signaling events. Antiox Redox Signal. 2004;6:41–52. doi: 10.1089/152308604771978336. [DOI] [PubMed] [Google Scholar]
- 21.Saitoh M. Nishitoh H. Fujii M. Takeda K. Tobiume K. Sawada Y. Kawabata M. Miyazono K. Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smart DK. Ortiz KL. Mattson D. Bradbury CM. Bisht KS. Sieck LK. Brechbiel MW. Gius D. Thioredoxin reductase as a potential molecular target for anticancer agents that induce oxidative stress. Cancer Res. 2004;64:6716–6724. doi: 10.1158/0008-5472.CAN-03-3990. [DOI] [PubMed] [Google Scholar]
- 23.Srinivasan U. Bala A. Jao S-C. Starke DW. Jordan TW. Mieyal JJ. Selective inactivation of glutaredoxin by sporidesmin and other epidithiopiperazinediones. Biochem. 2006;45:8978–8987. doi: 10.1021/bi060440o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vestweber D. Schatz G. Mitochondria can import artificial precursor proteins containing a branched polypeptide chain or a carboxy-terminal stilbene disulfonate. J Cell Biol. 1988;107:2045–2049. doi: 10.1083/jcb.107.6.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei SJ. Botero A. Hirota K. Bradbury CM. Markovina S. Laszlo A. Thioredoxin nuclear translocation and interaction with redox factor-1 activates the activator protein-1 transcription factor in response to ionizing radiation. Cancer Res. 2000;60:6688–6695. [PubMed] [Google Scholar]
- 26.Williams CH., Jr Arscott LD. Müller S. Lennon BW. Ludwig ML. Wang P-F. Veine DM. Becker K. Schirmer RH. Thioredoxin reductase: Two modes of catalysis have evolved. Eur J Biochem. 2000;267:6110–6117. doi: 10.1046/j.1432-1327.2000.01702.x. [DOI] [PubMed] [Google Scholar]