

Abstract
Repeated psychosocial or restraint stress causes atrophy of apical dendrites in CA3 pyramidal neurons of the hippocampus, accompanied by specific cognitive deficits in spatial learning and memory. Excitatory amino acids mediate this atrophy together with adrenal steroids and the neurotransmitter serotonin. Because the mossy fibers from dentate granule neurons provide a major excitatory input to the CA3 proximal apical dendrites, we measured ultrastructural parameters associated with the mossy fiber–CA3 synapses in control and 21-day restraint-stressed rats in an effort to find additional morphological consequences of stress that could help elucidate the underlying anatomical as well as cellular and molecular mechanisms. Although mossy fiber terminals of control rats were packed with small, clear synaptic vesicles, terminals from stressed animals showed a marked rearrangement of vesicles, with more densely packed clusters localized in the vicinity of active zones. Moreover, compared with controls, restraint stress increased the area of the mossy fiber terminal occupied by mitochondrial profiles and consequently, a larger, localized energy-generating capacity. A single stress session did not produce these changes either immediately after or the next day following the restraint session. These findings provide a morphological marker of the effects of chronic stress on the hippocampus that points to possible underlying neuroanatomical as well as cellular and molecular mechanisms for the ability of repeated stress to cause structural changes within the hippocampus.
The hippocampus is a target for adrenal steroids that provides a model for studying neurobiological consequences of stress (1). The human hippocampus undergoes atrophy in the aftermath of traumatic stress (2, 3), recurrent depression (4), and Cushing’s syndrome (5) as well as in some aging individuals (6). Prolonged psychosocial stress in monkeys is associated with loss of hippocampal neurons (7), whereas repeated psychosocial stress in a primitive primate, the tree shrew, and in rats causes hippocampal CA3 pyramidal neurons to undergo dendritic atrophy (8, 9).
A similar structural change is observed in rats subjected to a chronic repeated restraint stress paradigm. After 21 days of daily restraint stress, CA3 pyramidal neurons show a decrease in apical dendritic branching and total dendritic length, whereas basal dendrites remain unchanged (10). This highly specific atrophy, which results in deficits in spatial learning and memory (11, 12), is prevented by inhibiting glucocorticoids secretion or by interfering with excitatory amino acid neurotransmission (13) and by enhancing serotonin reuptake (14). Psychosocial stress-induced CA3 apical dendritic atrophy in the tree shrew was also shown to be dependent on excitatory amino acids (8).
The proximal segments of apical dendrites of CA3 pyramidal neurons are covered with complex spines or excrescences that receive mossy fiber input from granule neurons of the dentate gyrus (15). The dentate gyrus–CA3 pathway provides the major excitatory afferent to the hippocampal regio inferior, by using glutamate as its major neurotransmitter (16). CA3 pyramidal neurons have recurrent projections that excite other CA3 neurons (17, 18); moreover, neurons in the region CA3c closest to the hilus send some projections back to the dentate gyrus, providing another recurrent feedback loop (17–20); together, these pathways provide a means of exciting bursting activity, known as SPW (sharp waves) in the CA3 region (21–23). These findings suggested that a key to understanding the atrophy of apical dendrites of CA3 neurons might lie in identifying plasticity within the mossy fiber terminals (MFT) themselves. To learn more directly whether the mossy fiber synapses are involved in the response to chronic stress, we examined the ultrastructure of the MFT in stratum lucidum of control and 21-day restraint-stressed animals and quantitatively evaluated morphological parameters of the mossy fiber–CA3 interaction.
MATERIALS AND METHODS
Experimental Animals.
Adult male Sprague–Dawley rats (CD strain) weighing 250–300 g at the beginning of the experiment were housed in groups of three with ad libitum access to food and water. Animals were maintained in a temperature-controlled room, with a light/dark cycle of 12/12 h (lights on at 0700 h) and were handled daily for 1 week before being randomly assigned to experimental groups. Experiments were performed during the light period of the cycle and were conducted in accordance with the principles and procedures of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Control animals (n = 7) were left undisturbed, and stressed animals (n = 7) were subjected to daily restraint stress for 3 weeks in a separate room of the same animal facility. The sessions consisted of 6 h/day (10 a.m. to 4 p.m.) restraint of the rats in wire mesh restrainers secured at the head and tail ends with clips. Two additional control groups (n = 5 each) were subjected to a single restraint stress session. During the restraint sessions the rats were placed in their home cages.
Electron Microscopy.
