

1. Introduction
Alzheimer’s disease is defined pathologically by the presence of senile plaques, which consist primarily of extracellular aggregates of fibrillar Aβ peptide, and neurofibrillary tangles, which are abnormal, intracellular bundles of fibrillar tau protein. The advent of amyloid binding agents as diagnostic imaging probes for Alzheimer’s disease (AD) has made it imperative to understand at a molecular and disease level what these ligands are reporting. In addition to improving the accuracy of diagnosis, we argue that these selective ligands can serve as probes for molecular polymorphisms that may govern the pathogenicity of abnormal protein aggregates.
2. There is a disconnect between amyloid pathology and neurodegeneration/clinical symptoms
A definite histopathologic diagnosis of AD requires the presence of senile plaques. Whether they are directly linked to neurodegeneration or are an epiphenomenon of the pathogenic process, the presence of plaques must be explained by any proposed disease mechanism. Their utility as biomarkers, though, is hampered by the weak correlation of the amount and regional distribution of Aβ deposition with the extent of neurodegeneration that results in functional deficits [5,8,50]. Although some investigators interpret the poor correlation to mean that the senile plaques are secondary to the essential disease process, a great deal of genetic and biochemical data indicate that Aβ is a prime-mover in the pathogenic cascade [12]. The amended Amyloid Cascade Hypothesis (perhaps better designated the Aβ Cascade Hypothesis) holds that soluble, multimeric forms of misfolded Aβ (i.e., Aβ-oligomers) are the most toxic species, whereas insoluble fibrils, which comprise the bulk of senile plaque cores, serve as either a less toxic depot or as a sink in equilibrium with monomers and/or oligomers.
3. Animal models have substantial Aβ deposition but relatively little neurodegeneration
Any postulated Alzheimer’s disease mechanism must be reconciled with the stereotypical Aβ deposition disease hallmarks. It is critical to determine the association of Aβ deposits with the disease to validate the utility of imaging senile plaques in diagnosing AD and/or monitoring disease progression. Nonhuman species have proven to be only partial models of AD. Although APP-transgenic mice, nonhuman primates, and dogs all manifest age-related CNS Aβ deposition that is similar in distribution and quantity to that in humans with AD, these models lack the massive neurodegeneration and neurofibrillary tangles of AD [9,29]. Is it possible that the Aβ deposits in nonhuman animals are structurally different on a molecular/conformational level from AD Aβ deposits, and is this difference directly or indirectly associated with disease mechanism? Understanding the molecular diversity of Aβ multimers could provide important clues to the pathogenesis of AD, and could thereby suggest new therapeutic approaches to the disorder.
4. When is a misfolded protein deposit pathologic?
Insoluble protein deposits have classically been characterized by the presence or absence of fibrillar morphology and a condensed core, as well as by their staining properties with amyloid dyes such as Congo Red (Figure 1 [6]) and the Thioflavines (T and S, Figure 1 [1, 5]). Similar characteristics have been observed in deposits generated by animal models of Aβ deposition, yet neuronal loss is minimal in these instances, suggesting that some as yet unidentified property of the multimeric protein renders the molecule uniquely pathogenic in humans.
Figure 1.
