

Abstract
Background
Candida albicans is a commonly encountered fungal pathogen in humans. The formation of biofilm is a major virulence factor in C. albicans pathogenesis and is related to antidrug resistance of this organism. Although many factors affecting biofilm have been analyzed, molecular mechanisms that regulate biofilm formation still await to be elucidated.
Results
In this study, from the gene regulatory network perspective, we developed an efficient computational framework, which integrates different kinds of data from genome-scale analysis, for global screening of potential transcription factors (TFs) controlling C. albicans biofilm formation. S. cerevisiae information and ortholog data were used to infer the possible TF-gene regulatory associations in C. albicans. Based on TF-gene regulatory associations and gene expression profiles, a stochastic dynamic model was employed to reconstruct the gene regulatory networks of C. albicans biofilm and planktonic cells. The two networks were then compared and a score of relevance value (RV) was proposed to determine and assign the quantity of correlation of each potential TF with biofilm formation. A total of twenty-three TFs are identified to be related to the biofilm formation; ten of them are previously reported by literature evidences.
Conclusions
The results indicate that the proposed screening method can successfully identify most known biofilm-related TFs and also identify many others that have not been previously reported. Together, this method can be employed as a pre-experiment screening approach that reveals new target genes for further characterization to understand the regulatory mechanisms in biofilm formation, which can serve as the starting point for therapeutic intervention of C. albicans infections.
Background
Candida albicans, the most commonly isolated opportunistic human fungal pathogen, can cause skin and mucosal infections as well as life-threatening systemic infections [1,2]. In healthy individuals, C. albicans occurs as a dimorphic commensal colonizer of mucosal membranes in the oral cavity, gastrointestinal tract, urogenital mucosa, and vagina. In immunocompromised patients including those undergoing cancer chemotherapy, organ or bone marrow transplantation and those are AIDS sufferers, this organism can become pathogenic, resulting in proliferative growth on mucosal surfaces locally and systemically [3-5]. Candida infections, or candidiasis, are difficult to treat and create very serious challenge in medicine. Mortality rates among patients with candidiasis have been increasing and can be as high as 40% to 60%, especially for those who have bloodstream infections (candidemia) [6-8]. Therefore, to understand the molecular mechanisms underlying the pathogenicity of C. albicans is imperative for management of such infections.
Biofilm formation plays an important role in the pathogenicity of C. albicans. For example, biofilm can serve as reservoirs for the cells to continually seed infection. Moreover, C. albicans biofilm cells are much more resistant than free-living planktonic cells to many antifungal agents. As a result, the biofilm-specific property of C. albicans cells has prompted recent interests in the study of biofilm structure, physiology, and regulation, and research into the pathogenicity of Candida focusing on the prevention and management of biofilm development and antifungal resistance [6,9]. Biofilms are defined as surface-associated communities of cells surrounded by an extracellular matrix and displaying phenotypic features that differ from their planktonic counterparts [10,11]. The development of C. albicans biofilm can be divided into four sequential steps. First, the yeast cells adhere to a foreign substrate (host tissue or medical device). Second, the yeast cells proliferate across the substrate surface and pseudohyphae and hyphae begin to develop. Third, the extracellular matrix is produced and the network of pseudohyphae and hyphae cells is embedded within this matrix. Biofilm will then mature into a complex three-dimensional structure. Finally, the progeny biofilm cells disperse to enable remote surfaces to be populated [6,9,10]. Although previous studies have provided some insights, the details of molecular mechanisms that are responsible for biofilm formation still await to be elucidated.
Recently, the C. albicans genome for strain SC5314 was sequenced [2], revealing that almost two-thirds of its ~6000 open reading frames are orthologous to genes of Saccharomyces cerevisiae, a well-studied model organism and the first eukaryotic organism to have its entire genome sequenced [1,4,12]. In addition, the ease of genetic/molecular manipulation and the development of various tools for genome-wide functional analysis have led to accumulate a large amount of data from the study of S. cerevisiae. Since C. albicans and S. cerevisiae are closely related, i.e., both fall within the hemiascomycete group, the information from S. cerevisiae could be adapted and useful for our understanding in C. albicans biology and pathogenesis [1,13].
We are investigating the underlying molecular mechanisms that are responsible for biofilm formation in C. albicans. Specifically, it is aimed to unravel what makes the difference between biofilm and planktonic cells from the gene regulatory network point of view. Gene regulatory networking is achieved by the action of multiple transcription factors (TFs) binding to cis-regulatory DNA elements of the target genes, in response to different environmental signals. Since transcription factors are central to gene regulatory networks, in this study, we developed a computational framework for global screening of potential C. albicans biofilm-related TFs via network comparison (Figure 1). We integrated different kinds of data from genome-scale analysis, including gene expression profiles of biofilm formation from C. albicans [3], regulatory associations between TFs and genes adopted from S. cerevisiae [14,15], ortholog data between C. albicans and S. cerevisiae genes [16], and Gene Ontology [17]. By using this information, the gene regulatory networks for biofilm cells and planktonic cells were constructed separately. These gene regulatory networks were then compared based on the network structure to reveal their differences and to identify their relevance to biofilm formation for each TF via the so-called 'gain-of-function' and 'loss-of-function' subnetworks. The significance for the potential TFs was determined by statistical analysis. A total of twenty-three TFs are identified to be related to the biofilm formation; ten of them are previously reported by literature evidences. These results indicate that our approach can be useful to reveal TFs significant in biofilm formation and importantly, provide new targets for further studies to understand the regulatory mechanisms in biofilm formation and the fundamental difference between biofilm and planktonic cells.
Figure 1.

The flowchart of the proposed method for screening potential biofilm-related TFs in C. albicans.
Methods
Overview of the proposed screening method
The method of the global screening for biofilm-related TFs was divided into three key steps: (i) selection scheme for TFs and genes, (ii) scheme for gene regulatory network reconstruction, and (iii) comparison scheme between two networks of biofilm cells and planktonic cells. The output of the method is a score named relevance value (RV) for each TF. RV is computed to correlate the TF with regulation of biofilm formation. A higher score suggests that the particular TF is more likely involved in the regulatory network for C. albicans biofilm formation. Based on the RVs, the biofilm-related TFs are chosen. The whole process of the proposed screening method is shown in Figure 1. The data used and the details of each step are described in the following sections.