On day 22 control and chronically restrained rats were anesthetized with Metofane (Pitman–Moore, Mundelein, IL) and transcardially perfused with 100 ml of saline followed by 200 ml of a fixative containing 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M phosphate buffer (PB, pH 7.4). Rats subjected to a single stress session were perfused either as soon as they were removed from the restrainers or the next day. Two hours after the perfusions, brains were removed from the skulls and stored in the fixative solution overnight at 4°C. Coronal sections (100 μm thick) through the dorsal hippocampus were cut on a Vibratome and washed in several changes of PB. For electron microscopy, the sections were rinsed twice in PB and postfixed in 2% osmium tetroxide in PB for 2 h, rinsed again in PB, and partially dehydrated through an ascending series of ethanol concentrations (50% and 70%). Sections were stained with 2% uranyl acetate in 70% ethanol for 4 h, rinsed in 70% ethanol, and further dehydrated with 95% and 100% ethanol. Ethanol was then replaced with propylen oxide (two changes, 30 min each). Finally, six brain sections per experimental animal were flat embedded in Durcupan (Fluka). The mossy fiber termination zone (stratum lucidum) of three blocks per animal was trimmed (Fig. 1), and ultrathin silver sections were cut on a Reichert ultramicrotome and mounted on single slot copper grids coated with formvar film. Sections were counterstained with Reynolds’ lead citrate and the final preparations were examined and photographed with a Jeol 100 CX electron microscope. Photographs (10 per subject) were randomly taken from the stratum lucidum area at a primary magnification of ×5,400. Prints at a final magnification of ×13,500 were used to trace MFT as well as mitochondria, spine profiles, and area occupied by small clear vesicles. An average of 60 MFT per block per experimental animal were analyzed.
Figure 1.
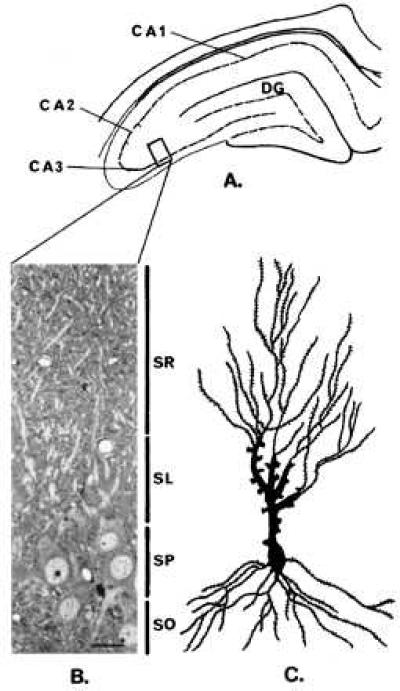
Localization of the hippocampal stratum lucidum area used for electron microscopic studies. (A) Durcupan-embedded blocks containing 100-μm-thick sections of the dorsal hippocampus were trimmed at the level of the CA3 region (see box). (B) Semithin sections (1.5 μm) stained with toluidine blue were used as a guide to further trim the blocks to an area containing the stratum lucidum (SL). (Bar = 17.5 μm.) (C) Camera lucida tracing of a representative pyramidal neuron impregnated with the “single” section Golgi method (13). DG, dentate gyrus, SO, stratum oriens, SP, stratum pyramidale, SL, stratum lucidum, SR stratum radiatum.
Data Analysis.
The total MFT area as well as the areas occupied by mitochondria and spine profiles, and by small clear synaptic vesicles, were measured by using a Zeiss Interactive Digitizing System. Because the majority of MFT contained mitochondria and were perforated by CA3 spine profiles, the net mossy fiber area was derived by subtracting the mitochondrial and the spine areas from the total MFT area. Only few MFT contained no mitochondria and were not invaginated by spine profiles. In the latter case the total and net MFT areas were the same. Finally, the number of mitochondria and spine profiles per MFT was also counted.
Synaptic vesicle density was estimated positioning an unbiased counting frame of known area (0.2 cm2) at the coordinates of a quadratic lattice superimposed on the micrographs containing the MFT. Vesicles were counted in the area of the terminals not associated with mitochondria or spines. Because mossy fibers from chronically stressed animals showed vesicles concentrated in clusters, the counting was performed within the limits of those aggregations because virtually no vesicles were found outside the boundaries of the clusters. The number of synaptic vesicles per unit area was adjusted to consider section thickness and lost caps by using a variation of Floderus’s equation (24):
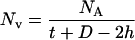 |
In this equation, Nv is the number of vesicles per unit volume, NA is the number of vesicles counted per unit area, and t is the average thickness of the ultrathin sections that was estimated by using the minimal fold technique (25). A mean section thickness of 65 nm was found. To estimate D, the mean vesicle diameter, a total of 10 MFT were randomly selected per experimental animal and the diameters of 50 vesicles per MFT was measured from prints at a final magnification of ×30,000. No significant stress effect was observed and the mean vesicle diameter was estimated to be 45 nm. Finally, h is a correction for lost caps (estimated to be 10% of the mean vesicle diameter).
Each variable was averaged across MFT to obtain a single mean value per animal and two-tailed unpaired Student’s t test was applied to assess statistical significance.
RESULTS
Repeated Restraint Stress Induces Ultrastructural Changes in Hippocampal MFT.
