

Abstract
Mutations in the mitochondrial genome are associated with a wide range of neurological symptoms, but many aspects of the basic neuronal pathology are not understood. One candidate mechanism, given the well-established role of mitochondria in calcium buffering, is a deficit in neuronal calcium homoeostasis. We therefore examined calcium responses in the neurons derived from various ‘cybrid’ embryonic stem cell lines carrying different mitochondrial DNA mutations. Brief (∼50 ms), focal glutamatergic stimuli induced a transient rise in intracellular calcium concentration, which was visualized by bulk loading the cells with the calcium dye, Oregon Green BAPTA-1. Calcium entered the neurons through N-methyl-d-aspartic acid and voltage-gated calcium channels, as has been described in many other neuronal classes. Intriguingly, while mitochondrial mutations did not affect the calcium transient in response to single glutamatergic stimuli, they did alter the responses to repeated stimuli, with each successive calcium transient decaying ever more slowly in mitochondrial mutant cell lines. A train of stimuli thus caused intracellular calcium in these cells to be significantly elevated for many tens of seconds. These results suggest that calcium-handling deficits are likely to contribute to the pathological phenotype seen in patients with mitochondrial DNA mutations.
Keywords: calcium, mitochondrial disease, SERCA, ATP, ageing, neurodegeneration
Introduction
The pathological mechanisms underlying mitochondrial disease are not understood, and yet there is increasing evidence of the importance of these disorders. The prevalence of clinical disease directly attributable to mitochondrial DNA mutations is in excess of 1 in 10 000 adults in the UK (Schaefer et al., 2008), while a study of 3000 sequential births showed that pathogenic mitochondrial DNA mutations are present at surprisingly high levels in the general population: one mitochondrial tRNA mutation, the m.3243A>G MTTL1 gene mutation, was present at a frequency of 0.14% (Elliott et al., 2008). Mitochondrial pathology may also be involved in Alzheimer's disease (Bender et al., 2008), Parkinson's disease (Bender et al., 2006), and in normal brain ageing (Bender et al., 2006; Kraytsberg et al., 2006), since all are associated with strikingly similar patterns of mitochondrial abnormalities and mitochondrial respiratory chain deficient cells.
Neuronal dysfunction and neurodegeneration are the most prominent and disabling features in patients with mitochondrial disease (DiMauro and Davidzon, 2005; DiMauro and Schon, 2008). An enduring puzzle about these pathologies is the extreme variability of presentation, ranging from focal deficits (e.g. optic neuropathy) to more global disease such as dementia and ataxia (McFarland et al., 2007). To address this issue, it is necessary to understand the basic pathological mechanisms. For instance, neuronal loss is a common feature of these pathologies (Sparaco et al., 1993), but it is possible that mitochondrial deficits could disrupt neuronal function even without neurodegeneration. These different pathological processes would indicate different strategies for treating or preventing the development of symptoms in patients with mitochondrial disease. The pathology underlying the various clinical presentations may thus be best served by very different therapies.
One possible pathological mechanism may involve the well-established role of mitochondria in regulating cytosolic Ca2+ levels. Calcium ions lie at the heart of neuronal function, transducing electrical activity into molecular signals. The specificity of the molecular response is thought to be set by the kinetics and localization of the Ca2+ transients (Berridge et al., 2003). Altered cytosolic Ca2+ regulation may also cause cells to enter into various cell death pathways. Consequently, factors which shape the Ca2+ transients are likely to assume great importance in normal neuronal function. A key role in this regard is played by mitochondria. The large electrochemical gradient across the mitochondrial inner membrane provides a powerful driving force to absorb Ca2+ from the cytosol (Vasington and Murphy, 1962; Carafoli et al., 1964; Crompton et al., 1978; Nicholls, 1978). Agents which uncouple the mitochondrial membrane potential (ΔΨm) prevent mitochondria from taking up Ca2+, and thus alter the time course of cytosolic Ca2+ transients (Werth and Thayer, 1994). The mitochondrial membrane potential is generated by the electron transfer chain (Mitchell and Moyle, 1967), but it is not known how subtle deficits in the electron transfer chain, caused by genetic mutations, might impact on Ca2+ homoeostasis in neurons. This information will be critical for us to understand how such mutations cause neurological disease.