Data used in the proposed screening method
In this study, four kinds of data are integrated-microarray gene expression profiles, regulatory associations between TFs and genes, ortholog data between C. albicans and S. cerevisiae genes, and Gene Ontology annotation information. The microarray data were obtained from Murillo et al. [3], in which genome-wide transcription analysis of biofilm formation are profiled using Affymetrix oligonucleotide GeneChips representative of the entire genome of C. albicans. Briefly, the DNA microarray includes 7116 ORFs and each microarray experiment was performed in duplicate [3]. The resulting time-course microarray data contain two sets of information for biofilm and planktonic cells, generated from early stages of biofilm formation (0-390 mins, 6 time points). The regulatory associations between TFs and genes were obtained from S. cerevisiae using YEASTRACT database http://www.yeastract.com/ and genome-wide location analysis of yeast TFs from Harbison et al. [14]. YEASTRACT (Yeast Search for Transcriptional Regulators And Consensus Tracking) deposits more than 34469 regulatory associations between TFs and target genes in S. cerevisiae, based on more than 1000 bibliographic references [15]. The genome-wide location analysis allows protein-DNA interactions to be monitored across the entire yeast genome by combing a modified Chromatin Immunoprecipitation (ChIP) procedure with DNA microarray analysis. In Harbison et al. [14], the genomic occupancy of 203 DNA-binding TFs in S. cerevisiae was determined. The p-value threshold for significant binding was selected as p ≦ 0.001 since their analysis indicated that the threshold maximizes inclusion of legitimate regulator-DNA interactions and minimizes false positives [14]. The ortholog data between C. albicans and S. cerevisiae genes were retrieved from Candida Genome Database or CGD http://www.candidagenome.org/[16]. Gene orthology and its best hit mappings were used to correlate S. cerevisiae genes with C. albicans genes using the InParanoid program [18]. The annotations for C. albicans genes were acquired from the Gene Ontology (GO) [17]. The GO annotations were facilitated to query for molecular function or biological process of a gene-of-interest in this study. The way we used these data for screening of biofilm-related TFs are further described in the following sections.
Selection scheme for transcription factors and target genes
To select TFs and genes for gene regulatory network reconstruction, we included as many TFs as possible in this step. Taking advantages of the fact that C. albicans and S. cerevisiae are closely related and S. cerevisiae is much better characterized than C. albicans, the information derived from S. cerevisiae was adopted and used in this study. If a S. cerevisiae TF has an ortholog found in C. albicans, the ortholog was assigned as a TF in C. albicans. An example is shown in Figure 2. Ste12 is a well-known transcription factor in S. cerevisiae and has a good sequence homologue (named Cph1) in C. albicans, this Cph1 protein is thus identified as a TF in C. albicans. In this way, TFs are pooled together and will be selected for biofilm-related TFs screening by the proposed method. Notably, some particular C. albicans TFs, which have either not been included in the microarray data or lack of association information with target genes, were excluded from the TF pool.
Figure 2.

An example for illustration of C. albicans TF-gene regulatory association inference.
As for the selection of target genes, GO annotations were used [17]. An assumption of the proposed screening method is that if a TF regulates gene expression in biofilm cells rather than in planktonic cells, this particular TF is more likely involved in the regulatory machinery that governs biofilm formation. Therefore, the genes annotated with GO terms such as biofilm formation, or those possibly related to different steps of biofilm formation and development, such as cell adhesion, and filamentous growth, were selected for further analysis. However, if the selected target genes of C. albicans are not included in gene expression profiles or have no ortholog mapping data with S. cerevisiae genes, they were excluded for the subsequent steps.
The regulatory associations between TFs and genes in S. cerevisiae from YEASTRACT database [15] and Harbison et al. [14] were used to infer the possible TF-gene regulatory associations in C. albicans. An example for this step is illustrated in Figure 2. Borneman et al. [19] identified Ste12-MUC1 association by chromatin immunoprecipitation (ChIP)-chip experiment with a p-value = 2e-15 and the result is deposited in the YEASTRACT database. According to CGD, the TF Ste12 and its target gene MUC1 in S. cerevisiae have orthologs Cph1 and HWP1 in C. albicans, respectively. Consequently, based on the experimental results from S. cerevisiae, the possible associations between Cph1 and HWP1 in C. albicans were inferred in our study.
Gene regulatory network reconstruction scheme
From the first step described above, we have selected TFs, their potential target genes, and their possible regulatory associations. This information was used to further constitute the candidate gene regulatory network [Additional file 1]. A stochastic dynamic model was then applied to prune the candidate network to obtain the gene regulatory networks independently for biofilm cells and planktonic cells, according to their respective data sets. For a target gene i in the candidate gene regulatory network, the gene was described using the stochastic discrete dynamic equation (1) [20].
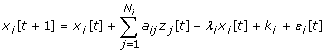 |
(1) |
where xi[t] represents the gene expression level at time t for the particular gene i, aij denotes the regulatory ability of the j-th TF toward the i-th target gene with a positive sign indicating gene activation and a negative sign indicating gene repression, zj[t] represents the regulation function of the j-th TF (the Ni TFs binding to the target gene i are retrieved from the candidate gene regulatory network in Additional file 1), λi indicates the degradation effect of the present time t on the next time t+1, ki represents the basal level of expression, εi[t] denotes the stochastic noise due to the model uncertainty and the fluctuation of the DNA microarray data. It has been shown that TF binding usually affects gene expression in a nonlinear fashion, that is, below some level of protein concentration a TF has no effect, while above a certain expression level the effect of the TF may become saturated [21,22]. Thus, the regulation function zj[t] was modeled as the sigmoid function, which is one kind of Hill function, of yj[t] (the protein concentration profiles of TF j) shown in equation (2) [20,22-24].
| (2) |
where fj denotes the sigmoid function, μj and σj represent the mean and standard deviation of protein concentration level of TF j. The biological implication of the equation (1) is that the gene expression of the target gene i at the next time t+1 is determined by the present gene expression, the present regulation function of Ni TFs binding to this target gene, the degradation effect of the present time, the basal level of gene expression, and some stochastic noises. For each target gene selected from the previous scheme, a stochastic dynamic model was constructed. Consequently, the stochastic dynamic equations for all the target genes constituted the mathematical model of the candidate gene regulatory network.