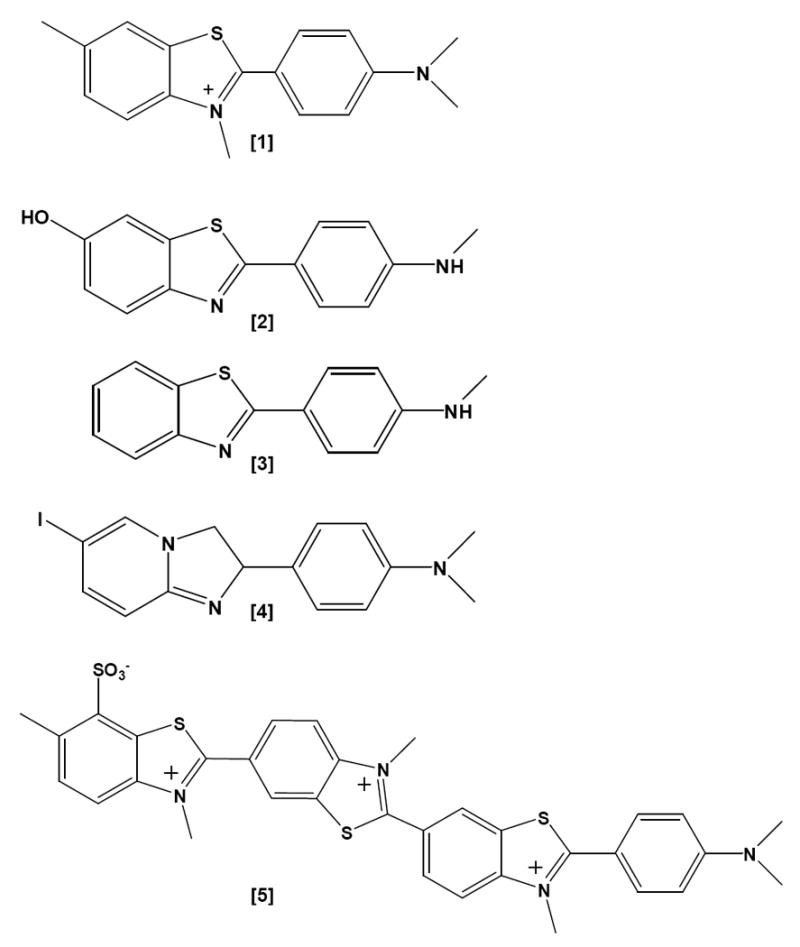
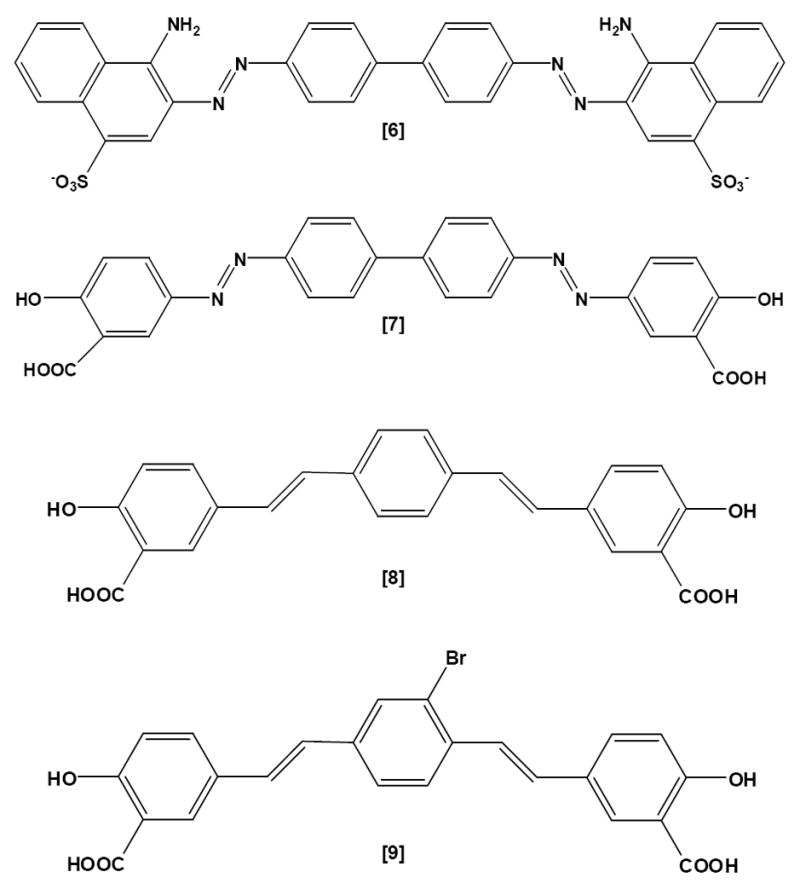
-
[1]Thioflavine T
-
[2]PIB1
-
[3]BTA-1
-
[4]IMPY
-
[5]Thioflavine S (mixture)
-
[6]Congo Red
-
[7]Chrysamine G
-
[8]X-34
-
[9]BSB
The prion diseases are the best characterized example of how conformational variants of the same primary amino acid sequence can lead to different disease phenotypes. During the pathogenesis of prionosis, the normal cellular conformation of the prion protein (PrPC) is converted into β-sheet-rich, pathogenic conformations (PrPSc) that are resistant to denaturation and proteolysis. Distinct polymorphic forms of this multimeric PrPSc, also called strains, are differently folded molecules that present pathologically different phenotypes and morphologies [1,7]. Diverse structural variants of prions also can be generated in vitro from the same amino acid sequence and starting conformation of prion protein by adjusting the assembly conditions (temperature, pH, agitation) [30,46]. Each strain of prion protein tends to elicit a particular lesion phenotype and clinical course of the individual prion diseases that is dependent on characteristics of both the host and the protein [44,47,48].