In accordance with previous descriptions (15, 26), the stratum lucidum of the hippocampus of unstressed rats contains unmyelinated mossy fibers with abundant boutons en passant (Fig. 2). At the ultrastructural level, the endings of these axons form the characteristic MFT (Fig. 3 A–C), containing numerous peripherally located mitochondria usually associated in groups. The net profile area of the terminals (minus mitochondria and spine profile areas) is tightly packed with small clear vesicles (40–50 nm in diameter) and contains only a few large dense core vesicles (70–150 nm in diameter). Giant spines or excrescences (see Fig. 2) from CA3 pyramidal neurons protrude into the MFT, and they establish multiple asymmetric synaptic contacts. A different type of membrane specialization that is nonsynaptic in nature because of its lack of association with synaptic vesicles [puncta adherentia (27)] is also found between the mossy fiber boutons and the CA3 apical dendritic shafts (Fig. 3).
Figure 2.
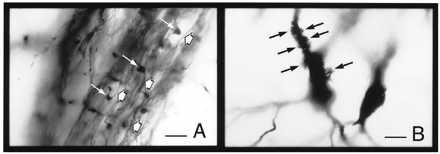
Pre- and postsynaptic elements of the mossy fiber–CA3 pyramidal neuron synapse in Golgi-impregnated material (for methodological details see ref. 13). (A) Mossy fiber axons (short arrows) and synaptic boutons en passant (long arrows) within the stratum lucidum. (B) Excrescences or complex spines in the proximal apical dendrites of a CA3 pyramidal neuron (arrows). (Bar = 30 μm.)
Figure 3.
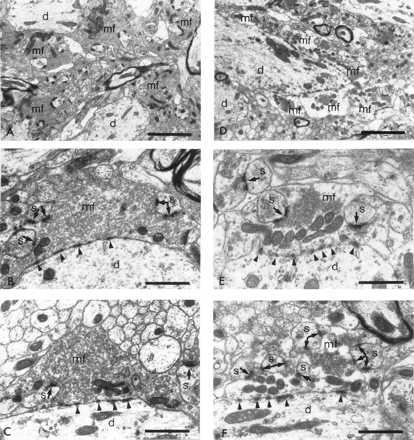
Ultrastructure of MFT (mf) in the stratum lucidum of the dorsal hippocampal CA3 region of control (A, B, and C) and restraint-stressed (D, E, and F) rats. (A) Control MFT filled with small clear vesicles are perforated with giant spines of CA3 proximal apical dendrites. (B and C) Higher magnification of two representative terminals from control nonstressed rats densely packed with small clear vesicles. The terminals form asymmetric synaptic contacts (arrows) with dendritic spines. Electron dense contacts with the dendritic shaft (d) are considered to be zones of adherence, (D) After 3 weeks of restraint stress MFT show a depletion of small clear vesicles. (E and F) Higher magnifications of MFT in stressed animals. Notice the compact clusters of vesicles accumulated near active zones. d, Dendritic shaft; mf, MFT; s, dendritic spine. [Bars = 2.5 μm (A and D) and 1 μm (B, C, E, and F).
In chronically stressed animals, an altered synaptic vesicle distribution pattern was observed along with an apparent depletion of vesicles. Compared with controls, the MFT of stressed animals were characterized by the presence of small clear vesicle clusters confined to restricted areas of the boutons. Most of the synaptic vesicle clusters were found in the vicinity of active zones (Fig. 3 D–F). Rats subjected to a single restraint stress that were killed either immediately after or the next day following the restraint session showed no differences in the ultrastructure of their MFT compared with control animals (Table 1). Moreover, we previously did not find any atrophy in CA3 apical dendrites after a single restraint stress (11).
Table 1.
Quantitation of ultrastructural variables in MFT in control, single restraint–stressed, and daily restraint–stressed rats for 21 days
| Morphological variable | Control | Single restraint
|
Repeated restraint
|
|
|---|---|---|---|---|
| Immediately after session | 16 h after session | 16 h after session 21 | ||
| MFT profile area, μm2 | 6.26 ± 0.37 | 6.02 ± 0.41 | 6.15 ± 0.35 | 6.43 ± 0.20 |
| Mitochondrial area, μm2 | 0.29 ± 0.01 | 0.30 ± 0.02 | 0.27 ± 0.03 | 0.44 ± 0.03* |
| Spine profile area, μm2 | 0.87 ± 0.06 | 0.90 ± 0.08 | 0.93 ± 0.09 | 0.91 ± 0.07 |
| Net MFT area, μm2 | 5.10 ± 0.33 | 4.97 ± 0.40 | 5.10 ± 0.49 | 5.08 ± 0.18 |
| Area covered by vesicles, μm2 | 5.05 ± 0.32 | 5.59 ± 0.45 | 5.01 ± 0.52 | 1.84 ± 0.15* |
| Number of mitochondria profiles/MFT profile | 8.29 ± 0.47 | 8.02 ± 0.51 | 8.25 ± 0.79 | 8.01 ± 0.48 |
| Number of spines/MFT profile | 3.67 ± 0.43 | 3.17 ± 0.32 | 3.41 ± 0.27 | 3.23 ± 0.23 |
| MFT perimeter occupied by active zones, % | 20.99 ± 0.49 | 19.80 ± 0.78 | 22.35 ± 0.90 | 21.13 ± 1.11 |
Animals perfused either immediately or 16 h after the single stress session showed similar quantitative results compared with controls. The area occupied by mitochondrial profiles and small clear vesicles was increased and decreased, respectively, in repeatedly stressed animals compared with controls (*P < 0.001, two-tailed unpaired Student’s t test).