After constructing the stochastic dynamic model of the candidate gene regulatory network, the microarray gene expression profiles were then overlaid to identify the regulatory parameters in equation (1). Since the DNA microarray data for gene expression profiles of biofilm and planktonic cells are collected separately, the gene regulatory networks of biofilm and planktonic cells can be independently reconstructed. The identification of the gene regulatory network was performed gene by gene, so that the process was not limited by the number of target genes. Due to the non-negativity of basal level of expression (ki ≥ 0 in equation (1)), the constrained least squares regression method was used to identify the regulatory parameters [25,26] (see Additional file 2 for details). Moreover, since there are no good data available for genome-wide protein concentration levels in C. albicans, gene expression profiles were used instead for identifying the regulatory parameters. Once the regulatory parameters were identified, the significant TF-gene interactions were determined based on the identified aij's. By means of Akaike Information Criterion (AIC) [27,28] and student's t-test [29,30], we determined the statistical significance of the interactions between TFs and genes, pruned the candidate gene regulatory network and reconstructed the gene regulatory networks for biofilm and planktonic cells (see Additional files 2, 3, 4 and 5 for details). The resulting biofilm and planktonic gene regulatory networks and the significant TF-gene interactions among them were then used for comparison scheme.
Comparison scheme between two networks of biofilm and planktonic cells
After the TF/target gene selection and gene regulatory network reconstruction, the gene regulatory networks of both biofilm and planktonic cells and their significant TF-gene interactions were obtained. This information allowed us to compare the networks of biofilm and planktonic cells, and compute the relevance value (RV) to identify TFs that are important in the regulation of biofilm formation. Regardless of the nature of each TF as an activator or a repressor toward its target genes in the gene regulatory networks, we compared the network structure between these two networks. The interactions between TFs and genes in these two networks were simplified as binary relation, in which '1' represents a significant interaction between the TF and the target gene (no matter activation or repression) and '0' denotes no significant interaction (see Figure 3(a) and Table 1 for illustration). As results, comparison of the biofilm with the planktonic gene regulatory network can generate two different subnetworks, one is called 'gain-of-function' subnetwork and the other is 'loss-of-function' subnetwork. If a significant interaction is detected in the biofilm but is absent in the planktonic gene regulatory network, such an interaction is classified into the gain-of-function subnetwork, which represents a subnetwork within the biofilm gene regulatory network. In contrast, if a significant interaction is detected in the planktonic but not in the biofilm gene regulatory network, this interaction is a part of the loss-of-function subnetwork, representing a subnetwork of the planktonic gene regulatory network [Additional files 6, 7 and 8]. Schematic diagrams and the corresponding binary description of TF-gene interactions for elucidation of gene regulatory network comparison are shown in Figure 3 and Table 1.
Figure 3.

Schematic diagrams for illustration of gene regulatory network comparison. (a) The biofilm gene regulatory network and the planktonic gene regulatory network. The regulatory abilities for TF-gene interactions are omitted for simplicity. (b) The gain-of-function and loss-of-function subnetworks after network structure comparison. aij,gain and aij,loss indicate the regulatory abilities of the j-th TF to the i-th target gene for gain-of-function and loss-of-function, respectively, in which a positive sign indicates activation and a negative sign indicates repression.
Table 1.
Construction of gain-of-function and loss-of-function subnetworks
| TF A | Biofilm | Planktonic | Gain-of-function | Loss-of-function |
|---|---|---|---|---|
| Gene 1 | 1 | 0 | 1 | 0 |
| Gene 2 | 1 | 1 | 0 | 0 |
| Gene 3 | 1 | 1 | 0 | 0 |
| Gene 4 | 1 | 0 | 1 | 0 |
| Gene 5 | 1 | 1 | 0 | 0 |
| Gene 6 | 1 | 0 | 1 | 0 |
| Gene 7 | 0 | 1 | 0 | 1 |
| Gene 8 | 0 | 1 | 0 | 1 |
The table demonstrates the gain-of-function and loss-of-function subnetworks construction shown in Figure 3. The gain-of-function and loss-of-function subnetworks were constructed by comparing the network structure of biofilm gene regulatory network with that of planktonic gene regulatory network via the comparison scheme.
Using the gain-of-function and loss-of-function subnetworks to distinguish the biofilm from planktonic gene regulatory network, we determined a score named relevance value (RV) to quantify the correlation of each TF in these subnetworks with the regulation of biofilm formation, and to identify potential C. albicans biofilm-related TFs. To determine the RV for each TF, two important issues are also taken into consideration. First, the magnitude of regulatory abilities aij's identified from the gene regulatory network reconstruction scheme denotes the significance of the TF in the transcriptional regulation for a specific target gene. Second, an assumption is made: if a TF regulates more biofilm-related genes in the gain-of-function and loss-of-function subnetworks, then the TF is more likely involved in the regulation for biofilm formation. Consequently, the RV was determined using the following equation, based on the regulatory abilities of TF in the gain-of-function and loss-of-function subnetworks.