5. Polymorphic forms of Aβ
Although the story is not as fully developed for Aβ, polymorphic forms of Aβ peptide fibrils also have been produced in vitro [40,49,52]. Distinct folded structures of two forms differing in hydrogen bonding across the β-turn between residues 25 and 28 have been determined by solid state NMR [40,41]. There is also evidence that pH similar to that in endosomal compartments influences epitope exposure in residues 8-14 of soluble monomeric Aβ peptide, reflecting a distinct, partially folded conformation [27,34]. In vivo, heterogeneous Aβ [3,51] deposits suggestive of Aβ strains have been induced by different exogenous Aβ seeds in APP-transgenic mice [35]. The molecular relationship between any of these molecular forms and the Aβ fibrils and oligomers found in AD brain remains to be determined. Another important question, given the strong correlation between neurofibrillary tangles and degree of dementia in AD [5,8,50], is whether certain Aβ strains might preferentially promote the development of neurofibrillary tangles.
Polymorphism in the molecular structure of Aβ multimers is a potential explanation for the observation that only humans have the severe neuronal loss, neurofibrillary tangles and cognitive decline of AD, and nonhuman species do not. Strain differences might also account for the fact that some cognitively normal humans have large amounts of cerebral Aβ [4], and for other features of the disorder such as inflammation and cerebral β-amyloid angiopathy (CAA) [3,56]. Furthermore, morphological and distributional variants of Aβ deposits coexist within an individual AD brain [51]. These lesions include dense-core senile plaques, CAA, and a multiplicity of diffuse, granular and ribbon-like deposits [13,45,51,53]. Whether these morphologically diverse lesions are the supramolecular manifestations of different Aβ strains remains to be established.
6. Recognizing Aβ Polymorphisms
Classical methods for detecting structural differences in cerebral Aβ deposits do not make fine distinctions at the molecular level. Widely used Aβ sequence-specific antibodies such as 6E10 and 4G8, and techniques such as amyloidotrophic Thioflavine- and Congo Red-staining are of limited utility in distinguishing different types of amyloid deposits. Conformation-specific antibodies that distinguish amyloid fibrils [18,26,36,39] and oligomers are not primary amino acid sequence-dependent (except see [26,36]). However, antibodies that distinguish among fibrillar Aβ strains, similar to those available for PrP, are not yet available. Some of the anti-oligomer antibodies may indeed recognize conformational variants, notably the A11, I11 and OC (officer) antibodies [17,26,55].
Putative structural varieties of Aβ can be distinguished biochemically by differential extraction of AD brain tissue. As AD progresses, it is possible to demonstrate characteristic shifts of Aβ into increasingly insoluble multimers, beginning with a buffer-soluble state, followed by alkali-soluble, SDS-soluble, and finally requiring formic acid to solubilize the peptide. Similar distributions of peptide solubility also are found in the animal models [16,25,37].
A series of fluorescent polythiophene anions and cations have recently been described [6] that report on the spatial (molecular?) heterogeneity of multimeric Aβ. Under appropriate conditions these agents bind the stacks of assembled β-sheets in Aβ and other amyloid fibrils in a sequence-independent fashion [2]. Their fluorescence emission spectra reflect intra- and inter-polythiophene chain contacts which shift to green for tightly packed, regularly oriented fibrils and to red for less organized, less compact fibrils [38]. Synthetic Aβ-amyloid fibrils formed under quiescent conditions fluoresce green with these reagents, while fibrils formed with agitation emit red. These compounds also recognize molecular heterogeneity in Aβ deposits formed in vivo [34]. The fibrillar Thioflavine-positive amyloid deposits in Tg APP/PS1 mice and AD brain are mosaics of the two fibril conformations, indicating that individual fibril deposits consist of polymorphic domains [38]. These compact lesions also are distinct from diffuse or non-fibrillar deposits, which are Thioflavine- and Congo Red-negative. Structural evidence supporting the potential for differential fibril organization comes from solid state NMR, which shows that the synthetic Aβ peptide folds differently in fibrils prepared under quiescent vs. agitated conditions [40].