Abnormal calcium homoeostasis has been reported in a variety of non-neuronal cell lines with oxidative phosphorylation defects. Fibroblasts from patients with m.3243A > G MTTL1 mutation showed increased basal Ca2+ levels and a sustained Ca2+ elevation in response to stimulation with 60 mM K+ compared with controls (Moudy et al., 1995). Likewise, fibroblasts from patients with complex I deficiency due to nuclear subunit mutations also showed a protracted period of elevated cytosolic Ca2+ in response to bradykinin (Visch et al., 2004, 2006; Willems et al., 2008). Another approach to understanding how pathogenic mtDNA mutations affect cellular Ca2+ homoeostasis has been to generate ‘cybrid’ cell lines, by fusing enucleated somatic cells harbouring pathogenic or neutral mtDNA mutations with a cell line in which the endogenous mtDNA had been chemically ablated (a ρ° cell line) (Brini et al., 1999). Cybrids derived from a patient with myoclonic epilepsy with ragged red fibres due to the m.8356T > C mutation showed no abnormality in cytosolic Ca2+, but did show a smaller than normal rise in mitochondrial Ca2+ in response to histamine (Brini et al., 1999). In contrast, cybrids from a patient with the neuropathy, ataxia and retinitis pigmentosa m.8993T > G mutation showed no Ca2+ abnormality. Intriguingly, in both the cybrid and fibroblast cell lines with abnormal Ca2+ responses, near-normal Ca2+ dynamics could be restored by inhibition of mitochondrial sodium/calcium exchange by the benzothiopene CGP37157 (Brini et al., 1999; Visch et al., 2004, 2006; Willems et al., 2008). A notable feature of all these studies though is that Ca2+ deficits in these cells are subtle.
Importantly however, there are currently no published studies of Ca2+ deficits in neurons with mtDNA mutations, and yet the brain is where the pathology has its major effect. Neuronal function places unique demands on Ca2+-handling pathways, and for this reason, fibroblast responses to G protein coupled receptor activation are likely to give only limited insights into how mtDNA mutations might impact on brain function. For instance, the major source of Ca2+ entering the cytosol in neurons is from the extracellular space through voltage-gated Ca2+ channels [including N-methyl-d-aspartic acid (NMDA) glutamate receptors], whereas in non-neuronal cells, the major source is the endoplasmic reticulum (Hille, 2002). Consequently, cytosolic Ca2+ in neurons can rise extremely quickly, and there is good evidence that the kinetics of the Ca2+ transient determine the cellular response (Nishiyama et al., 2000). A further difference is that store operated Ca2+ channels play a key role in Ca2+ homoeostasis in many non-neuronal cells, but not apparently in neurons. The recent demonstration that store operated Ca2+ channels are regulated by mitochondria in a Ca2+-dependent manner (Glitsch et al., 2002; Parekh, 2003, 2008) suggests another way in which mitochondrial dysfunction may affect neuronal and non-neuronal cells rather differently.
So far, neuronal mitochondrial studies have all used pharmacological agents to impair mitochondrial function, since fresh neuronal material possessing mtDNA mutations is not readily available. Neurosurgical biopsy is not indicated, either for diagnosis or treatment, and attempts to generate sustainable transgenic mice colonies carrying pathogenic mtDNA mutations have proven difficult (Fan et al., 2008). For this reason, we utilized the cybrid technology to generate mouse embryonic stem cell lines possessing different pathogenic and benign mtDNA mutations (Kirby et al., 2009). We showed previously that these embryonic stem-derived cybrids could be differentiated into mature neurons, which readily fire action potentials and form synaptically connected networks (Kirby et al., 2009). Our model system provides the opportunity to examine Ca2+ homoeostasis in neurons, the most clinically affected tissues in oxidative phosphorylation disorders.
We report here that in addition to their electrophysiological phenotype, these differentiated stem cells also show a conventional, transient Ca2+ response to physiological activation by glutamate. Surprisingly, cells possessing pathogenic mtDNA mutations had a normal baseline response to glutamate. Repeated stimulation, however, unveiled a progressive deficit resulting in markedly distorted Ca2+ transients in response to later stimuli in a train. We predict that these distorted Ca2+ transients may be misinterpreted by the various molecular transduction pathways, leading to a loss or change in specificity of Ca2+ signals. Furthermore, the greatly protracted Ca2+ transients in cell lines containing pathogenic mtDNA mutations may also increase the risk of triggering necrotic or apoptotic cell death (Clarke et al., 1993; Hardingham et al., 2002). Thus, pathogenic mtDNA mutations can cause changes in Ca2+ regulation that can explain the appearance of neuronal pathology with or without accompanying neuronal cell death.