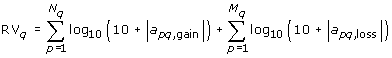 |
(3) |
where RVq denotes the relevance value for TF q, apq,gain and apq,loss, which are numerically obtained from the gene regulatory network reconstruction scheme, indicate the regulatory ability of TF q to control the target gene p in the gain-of-function subnetwork and loss-of-function subnetwork, respectively; Nq and Mq represent the numbers of target genes for the TF q identified from the gain-of-function and loss-of-function subnetworks, respectively. The implication of equation (3) is that RV quantifies the extent of the TF involved in the interactions with target genes that differentiate biofilm and planktonic gene regulatory networks. The measurement of RV is conceptually similar to the well-known 'graph edit distance' previously used to compare pathways structurally [31]. In the illustrated schematic diagram in Figure 3, the relevance value for TF A is calculated as:
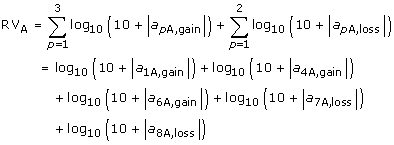 |
(4) |
For each TF, a corresponding RV was assigned and an empirical p-value was computed to determine the significance of the RV. To determine the p-value for an observed RV, a null distribution of RVs (Figure 4) was generated by repeatedly permuting the network structure of the candidate gene regulatory network and computing the RV for each random network structure. The permutation of the network structure was performed by keeping the network size, i.e., the target genes to which a particular TF associated were permuted without changing the total number of TF-gene regulatory associations of the network. Specifically, suppose there are A selected TFs, B target genes, and C TF-gene regulatory associations in the candidate gene regulatory network, the probability of a rewiring of a TF-gene association in the permuted random network is uniformly given by C/AB. We repeated this process 100000 times and estimated the p-value for the corresponding RV as the fraction of random network structures whose RV is at least as large as the RV of the real network structure. The p-values were then adjusted by Bonferroni correction to avoid multiple testing problem [29,30]. The RVs with adjusted p-value ≦ 0.05 were determined as significant RVs and the corresponding TFs were identified as the potential C. albicans biofilm-related TFs.
Figure 4.

Distribution of relevance values (RVs) of random network.
Results
Screening of potential C. albicans biofilm-related TFs
We applied the proposed method to analyze and compare data derived from C. albicans biofilm and planktonic cells for screening of potential C. albicans biofilm-related TFs. Among all C. albicans genes, 361 were selected as target genes since they are annotated by at least one of the GO terms, including biofilm formation, cell adhesion, and filamentous growth. By S. cerevisiae TF information and the orthologs between C. albicans and S. cerevisiae, we identified 220 C. albicans TFs which have expression profiles in the experiments comparing biofilm with planktonic cells. From the identified TFs and target genes, we further reconstructed the gene regulatory networks for biofilm and planktonic cells, in which 2149 and 2211 TF-gene interactions are included, respectively [Additional files 3, 4 and 5]. Among these two networks, excluding the 1442 common interactions, there are 707 interactions in gain-of-function subnetwork and 769 interactions in loss-of-function subnetwork [Additional files 6, 7 and 8]. We then used the regulatory abilities of TFs in the gain-of-function and loss-of-function subnetworks to compute the RVs for each TF and to determine the significance of these RVs. Consequently, 23 potential TFs related to C. albicans biofilm formation were identified and shown in Table 2.
Table 2.
Identification of potential C. albicans biofilm-related TFs
| Systematic name | TF* | RV | Adjusted p-value§ | Literature evidence |
|---|---|---|---|---|
| orf19.5953 | orf19.5953 | 117.6815 | < 1e-05 | |
| orf19.610 | Efg1 | 76.1153 | < 1e-05 | [32,34] |
| orf19.4433 | Cph1 | 75.4189 | < 1e-05 | [36] |
| orf19.5498 | Efh1 | 70.8097 | < 1e-05 | [39] |
| orf19.861 | orf19.861 | 68.2492 | < 1e-05 | |
| orf19.1773 | Rap1 | 59.4340 | < 1e-05 | [40] |
| orf19.837.1 | Ino4 | 53.3481 | < 1e-05 | |
| orf19.1069 | Rpn4 | 52.8505 | < 1e-05 | |
| orf19.2236 | orf19.2236 | 51.4651 | < 1e-05 | |
| orf19.5908 | Tec1 | 51.2743 | < 1e-05 | [41,42] |
| orf19.4545 | Swi4 | 49.9914 | < 1e-05 | |
| orf19.5041 | orf19.5041 | 45.9961 | < 1e-05 | |
| orf19.2054 | Fgr15 | 45.6984 | < 1e-05 | [43] |
| orf19.5312 | orf19.5312 | 44.3625 | < 1e-05 | |
| orf19.1358 | Gcn4 | 38.4428 | < 1e-05 | [45] |
| orf19.7046 | Met28 | 38.4261 | < 1e-05 | |
| orf19.4573 | Zcf26 | 35.9090 | 0.0022 | |
| orf19.971 | Skn7 | 35.7618 | 0.0022 | [46] |
| orf19.6121 | Mnl1 | 35.4126 | 0.0022 | |
| orf19.7025 | Mcm1 | 34.7227 | 0.0066 | [47] |
| orf19.952 | orf19.952 | 32.3603 | 0.0242 | |
| orf19.5975 | orf19.5975 | 31.8614 | 0.0286 | |
| orf19.2752 | Adr1 | 31.8191 | 0.0286 | [43] |
*The TF names are retrieved from CGD database http://www.candidagenome.org/.
§The adjusted p-values are obtained by Bonferroni correction.
The potential biofilm-related TFs
A total of 23 TFs were determined as potential C. albicans biofilm-related TFs (Table 2). To assure the effectiveness of our proposed screening method, we seek evidences from literature to validate the inferred functions in regulation of biofilm formation.
(1) Efg1, Cph1 and Efh1: Both cell adhesion and morphogenesis to form hyphae play important roles in biofilm formation and maturation [10]. Efg1 is a downstream transcription factor of Ras-protein kinase A signaling pathway and governs multiple different morphogenetic processes including phenotypic switching and filamentous growth [32-34]. Deletion of C. albicans EFG1 gene decreases the ability of the cell to adhere to oral epithelial cells in vitro [32].
C. albicans Cph1 is an ortholog of S. cerevisiae Ste12. In S. cerevisiae, the cells mate by responding to pheromones via the functions of mitogen-activated protein kinase (MAPK) cascade and its downstream TF, Ste12. C. albicans Cph1 is not only required for mating [35], but is also important for hyphal formation [36]. Finally, efg1/efg1 cph1/cph1 double mutant cannot form hyphae and is also defective in biofilm formation [37,38].
APSES proteins regulate fungal filamentation and differentiation. There are two APSES proteins in C. albicans, Efg1 and Efh1 [39]. C. albicans EFH1 gene deletion causes hyperfilamentation in an efg1 background under certain conditions, indicating that Efh1 modulates and supports the regulatory functions of Efg1 [39].