Quantitative Morphology.
As shown in Table 1, the quantitative morphology of the postsynaptic elements of the MFT showed that the area occupied by spine profiles as well as the number of spine head profiles per MFT profile was not altered by repeated restraint stress. Moreover, the average total cross-sectional area of MFT in controls was similar to that in restraint stressed rats. Likewise, stress did not affect the number of mitochondrial profiles per MFT bouton (Table 1). However, the total area occupied by mitochondrial profiles was increased after the stress paradigm (P < 0.001, two-tailed unpaired Student’s t test). This result implies that the mitochondria were somewhat larger, on average, in MFT of stressed rats.
Moreover, stressed animals showed a significantly decreased area occupied by small clear vesicles (P < 0.01, two-tailed unpaired Student’s t test). In control animals, the area occupied by vesicles represented 99% of the net MFT area, whereas in stressed animals, only 36% of the net MFT area was occupied with vesicles.
The quantitation of vesicle density (no. of vesicles/μm3) revealed that stressed rats showed a higher packing density of synaptic vesicles within the regions in which they are clustered (Fig. 4, P < 0.001, two-tailed unpaired Student’s t test) compared with controls. Thus, although repeated stress causes a reduction in the area occupied by synaptic vesicles of around 60%, the increased packing density of the remained vesicles results in a net decrease in vesicles on the order of 40% (i.e., 16,984.16 ± 2,100.01 vesicles per mossy fiber profile in controls vs. 10,647.13 ± 999.34 in stressed rats, on average).
Figure 4.
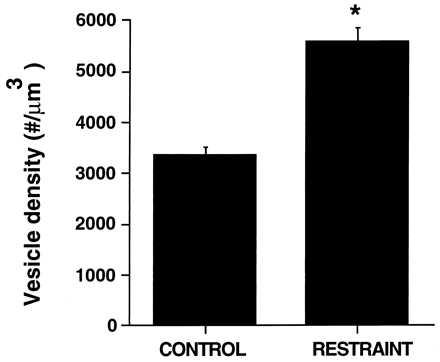
Effect of 3 weeks of daily restraint stress on vesicle density of MFT expressed as number of vesicles/μm3. The asterisk indicates P < 0.001 compared with controls (two-tailed unpaired Student’s t test). Bars represent means + SEM.
To determine whether the chronic restraint stress paradigm induced a change in the average length of active zones per terminal profile, we measured the percentage of MFT profile perimeter occupied by active zones. No significant differences were found between control and restraint-stressed rats (Table 1).
DISCUSSION
Repeated daily stress over 3 weeks induces a striking reorganization within MFT that involves the rearrangement of clear synaptic vesicles in the vicinity of the active zones between the mossy fiber boutons and the complex spines of CA3 pyramidal neurons. The stress-induced ultrastructural rearrangements were restricted to the presynaptic compartment of the MFT–CA3 pyramidal neuron synapse because no changes were observed in either the area occupied by, or the number of complex spine profiles embedded within, the mossy fiber boutons. A repeated stressful challenge is essential for the induction of MFT synaptic vesicle reorganization and clustering, just as it is required to cause atrophy of apical dendrites of CA3 pyramidal neurons, because we found that a single session of restraint stress failed to reorganize the synaptic apparatus of MFT or cause CA3 apical dendritic atrophy either immediately after or 1 day following the stress session. These findings provide a morphological marker of the effects of chronic stress on the hippocampus that points to underlying neuroanatomical as well as cellular and molecular mechanisms for the ability of repeated stress to cause structural changes within the hippocampus.
Neuroanatomical Relationships Between Morphological Changes Produced by Stress and Seizures.
Not only the MFT but their postsynaptic elements, the CA3 pyramidal neurons, are selectively targeted by repeated restraint stress. We previously reported that the apical dendritic tree of CA3 pyramidal neurons in rats and tree shrews, primitive primates, undergoes a decrease in the complexity of the branching of dendrites after chronic restraint stress and psychosocial stress (8, 13). Because the MFT reside on the proximal regions of the apical dendrites and are not reduced in number by chronic stress, the reduced number of CA3 apical dendritic branch points and total dendritic length observed in Golgi-impregnated material of repeatedly stressed rats and tree shrews might be interpreted as an adaptation to limit the enhanced excitatory input from recurrent axonal collaterals that are known to project from neighboring CA3 pyramidal neurons (17, 18). Not only is there a recurrent excitatory input between CA3 pyramidal neurons that can be activated by the mossy fiber system, pyramidal neurons in subregion CA3c that lies closest to the hilus send excitatory axons back to the dentate gyrus itself (see Introduction). Such a feedback loop can presumably reactivate the mossy fiber system and sustain CA3 excitation, as in the so-called SPW (sharp waves), and such an activation may drive the reorganization of vesicles within the MFT.