Materials and methods
Cell lines used in this study were described previously (Kirby et al., 2009). All cybrids were derived from ES-I (CC9.3.1). Control cell lines used for the calcium studies were the parental embryonic stem cell line ES-I and a cybrid (Cy1-I) with a benign polymorphic variant (m.9821Adel) in the mitochondrial transfer RNA for arginine (Mttr). Cybrids with altered electron transfer chain function were Cy2-I and Cy3-I. Cy2-I harbours the m.6589 T > C mutation in one of the three mtDNA encoded complex IV subunits (Mtco1) and has a mild complex IV deficit (∼40% residual complex IV activity). Cy3-I has two mutations, in different mtDNA encoded complex I subunits (m.13887Cins in Mtnd6 and m.12273G > A in Mtnd5) and has a severe complex I defect (<10% residual complex I activity).
Differentiation into neurons
Parental embryonic stem cells and cybrids were differentiated into neurons using the 4+/4− method (Bain et al., 1995) as described in Kirby et al. (2009). Cultures were maintained on poly-d-lysine/laminin coated coverslips. Calcium studies were performed on differentiated neurons 7–9 days post-plating.
Dye loading
Cultures were bulk loaded with Oregon Green 488 BAPTA-1 (OGB1)-acetoxymethyl ester as follows. OGB1-acetoxymethyl (50 μg vial, Molecular Probes) was mixed with 8 μl dimethyl sulphoxide and 2 µl pluronic acid F-127 solution (10% in dimethyl sulphoxide, Molecular Probes), and then further diluted by adding 90 μl of culture media [4:1 medium: 4:1 mixture of Neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen)] and Dulbecco’s modified Eagle’s medium:F12 (1:1) (Invitrogen) supplemented with modified N2 (Ying et al., 2003). Final mixture of 12–20 μl was then added to the well containing the culture plate bathed in 3 ml of 4:1 medium. The final concentrations were OGB1-acetoxymethyl ester, ∼12 µM; 0.1% dimethyl sulphoxide; 0.002% Pluronic F-127. For the majority of experiments, the cultures were incubated for 30–40 min at 37°C and then transferred to 4:1 medium without OGB1-acetoxymethyl for 20 min at 37°C prior to transferring to the recording chamber. For the control experiments using OGB2, the cultures were loaded up in exactly the same way, only substituting OGB2-acetoxymethyl for OGB1-acetoxymethyl.
To test the effect of changing dye concentrations, we performed one set of experiments with variable loading times (Supplementary Fig. S3). The coverslips, on which the cultures were grown, were fractured using a diamond knife, and the dye was added to the medium, and fragments of the coverslip were removed either at 30 min, or after 1–2 h loading. Imaging was performed after a 20 min wash period for both.
Imaging
The recording chamber was mounted on an upright Zeiss Axioskop FS microscope fitted with Luigs and Neumann micromanipulators. Two different imaging systems were used. The first used epifluorescence illumination (standard fluorescein filter blocks), with illumination controlled by a Master 8 pulse stimulator (A.M.P.I., Jerusalem, Israel) driving a Uniblitz shutter. Images were collected at 4 Hz, using a Hamamatsu camera connected to a Macintosh computer running IPlab software. The second system used a spinning disc confocal system (Visitech International, Sunderland, UK), collecting images at up to 30 Hz using a Hamamatsu C9100/13 EM charge-coupled device camera and Visitech software run on a Dell personal computer. Analysis was done offline using ImageJ (NIH, Bethesda), MATLAB (Mathworks) and Igor (Wavemetrics, Oregon) software.
Electrophysiology
In the recording chamber, the cultures were bathed in a continuously flowing stream (1–3 ml/min) of artificial CSF (in mM: NaCl, 125; NaHCO3, 26; glucose, 10; KCl, 3.5; CaCl2, 1.2; 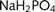 , 1.26; MgSO4, 1). Whole cell patch clamp recordings were made using 5–7 MΩ pipettes (borosilicate glass, Harvard). The pipette solution contained (in milli molar) K-methylsulphate, 125; K-Hepes, 10; Mg-ATP, 2.5; NaCl, 6; Na-GTP, 0.3 (corrected to pH 7.3–7.35 using KOH; 280 mOsm). Data were collected using Axopatch 1D amplifiers (Axon Instruments), filtered at 3 kHz, digitized at 5 kHz (AD board) and recorded on a Macintosh PowerMac computer using Axograph software. Recordings were done at room temperature.
, 1.26; MgSO4, 1). Whole cell patch clamp recordings were made using 5–7 MΩ pipettes (borosilicate glass, Harvard). The pipette solution contained (in milli molar) K-methylsulphate, 125; K-Hepes, 10; Mg-ATP, 2.5; NaCl, 6; Na-GTP, 0.3 (corrected to pH 7.3–7.35 using KOH; 280 mOsm). Data were collected using Axopatch 1D amplifiers (Axon Instruments), filtered at 3 kHz, digitized at 5 kHz (AD board) and recorded on a Macintosh PowerMac computer using Axograph software. Recordings were done at room temperature.