(2) Rap1 and Tec1: Rap1 is a transcription factor and telomere binding protein that is essential for cell viability in S. cerevisiae. Studies from C. albicans RAP1-deletion mutant shows that Rap1 is required for efficient repression of pseudohyphal growth under yeast-favoring conditions but is not essential for viability of C. albicans [40].
Tec1, a member of the TEA/ATTS family of transcription factors, is shown to regulate hyphal development and virulence in C. albicans. Insertion mutations of TEC1 cause severe defects in biofilm formation [41,42].
(3) Fgr15, Gcn4, Skn7, Mcm1 and Adr1: Fgr15 is a putative transcription factor with zinc finger DNA-binding motif. Transposon mutation of FGR15 affects filamentous growth [43]. Gcn4, like its ortholog in S. cerevisiae, activates the transcription of amino acid biosynthetic genes. In addition, C. albicans Gcn4 interacts with the Ras-cAMP pathway to promote filamentous growth in response to amino acid starvation [44]. C. albicans GCN4-deletion mutant reduces biofilm biomass, indicating that Gcn4 is required for normal biofilm growth [45].
Skn7, one of the response regulator proteins in C. albicans, is required for morphogenesis under some conditions and its mutant produces smooth colonies [46]. It is also required for adaptation under some types of oxidative stress in vitro [46]. Mcm1 is an essential gene in C. albicans whose protein levels are crucial for determination of cell morphology. It might be a mediator recruiting regulatory factors required for hyphal development in C. albicans [47]. Adr1, like Fgr15, is also a putative transcription factor with zinc finger DNA-binding motif and its mutant results in less filamentous growth [43].
(4) Other TFs identified: Of the 23 TFs indentified, as described above, 10 of them have been shown to relate to various processes of biofilm formation (e.g. filamentation and cell adhesion) or biofilm formation per se. Therefore, the remaining 13 TFs provide good candidates for further experiments to determine their regulatory roles in biofilm formation.
Statistical measurements of the performance
Among total 220 TFs selected for screening, 23 potential biofilm-related TFs with significant RVs were identified (Table 2). Of the other 197 TFs, we also check literature evidences to see if they are validated by experiments as biofilm-related TFs. Twenty-six out of 197 TFs which do not have significant RV were annotated with GO terms such as biofilm formation, cell adhesion, or filamentous growth. The sensitivity, specificity, positive predictive value, and negative predictive value of the proposed screening method were evaluated (see Additional file 2 for details). The proposed approach can identify potential C. albicans biofilm-related TFs with a low sensitivity of 27.78% and a high specificity of 92.93%. Moreover, our method is effective on determining the TFs that are not biofilm-related as the negative predictive value is 86.80%. The positive predictive value is 43.48%, enriching by 2.7-fold the likelihood of screening TFs that are biofilm-related since the biofilm-related prevalence among total 220 TFs is 16.36%. It is noteworthy that these statistics are evaluated based on the published literature evidences and GO annotations, suggesting that if more C. albicans biofilm-related TFs are validated by experiments, the statistics should be improved.
Discussion
The architecture of C. albicans biofilms and the correlation between biofilm and infection have been analyzed, but our understanding of the gene regulations that are responsible for the biofilm formation is still limited. Since transcription factors play an important role in gene regulatory networks, here, we develop a computational framework via network comparison to screen for C. albicans TFs that may be important for biofilm formation. The original idea is derived from the concept of comparative biology which commonly utilizes comparative approaches in the analysis of genomic sequences to reveal the functional similarities and differences among different species [48]. We extend the concept and compare the gene regulatory networks to explore what makes the difference between biofilm and planktonic cells in C. albicans. The advantage of the proposed screening method lies in the convenience and systematicity. Compared with the time- and labor-consuming experiments, we provide an efficient and rapid way for screening TFs by comparing two gene regulatory networks from the systematic point of view. Richard et al. [9] used a collection of insertion mutations in 197 C. albicans ORFs to screen those mutants that are defective in biofilm formation; however, only 4 such genes are identified. In this study, our computational method has a positive predictive value of 43.48% which is much higher than that shown by Richard et al. (~2.03%). Consequently, the proposed screening method can be useful for providing potential target genes for biologists to perform further experiments. It can be considered as a pre-experiment screening. In addition, our approach is not only capable of studying biofilm and planktonic cells, but can also be used to compare two physiological conditions as long as the adequate data are available. For example, this method can be used to screen TFs possibly involved in the cancer development process by comparing the normal cell and cancer cell and the TFs screened could serve as a starting point for therapeutic intervention [49].
Although our approach is shown to be useful, some drawbacks or improvements are still need to be taken in consideration. One assumption of the stochastic dynamic model in equation (1) is that the time delay of transcriptional regulation of the TF to the target gene is only one time unit (about seven minutes in this study), which is not always the case. Previous studies have shown from gene expression profiles that different time delays are required for different TFs to exert regulatory effects on their target genes [21,50,51]. However, since the time delays cannot be experimentally measurable for all the TFs and its potential target genes and the computationally predicted time delays are not completely reliable, the time delays are all set to one time unit when reconstructing the gene regulatory networks. In addition to the time-delay assumption, one important consideration is data accuracy from public domains. For example, based on the orthology information between C. albicans and S. cerevisiae, we adopt the information of regulatory associations between TFs and genes from S. cerevisiae to the study of C. albicans. The orthology mappings were performed at CGD using InParanoid software, which basically employs the computed sequence similarity to determine orthologs [18]. If the orthology mapping data is not perfectly accurate, it can result in the misinterpretation of regulatory associations between TFs and genes in C. albicans. To overcome the problem, it is better to acquire the TF-gene regulatory associations directly from the experiments (e.g. genome-wide ChIP-chip) using C. albicans. Recently, genome-wide location analysis by ChIP (chromatin immunoprecipitation)-chip has been developed for the study of C. albicans [52,53]. However, similar studies for biofilm-related TFs are still not available. Another shortage of the information from public domains is the lack of information related to S. cerevisiae TF-gene association in YEASTRACT and ChIP-chip data from Harbison et al. [14], although orthologs of the TF and target genes do exist in C. albicans. Consequently, it will not be able to reconstruct the corresponding gene regulatory network, thus the particular TF is being excluded from the TF pool. One can also solve this problem by performing C. albicans ChIP-chip experiments. Once the reliable C. albicans TF-gene regulatory associations are obtained, the performance of the proposed screening method can be improved and the reliable gene regulatory networks can be reconstructed.