7. Amyloid probes and polymorphic Aβ
A probe of fibrillar Aβ, the N- 11CH3-labeled benzothiazole ligand 6-OH-BTA-1 (Pittsburgh compound B; 11C-PIB) (Figure 1 [2]), a brain-penetrant analog of BTA-1 (Figure 1, [3]), is retained in Aβ-rich brain regions of living MCI and AD subjects at a high affinity binding site, but it washes rapidly out of unaffected regions or appropriate control brains [20]. Positron-Emission-Tomography (PET) images of these subjects can be processed into a map of putative Aβ-deposition in the living brain. Neurofibrillary tangles (hyperphosphorylated tau) [22] and Lewy Bodies (α-synuclein) [10], which also contain β-sheet structure, have low affinity for PIB, so they are not expected to contribute significantly to the signal at imaging concentrations of PIB used in AD subjects. Frontotemporal dementia (a primary tauopathy) gives detectable, but much weaker, PIB signals in some subjects [42], but the retention of label is distinguishable by its regional distribution. The possible contribution of a small amount of Aβ plaque pathology in these cases has not been excluded. PIB uptake is increased in disease-relevant brain regions in MCI subjects [11,19]. CAA, which is found to varying degrees in most AD subjects, also binds PIB [15]. When 3H-PIB is applied to AD histological sections, dense-core plaques, CAA and some diffuse Aβ deposits bind the agent, but NFTs do not [14,31]. Studies are underway to determine whether PIB retention can be used to follow the progression of AD and whether it will be useful for monitoring the effectiveness of therapeutic interventions.
8. PIB detects differences between animal models and AD
The sulfonated Congo Red [6] and Thioflavine S [5] dyes are used routinely to analyze AD brain pathology. Congo Red and its congeners (Chrysamine G, X-34, BSB; Figure 1 [7-9]), as well as Thioflavine S, bind to sites distinct from the high affinity site that is occupied by PIB [32,54]. In fibrils made with synthetic Aβ1-40 or Aβ1-42 peptide, and in plaque-rich transgenic mouse and AD brain, Congo Red binds fibrils in a 1:1 stoichiometry with Aβ [21,28]. PIB binding may be more selective on a molecular level than are Congo Red and Thioflavine S. The Klunk group made the seminal observation that when 11C-PIB of the same specific radioactivity and concentration (~nM) used for human studies was used to image APP/PS1 transgenic mice (Tg2576 X M146L-PS1) with an Aβ plaque load equal to or exceeding that in AD brain, the plaques did not bind enough radioligand to produce an image [21]. However, the murine Aβ plaque pathology was revealed by staining sections with 100 nM 6-CN-BTA-1 (a close analog of PIB [6-OH-BTA-1]), X-34, or with anti-Aβ antibodies. Subsequent quantitative binding studies comparing brain homogenates from AD, non-demented humans, and plaque-bearing transgenic mice revealed that the transgenic mice had a greatly reduced number of high affinity (nM) PIB binding sites per mole of Aβ peptide (< 1:100 compared to 1:2 in AD). These high affinity sites would be the only sites significantly labeled at the concentrations of 11C-PIB usually achieved in vivo, hence the weak signal in transgenic mice. The high affinity binding stoichiometry was even lower (1:370) on fibrils of Aβ1-40 or Aβ1-42 formed from synthetic peptide [21]. The structural basis at the molecular level for these differences remains to be elucidated.