Collectively, the results from the present and previous studies support a model of mossy fiber function in which stressors such as restraint activate the release of excitatory amino acids from mossy fiber synapses and promote serotonin release and adrenal steroid secretion that synergize to enhance the efficacy of stressful events in the hippocampus. We previously showed that the CA3 apical dendritic atrophy appears to be the result of the activation of N-methyl-d-aspartate (NMDA) receptors (13), and that this may be a reflection of the wider activation of CA3 neurons by the excitatory axon collaterals noted above as well as the recurrent activation of the mossy fiber system. The role of serotonin is less clear, but a likely mechanism may involve its ability to enhance the activity of NMDA receptors via a second messenger system (28). As a reversible process, the atrophy or remodeling of dendrites may be caused by a depolymerization of the dendritic cytoskeleton resulting from increased intracellular calcium (29).
CA3 pyramidal neurons display a high vulnerability not only to chronic stress but also to kainic acid administration, an effect that requires the integrity of the mossy fiber pathway (30). As in the case of chronic restraint stress, CA3 region vulnerability is mediated by endogenous glucocorticoids and excitatory amino acid receptors (13, 31) and exogenous glucocorticoids have a potentiating effect (32, 33).
The CA3 hippocampal subregion is also damaged by epileptogenic stimulation of the perforant path, which involves the activation of the dentate gyrus-MFT-CA3 pathway (34). Furthermore, in the epilepsy model, another parallel exists with the chronic stress model in the clustering of synaptic vesicles. It has been reported that synaptic vesicle clustering occurs in MFT of genetically prone epileptic gerbils, and this effect could be totally blocked by the disruption of the perforant pathway (35).
Accumulating data has linked epilepsy with excessive excitatory input, resulting in morphological alterations at the synaptic level. Well-characterized animals models of epilepsy, such as kindling and genetically epileptic mice, show that sprouting of mossy fibers generating a recurrent excitatory circuit involving aberrant granule cell–granule cell synapses (36–38). Although we have not yet tested the possibility of mossy fiber sprouting after repeated restraint stress, other characteristics of the CA3 pyramidal cells connections such as their numerous recurrent collaterals and feedback to the dentate gyrus itself (see above and Introduction) could account for an amplification of the excitatory input, and therefore, for their extreme sensitivity to the stress paradigm.
The Cellular and Molecular Dynamics of Synaptic Vesicles in Excitatory Nerve Terminals.
The synaptic vesicle reorganization in MFT provides insights into possible cellular and molecular mechanisms of the effects of stress and stress mediators. MFT produce large amplitude miniature excitatory postsynaptic currents (39) and show a robust long-term potentiation (40). Neurotransmitter release occurs at discrete active zones containing readily releasable pools of synaptic vesicles; this process requires fusion of vesicles with the presynaptic plasma membrane, and this membrane is retrieved and recycled for subsequent transmitter release (41). In MFT from stressed rats, the redistribution of vesicles that we found and their localization near the active zones suggest that more vesicles are available for glutamate release. This notion is based on the idea that more vesicles from the reserve pool become competent to fuse with the presynaptic membrane and empty their contents by being released from the cytoskeletal matrix, although this possibility must be tested directly by electrophysiology and microdialysis.
Within the presynaptic compartment, synaptic vesicle-associated phosphoproteins play a key role in regulating the efficiency of the neurotransmitter release from the nerve terminal. In particular, synapsin I, in its dephosphorylated state, keeps the pool of “reserve” synaptic vesicles anchored to the nerve terminal cytoskeleton. Upon cell activation, Ca2+/calmodulin-dependent protein kinase II mediates the phosphorylation of synapsin I, and the reserve pool of vesicles is freed from the cytoskeletal matrix, becoming available for neurotransmitter release (42–45).
In the adult rodent brain, high levels of synapsin I protein and mRNA levels are present in the hippocampal MFT zone and granule cell neurons, respectively (46, 47). It is possible that the effect of repeated stress to cause clustering of vesicles close to the active zones could involve an increased expression of phosphorylated synapsin I. In fact, increases in synapsin I phosphorylation that correlate with enhanced neurotransmitter release have been observed in the hippocampus and parietal cortex of rats subjected to septal kindling, a model of synaptic plasticity that involves an increase in synaptic efficiency in response to repetitive local stimulations (48). Although changes in the levels of phosphorylated synapsin I seem to influence the ultrastructure of the presynaptic synaptic apparatus and, as a consequence, the efficiency of neurotransmitter release, brains of mice lacking the synapsin I gene show smaller MFT and a significant reduction in the number of synaptic vesicles that are not clustered but dispersed throughout the terminal (49).