Numerous factors can affect C. albicans biofilm formation, including supporting substrate, growth medium, and C. albicans strains [6,9]. Given the complex conditions that affect the kinetics of biofilm formation process and the huge amounts of data generated by post-genomic approaches under different experimental conditions, we can now investigate the most significant TFs that are responsible for the biofilm formation. The screening of biofilm-related TFs is the initial step to elucidate the whole gene regulatory network that governs biofilm formation. Lu and Collins [54] have successfully demonstrated that synthetic biology techniques are feasible to engineer bacteriophage to express DspB, an enzyme that hydrolyzes the crucial biofilm formation adhesin (β-1,6-N-acetyl-D-glucosamine) encoded by genes pgaABCD in E. coli [55,56], therefore reducing bacterial biofilms. As a result, by combining the systems biology approaches to gain more insight into the molecular mechanisms for biofilm formation with the synthetic biology techniques to engineer the enzyme needed, we may develop new therapeutic strategies to combat the recalcitrant infections caused by C. albicans and other microbial pathogens.
Conclusions
Biofilm formation is a major virulence factor in C. albicans pathogenesis and is related to antidrug resistance of this organism. However, little is known about the molecular mechanisms that regulate biofilm formation. In this study, we developed an efficient computational framework for global screening of potential transcription factors controlling C. albicans biofilm formation. S. cerevisiae information was used to infer the possible TF-gene regulatory associations in C. albicans. Gene regulatory networks of C. albicans biofilm and planktonic cells were compared to identify the transcription factors involved in biofilm formation and maintenance. A total of twenty-three TFs are identified; ten of them are previously reported to be involved in biofilm formation. Literature evidences indicate that our approach can be useful to reveal TFs significant in biofilm formation and importantly, provide new targets for further studies to understand the regulatory mechanisms in biofilm formation and the fundamental difference between biofilm and planktonic cells, which can serve as the starting point for therapeutic intervention of C. albicans infections.
Authors' contributions
YCW developed the method, performed the analysis, evaluated the results and wrote the manuscript. CYL evaluated the results and revised the manuscript. WPH participated in the statistical analysis and revised the manuscript. LAM and NA provided essential guidance. BSC provided essential guidance and revised the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Supplementary Table S1. Supplementary table S1 lists the TF-gene regulatory associations in the candidate gene regulatory network.
Supplementary Methods.
Supplementary Table S2. Supplementary table S2 comprises the significant TF-gene regulatory associations in the biofilm and planktonic gene regulatory networks, respectively.
Supplementary Figure S1. Supplementary figure S1 shows the schematic view of the biofilm regulatory network.
Supplementary Figure S2. Supplementary figure S2 displays the schematic view of the planktonic regulatory network.
Supplementary Table S3. Supplementary table S3 consists of the TF-gene regulatory associations in the gain-of-function and loss-of-function subnetworks, respectively.
Supplementary Figure S3. Supplementary figure S3 illustrates the schematic view of the gain-of-function subnetwork.
Supplementary Figure S4. Supplementary figure S4 demonstrates the schematic view of the loss-of-function subnetwork.
Contributor Information
Yu-Chao Wang, Email: lancewang@moti.ee.nthu.edu.tw.
Chung-Yu Lan, Email: cylan@life.nthu.edu.tw.
Wen-Ping Hsieh, Email: wphsieh@stat.nthu.edu.tw.
Luis A Murillo, Email: Luis.Murillo@ucsf.edu.
Nina Agabian, Email: nina.agabian@ucsf.edu.
Bor-Sen Chen, Email: bschen@ee.nthu.edu.tw.
Acknowledgements
The work was supported by the National Science Council of Taiwan under grants NSC 97-2627-B-007-001, NSC 98-2627-B-007-014 (to B.-S. Chen) and NSC 97-2627-B-007-002, NSC 98-2627-B-007-015 (to C.-Y. Lan).
References
- Ihmels J, Bergmann S, Berman J, Barkai N. Comparative gene expression analysis by differential clustering approach: application to the Candida albicans transcription program. PLoS Genet. 2005;1:e39. doi: 10.1371/journal.pgen.0010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T, Federspiel NA, Chibana H, Dungan J, Kalman S, Magee BB, Newport G, Thorstenson YR, Agabian N, Magee PT. The diploid genome sequence of Candida albicans. Proc Natl Acad Sci USA. 2004;101:7329–7334. doi: 10.1073/pnas.0401648101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murillo LA, Newport G, Lan CY, Habelitz S, Dungan J, Agabian NM. Genome-wide transcription profiling of the early phase of biofilm formation by Candida albicans. Eukaryot Cell. 2005;4:1562–1573. doi: 10.1128/EC.4.9.1562-1573.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Enfert C, Hube B. Candida: comparative and functional genomics. Wymondham: Caister Academic; 2007. [Google Scholar]
- Calderone RA. Candida and candidiasis. Washington, D.C.: ASM Press; 2002. [Google Scholar]
- Seneviratne CJ, Jin L, Samaranayake LP. Biofilm lifestyle of Candida: a mini review. Oral Dis. 2008;14:582–590. doi: 10.1111/j.1601-0825.2007.01424.x. [DOI] [PubMed] [Google Scholar]
- Warnock DW. Trends in the epidemiology of invasive fungal infections. Nippon Ishinkin Gakkai Zasshi. 2007;48:1–12. doi: 10.3314/jjmm.48.1. [DOI] [PubMed] [Google Scholar]