9. Implications
An intriguing possibility is that the high affinity PIB binding site represents a molecular polymorph of Aβ that constitutes a high proportion of deposits in AD brain, but a low proportion in APP transgenic mouse brain and in fibrils of synthetic peptide. Support for this hypothesis comes from imaging studies with ten-fold higher specific activity 11C-PIB, which is sufficient to detect the lower percentage of high affinity binding sites in the APP23 Tg mouse and in histological sections of Tg2576 and Tg2576 X A260V-PS1 brains [33]. In vivo binding, homogenate binding, and autoradiographic studies of the Tg2576 X M146L-PS1 mice with 125I-IMPY ([4], an analog of PIB) of similar high specific radioactivity also detect a low abundance, high affinity binding site that is associated with plaques and cerebrovascular amyloid fibrils, but only minimally with neurofibrillary tangles [23,24].
Whether the material in brain that is selectively labeled by PIB is a cause or effect of the pathogenic process in AD, it appears to be a marker for an aspect of the disease that is only minimally recapitulated by synthetic peptides and current APP transgenic mouse models. Aged nonhuman primates and dogs naturally deposit human-sequence Aβ in the brain and display some behavioral dysfunction, but, like APP-transgenic mice, they do not exhibit extensive neuronal loss or dementia. Our preliminary studies of aged nonhuman primates with heavy Aβ-amyloidosis [43] support the hypothesis that PIB recognizes an enriched human molecular morphotype of Aβ. PIB binding, along with other new molecular probes, could be useful tools for assessing efforts to expand current models beyond recreating Alzheimer’s histopathology to more accurately phenocopying Alzheimer’s disease.
Acknowledgments
Support is acknowledged from the Woodruff Foundation, NIH RR-00165, P01 AG026423-01A2, and by the Sanders-Brown Center on Aging and Chandler Medical Center of the University of Kentucky.
Footnotes
The authors declare that there are no actual or potential conflicts of interest for the views expressed in this manuscript
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8(7):552–61. doi: 10.1038/nrm2204. [DOI] [PubMed] [Google Scholar]
- 2.Aslund A, Herland A, Hammarstrom P, Nilsson KP, Jonsson BH, Inganas O, Konradsson P. Studies of Luminescent Conjugated Polythiophene Derivatives: Enhanced Spectral Discrimination of Protein Conformational States. Bioconjug Chem. 2007 doi: 10.1021/bc700180g. [DOI] [PubMed] [Google Scholar]
- 3.Attems J, Lintner F, Jellinger KA. Amyloid beta peptide 1-42 highly correlates with capillary cerebral amyloid angiopathy and Alzheimer disease pathology. Acta Neuropathol (Berl) 2004;107(4):283–91. doi: 10.1007/s00401-004-0822-6. [DOI] [PubMed] [Google Scholar]
- 4.Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, Wilson RS. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66(12):1837–44. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 5.Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL, Perl DP. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch Neurol. 1995;52(1):81–8. doi: 10.1001/archneur.1995.00540250089017. [DOI] [PubMed] [Google Scholar]
- 6.Chandra R, Kung MP, Kung HF. Design, synthesis, and structure-activity relationship of novel thiophene derivatives for beta-amyloid plaque imaging. Bioorg Med Chem Lett. 2006;16(5):1350–2. doi: 10.1016/j.bmcl.2005.11.055. [DOI] [PubMed] [Google Scholar]
- 7.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318(5852):930–6. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 8.Crystal H, Dickson D, Fuld P, Masur D, Scott R, Mehler M, Masdeu J, Kawas C, Aronson M, Wolfson L. Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology. 1988;38(11):1682–7. doi: 10.1212/wnl.38.11.1682. [DOI] [PubMed] [Google Scholar]