Alternative Views of Synaptic Vesicle Rearrangement After Repeated Stress.
Because the synaptic vesicle recycling process is essential for the maintenance of a sustained neurotransmitter release in response to a repetitive stimulus, it could be argued that, rather than an increase in the efficiency of the neurotransmission, the relative depletion of clear synaptic vesicles within the MFT of chronically stressed animals could reflect a state of exhaustion, possibly caused by saturation of the recycling mechanism. In other words, the repetitive stimulation of MFT might result in reduced vesicle availability, so that the release of glutamate would become rate limiting.
This possibility seems unlikely for several reasons. First, in unpublished experiments, we observed that MFT of rats subjected to longer periods of daily restraint stress (namely, 5 weeks instead of the 3 weeks employed in the present work) show a similar vesicle clustering pattern after both durations of stress as well as an even greater apical dendritic atrophy of CA3 neurons at 5 weeks vs. 3 weeks, indicating a further progression of the atrophy process (A.M.M. and B.S.M., unpublished data). We note that MFT are equipped with a strikingly large reservoir of synaptic vesicles that could account for the ability of these terminals to readily respond to repetitive chronic stimuli and maintain that response. Second, the MFT area occupied by mitochondrial profiles is significantly increased after chronic stress, suggesting greater energy availability. Dynamic changes in mitochondrial volume have been shown to correlate with changes in cellular energy demands (50, 51).
Further evidence supports the hypothesis that stress may be increasing the efficiency of MFT glutamate release. Restraint stress is known to activate the release of excitatory amino acids in the hippocampus (52, 53), and this release is dependent, at least in part, on glucocorticoids because adrenalectomized animals show a reduced stress-induced excitatory amino acid release in the hippocampus (53–55). Furthermore, adrenal steroids regulate expression of the presynaptic kainate autoreceptor on mossy fiber terminals (56, 57), an effect that may enhance the positive feedback of glutamate release upon itself (58).
In conclusion, the application of repeated daily stress to adult male rats over 3 weeks results in a reorganization of the distribution of synaptic vesicles accompanied by both an increased packing density and a net depletion of vesicles. This finding provides a potentially informative endpoint for the effects of chronic stress on hippocampal morphology. Moreover, the association of synaptic vesicle clusters with active synaptic zones, packed more densely together, along with the increased terminal volume occupied by mitochondria, suggests as an interpretation that the mossy fiber synapses of repeatedly stressed animals may be more efficient. According to this interpretation, atrophy of dendrites would be a compensatory reaction to the elevated excitatory amino acid release. Future studies will be directed to address this hypothesis.
Acknowledgments
We thank Dr. Joseph Pierce (Cornell University College of Medicine) for his advice on quantitative stereology and his critical reading of the manuscript. We also acknowledge the excellent technical assistance of Alfonso Valverde Navarro. This research was supported by National Institutes of Health Grant MH 41256, the Health Foundation (New York), and Servier (France).
ABBREVIATION
- MFT
mossy fiber terminal(s)
References
- 1.McEwen B, Sapolsky R. Curr Opin Neurobiol. 1995;5:205–216. doi: 10.1016/0959-4388(95)80028-x. [DOI] [PubMed] [Google Scholar]
- 2.Bremner D J, Randall P, Scott T M, Bronen R A, Seibyl J P, Southwick S M, Delaney R C, McCarthy G, Charney D S, Innis R B. Am J Psychiatry. 1995;152:973–981. doi: 10.1176/ajp.152.7.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gurvits T V, Shenton M E, Hokama H, Ohta H, Lasko N B, Gilbertson M W, Orr S P, Kikinis R, Jolesz F A, McCarley R W, Pitman R K. Biol Psychiatry. 1996;40:1091–1099. doi: 10.1016/S0006-3223(96)00229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheline Y I, Wang P W, Gado M H, Csernansky J G, Vannier M W. Proc Natl Acad Sci USA. 1996;93:3908–3913. doi: 10.1073/pnas.93.9.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Starkman M N, Gebarski S S, Berent S, Schteingart D E. Biol Psychiatry. 1992;32:756–765. doi: 10.1016/0006-3223(92)90079-f. [DOI] [PubMed] [Google Scholar]