- Barnes RA. Early diagnosis of fungal infection in immunocompromised patients. J Antimicrob Chemother. 2008;61(Suppl 1):i3–6. doi: 10.1093/jac/dkm424. [DOI] [PubMed] [Google Scholar]
- Richard ML, Nobile CJ, Bruno VM, Mitchell AP. Candida albicans biofilm-defective mutants. Eukaryot Cell. 2005;4:1493–1502. doi: 10.1128/EC.4.8.1493-1502.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship JR, Mitchell AP. How to build a biofilm: a fungal perspective. Curr Opin Microbiol. 2006;9:588–594. doi: 10.1016/j.mib.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Costerton JW, Lewandowski Z, Caldwell DE, Korber DR, Lappin-Scott HM. Microbial biofilms. Annu Rev Microbiol. 1995;49:711–745. doi: 10.1146/annurev.mi.49.100195.003431. [DOI] [PubMed] [Google Scholar]
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M. Life with 6000 genes. Science. 1996;274:546, 563–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- Heckman DS, Geiser DM, Eidell BR, Stauffer RL, Kardos NL, Hedges SB. Molecular evidence for the early colonization of land by fungi and plants. Science. 2001;293:1129–1133. doi: 10.1126/science.1061457. [DOI] [PubMed] [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira MC, Monteiro P, Jain P, Tenreiro S, Fernandes AR, Mira NP, Alenquer M, Freitas AT, Oliveira AL, Sa-Correia I. The YEASTRACT database: a tool for the analysis of transcription regulatory associations in Saccharomyces cerevisiae. Nucleic Acids Res. 2006;34:D446–451. doi: 10.1093/nar/gkj013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud MB, Costanzo MC, Skrzypek MS, Shah P, Binkley G, Lane C, Miyasato SR, Sherlock G. Sequence resources at the Candida Genome Database. Nucleic Acids Res. 2007;35:D452–456. doi: 10.1093/nar/gkl899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund AC, Sjolund E, Ostlund G, Sonnhammer EL. InParanoid 6: eukaryotic ortholog clusters with inparalogs. Nucleic Acids Res. 2008;36:D263–266. doi: 10.1093/nar/gkm1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borneman AR, Zhang ZD, Rozowsky J, Seringhaus MR, Gerstein M, Snyder M. Transcription factor binding site identification in yeast: a comparison of high-density oligonucleotide and PCR-based microarray platforms. Funct Integr Genomics. 2007;7:335–345. doi: 10.1007/s10142-007-0054-7. [DOI] [PubMed] [Google Scholar]
- Chang YH, Wang YC, Chen BS. Identification of transcription factor cooperativity via stochastic system model. Bioinformatics. 2006;22:2276–2282. doi: 10.1093/bioinformatics/btl380. [DOI] [PubMed] [Google Scholar]
- Wu WS, Li WH, Chen BS. Identifying regulatory targets of cell cycle transcription factors using gene expression and ChIP-chip data. BMC Bioinformatics. 2007;8:188. doi: 10.1186/1471-2105-8-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon U. An introduction to systems biology: design principles of biological circuits. Boca Raton, FL: Chapman & Hall/CRC; 2007. [Google Scholar]
- Chen KC, Wang TY, Tseng HH, Huang CY, Kao CY. A stochastic differential equation model for quantifying transcriptional regulatory network in Saccharomyces cerevisiae. Bioinformatics. 2005;21:2883–2890. doi: 10.1093/bioinformatics/bti415. [DOI] [PubMed] [Google Scholar]
- Chen HC, Lee HC, Lin TY, Li WH, Chen BS. Quantitative characterization of the transcriptional regulatory network in the yeast cell cycle. Bioinformatics. 2004;20:1914–1927. doi: 10.1093/bioinformatics/bth178. [DOI] [PubMed] [Google Scholar]
- Gill PE, Murray W, Wright MH. Practical optimization. London; New York: Academic Press; 1981. [Google Scholar]
- Coleman TF, Hulbert LA. A direct active set algorithm for large sparse quadratic programs with simple bounds. Mathematical Programming. 1989;45:373–406. doi: 10.1007/BF01589112. [DOI] [Google Scholar]
- Akaike H. A new look at the statistical model identification. IEEE Transactions on Automatic Control. 1974;19:716–723. doi: 10.1109/TAC.1974.1100705. [DOI] [Google Scholar]
- Johansson R. System modeling and identification. Englewood Cliffs, NJ: Prentice Hall; 1993. [Google Scholar]
- Mendenhall W, Sincich T. Statistics for engineering and the sciences. 4. Englewood Cliffs, N.J.: Prentice-Hall; 1995. [Google Scholar]
- Pagano M, Gauvreau K. Principles of biostatistics. 2. Pacific Grove, CA: Duxbury; 2000. [Google Scholar]
- Emmert-Streib F. The chronic fatigue syndrome: a comparative pathway analysis. J Comput Biol. 2007;14:961–972. doi: 10.1089/cmb.2007.0041. [DOI] [PubMed] [Google Scholar]
- Park H, Myers CL, Sheppard DC, Phan QT, Sanchez AA, J EE, Filler SG. Role of the fungal Ras-protein kinase A pathway in governing epithelial cell interactions during oropharyngeal candidiasis. Cell Microbiol. 2005;7:499–510. doi: 10.1111/j.1462-5822.2004.00476.x. [DOI] [PubMed] [Google Scholar]
- Sonneborn A, Tebarth B, Ernst JF. Control of white-opaque phenotypic switching in Candida albicans by the Efg1p morphogenetic regulator. Infect Immun. 1999;67:4655–4660. doi: 10.1128/iai.67.9.4655-4660.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo HJ, Kohler JR, DiDomenico B, Loebenberg D, Cacciapuoti A, Fink GR. Nonfilamentous C. albicans mutants are avirulent. Cell. 1997;90:939–949. doi: 10.1016/S0092-8674(00)80358-X. [DOI] [PubMed] [Google Scholar]
- Chen J, Lane S, Liu H. A conserved mitogen-activated protein kinase pathway is required for mating in Candida albicans. Mol Microbiol. 2002;46:1335–1344. doi: 10.1046/j.1365-2958.2002.03249.x. [DOI] [PubMed] [Google Scholar]
- Liu H, Kohler J, Fink GR. Suppression of hyphal formation in Candida albicans by mutation of a STE12 homolog. Science. 1994;266:1723–1726. doi: 10.1126/science.7992058. [DOI] [PubMed] [Google Scholar]
- Ramage G, VandeWalle K, Lopez-Ribot JL, Wickes BL. The filamentation pathway controlled by the Efg1 regulator protein is required for normal biofilm formation and development in Candida albicans. FEMS Microbiol Lett. 2002;214:95–100. doi: 10.1111/j.1574-6968.2002.tb11330.x. [DOI] [PubMed] [Google Scholar]