Glutamate application
Glutamate (1 mM in artificial CSF) was applied directly from patch pipettes using a picospritzer pressure application system (10 ms pulses; pressure = 20 psi). The timing of the pressure applications was controlled using the Master 8 pulse stimulator. The pipette tip was ∼10–50 μm from the cells and, unless the pipette was broken, we never detected any movement artefact during the glutamate applications, indicating that this means of stimulating cells was relatively atraumatic. We discarded data if the pipette was broken. We calculated that the average bolus for a 10 ms pressure application through these pipettes was about 1.6 μl by calculating the volume of solution extruded from the pipette over 1000 pressure pulses.
A23187 application
To assess the functional range of the Ca2+ dye, we applied the Ca2+ ionophore, A23187 (non-brominated form, also known as calcimycin; Invitrogen) directly to the imaged cells using the same picospritz method as for glutamate applications. External Ca2+ concentration, as for other experiments, was 2 mM, the patch pipette A23187 concentration was 1 mM and 10 ms pressure pulses were applied at 10 Hz continually until the fluorescence signal peaked (∼10–20 s; Supplementary movie 5).
Other drugs [NBQX, APV (both from Tocris, UK), NiCl2 and CdCl2 (both Sigma, UK)] were added to the entire bathing medium.
Results
Neuronal Ca2+ transients triggered by brief glutamatergic stimuli
We examined the neuronal responses to the excitatory neurotransmitter glutamate in differentiated neurons derived from embryonic stem cell lines with different mitochondrial genotypes. Specifically, we examined Ca2+ transients in these cells, as assessed by changes in fluorescence in cells bulk labelled with the Ca2+ dye, OGB1. Our intention was to stimulate the cultures in a relatively physiological way. To this end, we made brief glutamate applications at a concentration close to that found in the synaptic cleft (∼1 mM) (Clements et al., 1992). We delivered the glutamate as a single, 10 ms pressure puff (singlet), or a short train of up to 10 puffs (each 10 ms, delivered at between 1 and 0.1 Hz), giving the cells adequate time between trials to recover to baseline fluorescence levels (∼2 min for singlets, and 5–8 min for longer trains). The time course of the picospritz stimulus was visualized by filling the pipette with rhodamine and imaging the fluorescence change in front of the pipette (see Supplementary movie 1). The half-width of the fluorescence pulse was ∼50 ms, less than a single time frame (67 ms; imaging speed, 15 Hz).
We recorded two distinct patterns of Ca2+ transients in response to brief glutamate applications (Fig. 1; Supplementary Movies 2 and 3). The first pattern had slow kinetics, was very variable from trial to trial, and consequently was not obviously time-locked to the stimuli; cells showing this pattern were assumed to be non-neuronal. The second, more common pattern was a rapidly rising fluorescence that occurred simultaneously in the soma and all the extended cellular processes (beyond the temporal resolution of the imaging at 4 Hz). The responses were tightly locked to the pressure pulse, and very reproducible over as many as 30 repeated trials, indicating that this low rate of stimulation was indeed reasonably physiological, producing very little, if any, deterioration in any cell line. When these latter cells were targeted for patch clamp recordings, they could always be driven to fire action potentials, thus confirming their neuronal identity (Fig. 1B).
Figure 1.

Neuronal responses to exogenously applied glutamate. (A–C) Different recording modes showing equivalent responses of a neuron to 1 mM glutamate delivered from a distance of about 25 μm from a patch pipette subsequent to a 10 ms pressure pulse applied to the pipette. All three recording modes are shown at the same time scale for ease of comparison. The culture was bulk loaded with OGB1, and the calcium response was recorded first, prior to patching (whole cell patching causes the bulk-loaded dye to be washed out); this cell had a robust and relatively constant (<5% variability) Ca2+ fluorescence response, thus allowing an appropriate correspondence with the subsequently recorded electrophysiology. The voltage clamp response was recorded at −70 mV. The sharp event is likely to be a breakthrough action potential. The current clamp recording was made with a small (8 pA) hyperpolarizing holding current to keep the baseline Em at −70 mV. Note the different decay rates of the current, the membrane potential and the Ca2+ dye fluorescence signal [since this greatly exceeds the decay in other preparations (Trevelyan et al., 2006), this is presumed to reflect the cytosolic concentration]. The responses shown were from a cell with a severe mitochondrial deficit. (D) Photomicrograph showing a cluster of neurons loaded with the Ca2+ dye, OGB1 (epi-fluorescence illumination and with low light transillumination). The pipette (at 7 o’clock) contains 1 mM glutamate in artificial CSF, from which brief pulses of glutamate are delivered on to the cultured neurons. (E) The same field viewed using differential interference microscopy, and also showing a patch pipette (5 o’clock) onto one of the OGB labelled neurons seem in D. (F) Change in fluorescence ∼25 mm in front of the picospritzing pipette when filled with a dilute concentration of rhodamine. This allows a visualization of the time course of the local glutamate concentration in these picospritzing experiments. Pulses were delivered at 2 s intervals for this visualization, although the cell behaviour was studied using lower frequency stimuli. (G) Pulsatile Ca2+ fluorescence in response to a 0.1 Hz train of glutamate applications.