- 6.Golomb J, Kluger A, de Leon M J, Ferris S H, Convit A, Mittelman M S, Cohen J, Rusinek H, De Santi S, George A E. Learn Mem. 1994;1:45–54. [PubMed] [Google Scholar]
- 7.Uno H, Ross T, Else J, Suleman M, Sapolsky R. J Neurosci. 1989;9:1705–1711. doi: 10.1523/JNEUROSCI.09-05-01705.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magariños A M, McEwen B S, Flügge G, Fuchs E. J Neurosci. 1996;16:3534–3540. doi: 10.1523/JNEUROSCI.16-10-03534.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKittrick C R, Magariños A M, Blanchard D C, Blanchard R J, McEwen B S, Sakai R R. Soc Neurosci Abstr. 1996;22:2060. [Google Scholar]
- 10.Watanabe Y, Gould E, McEwen B S. Brain Res. 1992;588:341–345. doi: 10.1016/0006-8993(92)91597-8. [DOI] [PubMed] [Google Scholar]
- 11.Luine V, Martinez C, Villegas M, Magariños A M, McEwen B S. Physiol Behav. 1996;59:27–32. doi: 10.1016/0031-9384(95)02016-0. [DOI] [PubMed] [Google Scholar]
- 12.Conrad C D, Galea L, Kuroda Y, McEwen B S. Behav Neurosci. 1996;110:1321–1334. doi: 10.1037//0735-7044.110.6.1321. [DOI] [PubMed] [Google Scholar]
- 13.Magariños A M, McEwen B S. Neuroscience. 1995;69:89–98. doi: 10.1016/0306-4522(95)00259-l. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe Y, Gould E, Daniels D, Cameron H, McEwen B S. Eur J Pharmacol. 1992c;588:341–345. doi: 10.1016/0014-2999(92)90830-w. [DOI] [PubMed] [Google Scholar]
- 15.Blackstad T W, Kjarheim A. J Comp Neurol. 1961;11:133–146. doi: 10.1002/cne.901170202. [DOI] [PubMed] [Google Scholar]
- 16.Storm-Mathisen J, Leknes A K, Bore A T, Vaaland J L, Edminson P, Haug F S, Ottersen O P. Nature (London) 1983;301:517–520. doi: 10.1038/301517a0. [DOI] [PubMed] [Google Scholar]
- 17.Ishizuka N, Weber J, Amaral D G. J Comp Neurol. 1990;295:580–623. doi: 10.1002/cne.902950407. [DOI] [PubMed] [Google Scholar]
- 18.Li X G, Somogy P, Ylinen A, Buzsaki G. J Comp Neurol. 1994;339:181–208. doi: 10.1002/cne.903390204. [DOI] [PubMed] [Google Scholar]
- 19.Kneisler T B, Dingledine R. J Physiol (London) 1995;481:125–146. doi: 10.1113/jphysiol.1995.sp020866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scharfman H E. J Neurophysiol. 1994;72:2167–2180. doi: 10.1152/jn.1994.72.5.2167. [DOI] [PubMed] [Google Scholar]
- 21.Buzsaki G. Brain Res. 1986;398:242–252. doi: 10.1016/0006-8993(86)91483-6. [DOI] [PubMed] [Google Scholar]
- 22.Ylinen A, Bragin A, Nadasdy Z, Jando G, Szabo I, Sik A, Buzsaki G. J Neurosci. 1995;15:30–46. doi: 10.1523/JNEUROSCI.15-01-00030.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Penttonen M, Kamondi A, Sik A, Acsady L, Buzsaki G. Hippocampus. 1997;7:437–450. doi: 10.1002/(SICI)1098-1063(1997)7:4<437::AID-HIPO9>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 24.Pierce J P, Mendell L M. J Neurosci. 1993;13:4748–4763. doi: 10.1523/JNEUROSCI.13-11-04748.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Small J V. In: Proceedings of the Fourth European Congress Electron Microscopy. Bocciarelli, Daria, Steve, editors. Rome: Tipografia Poliglotta Vaticana; 1968. p. 609. [Google Scholar]
- 26.Amaral D G, Dent J A. J Comp Neurol. 1981;195:51–86. doi: 10.1002/cne.901950106. [DOI] [PubMed] [Google Scholar]
- 27.Peters A, Palay S L, Webster H d. The Fine Structure of the Nervous System: Neurons and Their Supporting Cells. New York: Oxford Univ. Press; 1991. [Google Scholar]
- 28.Rahman S, Neuman R. Eur J Pharmacol. 1993;231:347–354. doi: 10.1016/0014-2999(93)90109-u. [DOI] [PubMed] [Google Scholar]
- 29.Elliott E M, Mattson M P, Vanderklish P, Lynch G, Chang I, Sapolsky R M. J Neurochem. 1993;61:57–67. doi: 10.1111/j.1471-4159.1993.tb03537.x. [DOI] [PubMed] [Google Scholar]
- 30.Nadler J V, Cuthbertson G J. Brain Res. 1980;195:47–56. doi: 10.1016/0006-8993(80)90865-3. [DOI] [PubMed] [Google Scholar]