- 9.Eriksen JL, Janus CG. Plaques, tangles, and memory loss in mouse models of neurodegeneration. Behavior genetics. 2007;37(1):79–100. doi: 10.1007/s10519-006-9118-z. [DOI] [PubMed] [Google Scholar]
- 10.Fodero-Tavoletti MT, Smith DP, McLean CA, Adlard PA, Barnham KJ, Foster LE, Leone L, Perez K, Cortes M, Culvenor JG, Li QX, Laughton KM, Rowe CC, Masters CL, Cappai R, Villemagne VL. In vitro characterization of Pittsburgh compound-B binding to Lewy bodies. J Neurosci. 2007;27(39):10365–71. doi: 10.1523/JNEUROSCI.0630-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forsberg A, Engler H, Almkvist O, Blomquist G, Hagman G, Wall A, Ringheim A, Langstrom B, Nordberg A. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 12.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 13.Ikeda S, Allsop D, Glenner GG. Morphology and distribution of plaque and related deposits in the brains of Alzheimer’s disease and control cases. An immunohistochemical study using amyloid beta-protein antibody. Lab Invest. 1989;60(1):113–22. [PubMed] [Google Scholar]
- 14.Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, Dekosky ST. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008 doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK, Smith EE, Rosand J, Rentz DM, Klunk WE, Mathis CA, Price JC, Dekosky ST, Fischman AJ, Greenberg SM. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol. 2007 doi: 10.1002/ana.21164. [DOI] [PubMed] [Google Scholar]
- 16.Kalback W, Watson MD, Kokjohn TA, Kuo YM, Weiss N, Luehrs DC, Lopez J, Brune D, Sisodia SS, Staufenbiel M, Emmerling M, Roher AE. APP transgenic mice Tg2576 accumulate Abeta peptides that are distinct from the chemically modified and insoluble peptides deposited in Alzheimer’s disease senile plaques. Biochemistry. 2002;41(3):922–8. doi: 10.1021/bi015685+. [DOI] [PubMed] [Google Scholar]
- 17.Kayed R, Glabe CG. Conformation-dependent anti-amyloid oligomer antibodies. Methods Enzymol. 2006;413:326–44. doi: 10.1016/S0076-6879(06)13017-7. [DOI] [PubMed] [Google Scholar]
- 18.Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, Glabe CG. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. 2007;2(1):18. doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kemppainen NM, Aalto S, Wilson IA, Nagren K, Helin S, Bruck A, Oikonen V, Kailajarvi M, Scheinin M, Viitanen M, Parkkola R, Rinne JO. PET amyloid ligand [11C]PIB uptake is increased in mild cognitive impairment. Neurology. 2007;68(19):1603–6. doi: 10.1212/01.wnl.0000260969.94695.56. [DOI] [PubMed] [Google Scholar]
- 20.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55(3):306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 21.Klunk WE, Lopresti BJ, Debnath ML, Holt DP, Wang Y, Huang G-F, Shao L, Lefterov I, Koldamova R, Ikonomovic M, DeKosky ST, Mathis CA. Amyloid deposits in transgenic PS1/APP mice do not bind the amyloid PET tracer, PIB, in the same manner as human brain amyloid. Neurobiology of Aging. 2004;25(S2):232. [Google Scholar]
- 22.Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Shao L, Hamilton RL, Ikonomovic MD, DeKosky ST, Mathis CA. The binding of 2-(4’-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci. 2003;23(6):2086–92. doi: 10.1523/JNEUROSCI.23-06-02086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kung MP, Hou C, Zhuang ZP, Cross AJ, Maier DL, Kung HF. Characterization of IMPY as a potential imaging agent for beta-amyloid plaques in double transgenic PSAPP mice. Eur J Nucl Med Mol Imaging. 2004 doi: 10.1007/S00259-004-1487-z. [DOI] [PubMed] [Google Scholar]
- 24.Kung MP, Hou C, Zhuang ZP, Zhang B, Skovronsky D, Trojanowski JQ, Lee VM, Kung HF. IMPY: an improved thioflavin-T derivative for in vivo labeling of beta-amyloid plaques. Brain Res. 2002;956(2):202–10. doi: 10.1016/s0006-8993(02)03436-4. [DOI] [PubMed] [Google Scholar]