- Lewis RE, Lo HJ, Raad II, Kontoyiannis DP. Lack of catheter infection by the efg1/efg1 cph1/cph1 double-null mutant, a Candida albicans strain that is defective in filamentous growth. Antimicrob Agents Chemother. 2002;46:1153–1155. doi: 10.1128/AAC.46.4.1153-1155.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedt T, Krishnamurthy S, Bockmuhl DP, Tebarth B, Stempel C, Russell CL, Brown AJ, Ernst JF. APSES proteins regulate morphogenesis and metabolism in Candida albicans. Mol Biol Cell. 2004;15:3167–3180. doi: 10.1091/mbc.E03-11-0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas K, Rieger KJ, Morschhauser J. Functional analysis of CaRAP1, encoding the Repressor/activator protein 1 of Candida albicans. Gene. 2003;307:151–158. doi: 10.1016/S0378-1119(03)00456-6. [DOI] [PubMed] [Google Scholar]
- Nobile CJ, Mitchell AP. Regulation of cell-surface genes and biofilm formation by the C. albicans transcription factor Bcr1p. Curr Biol. 2005;15:1150–1155. doi: 10.1016/j.cub.2005.05.047. [DOI] [PubMed] [Google Scholar]
- Schweizer A, Rupp S, Taylor BN, Rollinghoff M, Schroppel K. The TEA/ATTS transcription factor CaTec1p regulates hyphal development and virulence in Candida albicans. Mol Microbiol. 2000;38:435–445. doi: 10.1046/j.1365-2958.2000.02132.x. [DOI] [PubMed] [Google Scholar]
- Uhl MA, Biery M, Craig N, Johnson AD. Haploinsufficiency-based large-scale forward genetic analysis of filamentous growth in the diploid human fungal pathogen C. albicans. EMBO J. 2003;22:2668–2678. doi: 10.1093/emboj/cdg256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi G, Wiltshire C, Macaskill S, Tournu H, Budge S, Brown AJ. Gcn4 co-ordinates morphogenetic and metabolic responses to amino acid starvation in Candida albicans. EMBO J. 2002;21:5448–5456. doi: 10.1093/emboj/cdf507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sanchez S, Aubert S, Iraqui I, Janbon G, Ghigo JM, d'Enfert C. Candida albicans biofilms: a developmental state associated with specific and stable gene expression patterns. Eukaryot Cell. 2004;3:536–545. doi: 10.1128/EC.3.2.536-545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Chauhan N, Ghosh A, Dixon F, Calderone R. SKN7 of Candida albicans: mutant construction and phenotype analysis. Infect Immun. 2004;72:2390–2394. doi: 10.1128/IAI.72.4.2390-2394.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottmann M, Dieter S, Brunner H, Rupp S. A screen in Saccharomyces cerevisiae identified CaMCM1, an essential gene in Candida albicans crucial for morphogenesis. Mol Microbiol. 2003;47:943–959. doi: 10.1046/j.1365-2958.2003.03358.x. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Bilu Y, Barkai N. Comparative biology: beyond sequence analysis. Curr Opin Biotechnol. 2007;18:371–377. doi: 10.1016/j.copbio.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Cuccato G, Della G Gatta, di Bernardo D. Systems and Synthetic biology: tackling genetic networks and complex diseases. Heredity. 2009;102:527–532. doi: 10.1038/hdy.2009.18. [DOI] [PubMed] [Google Scholar]
- Qian J, Dolled-Filhart M, Lin J, Yu H, Gerstein M. Beyond synexpression relationships: local clustering of time-shifted and inverted gene expression profiles identifies new, biologically relevant interactions. J Mol Biol. 2001;314:1053–1066. doi: 10.1006/jmbi.2000.5219. [DOI] [PubMed] [Google Scholar]
- Ji L, Tan KL. Identifying time-lagged gene clusters using gene expression data. Bioinformatics. 2005;21:509–516. doi: 10.1093/bioinformatics/bti026. [DOI] [PubMed] [Google Scholar]
- Liu TT, Znaidi S, Barker KS, Xu L, Homayouni R, Saidane S, Morschhauser J, Nantel A, Raymond M, Rogers PD. Genome-wide expression and location analyses of the Candida albicans Tac1p regulon. Eukaryot Cell. 2007;6:2122–2138. doi: 10.1128/EC.00327-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Znaidi S, Barker KS, Weber S, Alarco AM, Liu TT, Boucher G, Rogers PD, Raymond M. Identification of the Candida albicans Cap1p regulon. Eukaryot Cell. 2009;8:806–820. doi: 10.1128/EC.00002-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TK, Collins JJ. Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci USA. 2007;104:11197–11202. doi: 10.1073/pnas.0704624104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Preston JF, Romeo T. The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J Bacteriol. 2004;186:2724–2734. doi: 10.1128/JB.186.9.2724-2734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Wang X, Hinnebusch BJ, Preston JF, Romeo T. Depolymerization of beta-1,6-N-acetyl-D-glucosamine disrupts the integrity of diverse bacterial biofilms. J Bacteriol. 2005;187:382–387. doi: 10.1128/JB.187.1.382-387.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Supplementary table S1 lists the TF-gene regulatory associations in the candidate gene regulatory network.
Supplementary Methods.
Supplementary Table S2. Supplementary table S2 comprises the significant TF-gene regulatory associations in the biofilm and planktonic gene regulatory networks, respectively.
Supplementary Figure S1. Supplementary figure S1 shows the schematic view of the biofilm regulatory network.
Supplementary Figure S2. Supplementary figure S2 displays the schematic view of the planktonic regulatory network.
Supplementary Table S3. Supplementary table S3 consists of the TF-gene regulatory associations in the gain-of-function and loss-of-function subnetworks, respectively.
Supplementary Figure S3. Supplementary figure S3 illustrates the schematic view of the gain-of-function subnetwork.
Supplementary Figure S4. Supplementary figure S4 demonstrates the schematic view of the loss-of-function subnetwork.