- 31.Armanini M, Hutchins C, Stein B, Sapolsky R M. Brain Res. 1990;532:7–14. doi: 10.1016/0006-8993(90)91734-x. [DOI] [PubMed] [Google Scholar]
- 32.Sapolsky R M. J Neurosci. 1986;6:2240–2244. doi: 10.1523/JNEUROSCI.06-08-02240.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Packan D, Sapolsky R M. Neuroendocrinology. 1990;51:613–618. doi: 10.1159/000125400. [DOI] [PubMed] [Google Scholar]
- 34.Sloviter R S. Brain Res Bull. 1993;10:675–697. doi: 10.1016/0361-9230(83)90037-0. [DOI] [PubMed] [Google Scholar]
- 35.Farias P A, Low S Q, Peterson G M, Ribak C E. Hippocampus. 1992;2:229–245. doi: 10.1002/hipo.450020304. [DOI] [PubMed] [Google Scholar]
- 36.Okazaki M M, Evenson D A, Nadler J V. J Comp Neurol. 1995;352:515–534. doi: 10.1002/cne.903520404. [DOI] [PubMed] [Google Scholar]
- 37.Sutula T, Koch J, Golarai G, Watanabe Y, McNamara J O. J Neurosci. 1996;16:7398–7406. doi: 10.1523/JNEUROSCI.16-22-07398.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parent J M, Yu T W, Leibowitz R T, Geschwind D H, Sloviter R S, Lowenstein D H. J Neurosci. 1997;17:3727–3728. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henze D A, Card J P, Barrionuevo G, Ben-Ari Y. J Neurophysiol. 1997;77:1075–1086. doi: 10.1152/jn.1997.77.3.1075. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto C, Chujo T. Exp Neurol. 1978;58:242–250. doi: 10.1016/0014-4886(78)90137-1. [DOI] [PubMed] [Google Scholar]
- 41.Bajjalieh S M, Scheller R H. J Biol Chem. 1995;270:1971–1974. doi: 10.1074/jbc.270.5.1971. [DOI] [PubMed] [Google Scholar]
- 42.Llinas R, McGuinness T L, Leonard C S, Sugimori M, Greengard P. Proc Natl Acad Sci USA. 1985;82:3035–3039. doi: 10.1073/pnas.82.9.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greengard P, Valtorta F, Czernik A J, Benfenati F. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- 44.Ceccaldi P E, Grohovaz F, Benfenati F, Chieregatti E, Greengard P, Valtorta F. J Cell Biol. 1995;128:905–912. doi: 10.1083/jcb.128.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pieribone V A, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernic A J, Greengard P. Nature (London) 1996;375:493–496. doi: 10.1038/375493a0. [DOI] [PubMed] [Google Scholar]
- 46.De Camilli P, Greengard P. Biochem Pharmacol. 1986;35:4349–4357. doi: 10.1016/0006-2952(86)90747-1. [DOI] [PubMed] [Google Scholar]
- 47.Melloni R H J, Hemmendinger L M, Hamos J E, DeGennaro L J. J Comp Neurol. 1993;327:507–520. doi: 10.1002/cne.903270404. [DOI] [PubMed] [Google Scholar]
- 48.Yamagata Y, Obata K, Greengard P, Czernik A J. Neuroscience. 1995;64:1–4. doi: 10.1016/0306-4522(94)00492-n. [DOI] [PubMed] [Google Scholar]
- 49.Takei Y, Harada A, Takeda S, Kobayashi K, Terada S, Noda T, Takahashi T, Kirokawa N. J Cell Biol. 1995;131:1789–1800. doi: 10.1083/jcb.131.6.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith R, Ord M J. Int Rev Cytol. 1983;83:63–164. doi: 10.1016/s0074-7696(08)61686-1. [DOI] [PubMed] [Google Scholar]
- 51.Lnenicka G A, Atwood H L, Marin L. J Neurosci. 1986;6:2252–2258. doi: 10.1523/JNEUROSCI.06-08-02252.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moghaddam B. J Neurochem. 1993;60:1650–1657. doi: 10.1111/j.1471-4159.1993.tb13387.x. [DOI] [PubMed] [Google Scholar]
- 53.Lowy M T, Gault L, Yamamoto B K. J Neurochem. 1993;61:1957–1960. doi: 10.1111/j.1471-4159.1993.tb09839.x. [DOI] [PubMed] [Google Scholar]
- 54.Krugers H J, Jaarsma D, Korf J. J Neurochem. 1992;58:826–830. doi: 10.1111/j.1471-4159.1992.tb09331.x. [DOI] [PubMed] [Google Scholar]
- 55.Moghaddam B, Bolinao M L, Stein-Beherens B, Sapolsky R M. Brain Res. 1994;655:251–4. doi: 10.1016/0006-8993(94)91622-5. [DOI] [PubMed] [Google Scholar]
- 56.Watanabe Y, Weiland N G, McEwen B S. Brain Res. 1995;680:217–225. doi: 10.1016/0006-8993(95)00235-i. [DOI] [PubMed] [Google Scholar]
- 57.Joels M, Bosma A, Hendriksen H, Diegenbach P, Kamphuis W. Mol Brain Res. 1996;37:15–20. doi: 10.1016/0169-328x(95)00267-v. [DOI] [PubMed] [Google Scholar]
- 58.Chittajallu R, Vignes M, Dev K K, Barnes J M, Collingridge G L, Henley J M. Nature (London) 1996;379:78–81. doi: 10.1038/379078a0. [DOI] [PubMed] [Google Scholar]