- 25.Kuo YM, Kokjohn TA, Beach TG, Sue LI, Brune D, Lopez JC, Kalback WM, Abramowski D, Sturchler Pierrat C, Staufenbiel M, Roher AE. Comparative analysis of amyloid-beta chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer’s disease brains. J Biol Chem. 2001;276(16):12991–8. doi: 10.1074/jbc.M007859200. [DOI] [PubMed] [Google Scholar]
- 26.Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, Khuon D, Gong Y, Bigio EH, Shaw P, De Felice FG, Krafft GA, Klein WL. Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem. 2007;100(1):23–35. doi: 10.1111/j.1471-4159.2006.04157.x. [DOI] [PubMed] [Google Scholar]
- 27.LeVine H., III Y10W beta(1-40) fluorescence reflects epitope exposure in conformers of Alzheimer’s beta-peptide. Arch Biochem Biophys. 2003;417(1):112–22. doi: 10.1016/s0003-9861(03)00322-9. [DOI] [PubMed] [Google Scholar]
- 28.LeVine H., III Multiple ligand binding sites on A beta(1-40) fibrils. Amyloid. 2005;12(1):5–14. doi: 10.1080/13506120500032295. [DOI] [PubMed] [Google Scholar]
- 29.LeVine H, III, Walker LC. Chapter 11. Models of Alzheimer’s Disease. In: Conn MP, editor. Handbook of Models for Human Aging. New York, NY: Academic Press/Elsevier; 2006. pp. 121–34. [Google Scholar]
- 30.Lim KH, Nguyen TN, Damo SM, Mazur T, Ball HL, Prusiner SB, Pines A, Wemmer DE. Solid-state NMR structural studies of the fibril form of a mutant mouse prion peptide PrP89-143(P101L) Solid state nuclear magnetic resonance. 2006;29(13):183–90. doi: 10.1016/j.ssnmr.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 31.Lockhart A, Lamb JR, Osredkar T, Sue LI, Joyce JN, Ye L, Libri V, Leppert D, Beach TG. PIB is a non-specific imaging marker of amyloid-beta (A{beta}) peptide-related cerebral amyloidosis. Brain. 2007 doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- 32.Lockhart A, Ye L, Judd DB, Merritt AT, Lowe PN, Morgenstern JL, Hong G, Gee AD, Brown J. Evidence for the presence of three distinct binding sites for the thioflavin T class of Alzheimer’s disease PET imaging agents on beta-amyloid peptide fibrils. J Biol Chem. 2005;280(9):7677–84. doi: 10.1074/jbc.M412056200. [DOI] [PubMed] [Google Scholar]
- 33.Maeda J, Ji B, Irie T, Tomiyama T, Maruyama M, Okauchi T, Staufenbiel M, Iwata N, Ono M, Saido TC, Suzuki K, Mori H, Higuchi M, Suhara T. Longitudinal, quantitative assessment of amyloid, neuroinflammation, and anti-amyloid treatment in a living mouse model of Alzheimer’s disease enabled by positron emission tomography. J Neurosci. 2007;27(41):10957–68. doi: 10.1523/JNEUROSCI.0673-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsunaga Y, Saito N, Fujii A, Yokotani J, Takakura T, Nishimura T, Esaki H, Yamada T. A pH-dependent conformational transition of Abeta peptide and physicochemical properties of the conformers in the glial cell. Biochem J. 2002;361(Pt 3):547–56. doi: 10.1042/0264-6021:3610547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313(5794):1781–4. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 36.Miller DL, Currie JR, Mehta PD, Potempska A, Hwang YW, Wegiel J. Humoral Immune Response to Fibrillar beta-Amyloid Peptide. Biochemistry. 2003;42(40):11682–92. doi: 10.1021/bi030100s. [DOI] [PubMed] [Google Scholar]
- 37.Murphy MP, Beckett TL, Ding Q, Patel E, Markesbery WR, Clair DKS, LeVine H, Keller JN. Aβ solubility and deposition during AD progression and in APPXPS1 Knock-in mice. Neurobiology of Disease. 2007 doi: 10.1016/j.nbd.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 38.Nilsson KP, Aslund A, Berg I, Nystrom S, Konradsson P, Herland A, Inganas O, Stabo-Eeg F, Lindgren M, Westermark GT, Lannfelt L, Nilsson LN, Hammarstrom P. Imaging distinct conformational states of amyloid-beta fibrils in Alzheimer’s disease using novel luminescent probes. ACS Chem Biol. 2007;2(8):553–60. doi: 10.1021/cb700116u. [DOI] [PubMed] [Google Scholar]
- 39.O’Nuallain B, Wetzel R. Conformational Abs recognizing a generic amyloid fibril epitope. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(3):1485–90. doi: 10.1073/pnas.022662599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005;307(5707):262–5. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 41.Petkova AT, Yau WM, Tycko R. Experimental Constraints on Quaternary Structure in Alzheimer’s beta-Amyloid Fibrils. Biochemistry. 2006;45(2):498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rabinovici GD, Furst AJ, O’Neil JP, Racine CA, Mormino EC, Baker SL, Chetty S, Patel P, Pagliaro TA, Klunk WE, Mathis CA, Rosen HJ, Miller BL, Jagust WJ. 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2007;68(15):1205–12. doi: 10.1212/01.wnl.0000259035.98480.ed. [DOI] [PubMed] [Google Scholar]
- 43.Rosen RF, LeVine H, Walker LC. PIB Binding in Alzheimer’s Disease and Nonhuman Primate Brain. 11th International Conference on Alzheimer’s Disease and Related Disorders; Chicago, IL. 2008. [Google Scholar]
- 44.Somerville RA. Host and transmissible spongiform encephalopathy agent strain control glycosylation of PrP. J Gen Virol. 1999;80(Pt 7):1865–72. doi: 10.1099/0022-1317-80-7-1865. [DOI] [PubMed] [Google Scholar]
- 45.Tagliavini F, Giaccone G, Frangione B, Bugiani O. Preamyloid deposits in the cerebral cortex of patients with Alzheimer’s disease and nondemented individuals. Neurosci Lett. 1988;93(23):191–6. doi: 10.1016/0304-3940(88)90080-8. [DOI] [PubMed] [Google Scholar]
- 46.Toyama BH, Kelly MJ, Gross JD, Weissman JS. The structural basis of yeast prion strain variants. Nature. 2007;449(7159):233–7. doi: 10.1038/nature06108. [DOI] [PubMed] [Google Scholar]
- 47.Walker L, LeVine H, Jucker M. Koch’s postulates and infectious proteins. Acta Neuropathol. 2006;112(1):1–4. doi: 10.1007/s00401-006-0072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weissmann C. The state of the prion. Nat Rev Microbiol. 2004;2(11):861–71. doi: 10.1038/nrmicro1025. [DOI] [PubMed] [Google Scholar]
- 49.Wetzel R, Shivaprasad S, Williams AD. Plasticity of Amyloid Fibrils. Biochemistry. 2007;46(1):1–10. doi: 10.1021/bi0620959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilcock GK, Esiri MM. Plaques, tangles and dementia. A quantitative study. J Neurol Sci. 1982;56(23):343–56. doi: 10.1016/0022-510x(82)90155-1. [DOI] [PubMed] [Google Scholar]
- 51.Wisniewski HM, Bancher C, Barcikowska M, Wen GY, Currie J. Spectrum of morphological appearance of amyloid deposits in Alzheimer’s disease. Acta Neuropathol (Berl) 1989;78(4):337–47. doi: 10.1007/BF00688170. [DOI] [PubMed] [Google Scholar]
- 52.Yagi H, Ban T, Morigaki K, Naiki H, Goto Y. Visualization and Classification of Amyloid beta Supramolecular Assemblies. Biochemistry. 2007;46(51):15009–17. doi: 10.1021/bi701842n. [DOI] [PubMed] [Google Scholar]
- 53.Yamaguchi H, Hirai S, Morimatsu M, Shoji M, Ihara Y. A variety of cerebral amyloid deposits in the brains of the Alzheimer-type dementia demonstrated by beta protein immunostaining. Acta Neuropathol. 1988;76(6):541–9. doi: 10.1007/BF00689591. [DOI] [PubMed] [Google Scholar]
- 54.Ye L, Morgenstern JL, Gee AD, Hong G, Brown J, Lockhart A. Delineation of PET imaging agent binding sites on beta -amyloid peptide fibrils. J Biol Chem. 2005;280(25):23599–604. doi: 10.1074/jbc.M501285200. [DOI] [PubMed] [Google Scholar]
- 55.Yoshiike Y, Kayed R, Milton SC, Takashima A, Glabe CG. Pore-forming proteins share structural and functional homology with amyloid oligomers. Neuromolecular Med. 2007;9(3):270–5. doi: 10.1007/s12017-007-0003-6. [DOI] [PubMed] [Google Scholar]
- 56.Zhang-Nunes SX, Maat-Schieman ML, van Duinen SG, Roos RA, Frosch MP, Greenberg SM. The cerebral beta-amyloid angiopathies: hereditary and sporadic. Brain Pathol. 2006;16(1):30–9. doi: 10.1111/j.1750-3639.2006.tb00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]