The glutamate applications induced a large inward current when the cells were held at −70 mV (Fig. 1A). The current decayed back to baseline with a time constant of 1.35 ± 0.92 s (mean ± SD; n = 5; best fit single exponential). Current clamp recordings showed that this current strongly depolarized the cells, triggering a barrage of action potentials riding on the crest of the depolarization. The membrane potential also decayed slowly (2.62 ± 1.84 s; best fit single exponential). Electrophysiological responses to picospritz glutamate applications were considerably more prolonged than estimates of individual post-synaptic current (τpsc = 1.8 ms; best fit single exponential) or post-synaptic potential decays (τpsp = 17.8 ms; both measures averages of 3; best fit single exponential) (Kirby et al., 2009).
These different time courses provided insight into the level of activation of glutamatergic receptors by the picospritz application. The profile of the rhodamine visualization of the picospritz event (Fig. 1D and Supplementary movie 1) indicates a very brief glutamate concentration peak (a few tens of milliseconds), while the neuronal response clearly extends for a much longer period, far longer than synaptic events in these cells (Kirby et al., 2009). This suggests that for picospritz glutamate applications, the glutamatergic receptors continue to be activated long after the peak glutamate concentration occurs, when the glutamate concentration has dropped to a fraction of the peak level. We can conclude from this that these cultured neurons are responsive to levels of glutamate considerably lower than 1 mM (concentration of glutamate within the pipette). Indeed, the uniform amplitude of the electrophysiological events to successive stimuli (Supplementary Fig. 1), in stark contrast to the incremental peaks in glutamate concentration (Fig. 1F), strongly suggests that glutamatergic receptors close to the puffer electrode were saturated, even from the first pulse (the rhodamine visualization of picospritzing suggests that the first pressure puff in a train produces a smaller glutamate concentration—Fig. 1F—presumably because of the diluting effect of diffusion of bath medium into the tip during the period prior to the first pressure pulse, which is very long relative to the periods prior to subsequent pulses in a train). Saturation of glutamatergic receptors may contribute to the low variability of responses that we utilized in the later experiments.
The Ca2+ transient was about half an order of magnitude slower than the current clamp responses (Table 1; best fit single exponential). Pharmacological analysis of the Ca2+ transients (Fig. 2) indicated that these relatively immature, cultured neurons had a very conventional neuronal phenotype with regard to the source of the increase in cytosolic Ca2+. The signal was abolished by the removal of extracellular Ca2+, indicating that intracellular stores make a negligible contribution to the transient signal. The signal was greatly reduced by the selective NMDA antagonist APV (50 μM), indicating that a substantial current passes through NMDA glutamatergic receptors. The residual signal was then abolished either by blockade of α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)/kainate glutamatergic receptors (10 μM NBQX), or by blockade of voltage-gated Ca2+ channels (100 μM Ni2+/100 μM Cd2+). The fact that blockade of the voltage-gated Ca2+ channels in isolation reduced the signal by ∼80% suggests that the major source of Ca2+ was through these channels. The large drop caused by blockade of NMDA channels (∼60%) can be understood simply as a 2-fold effect, with a loss of Ca2+ influx through these channels, and a reduced depolarizing drive also leading to reduced Ca2+entry through voltage-gated Ca2+ channels. The simplest interpretation of these data is that exogenously applied glutamate activates NMDA and AMPA/kainate receptors, causing a large depolarization, and frequently triggering action potentials, with Ca2+ entering through both NMDA receptors and voltage-gated Ca2+ channels.
Table 1.
Kinetics of fluorescence signal in response to single transient glutamate stimuli in different cell lines
| Cell lines | Ca2+ dye | n | 10–90% rise time | τdecay | |
|---|---|---|---|---|---|
| Mean ± SD (s) | Mean ± SD (s) | ||||
| Control | |||||
| ES-I | Parental line | OGB1 | 8 | 1.34 ± 0.87 | 7.27 ± 1.98 |
| ES-I | Parental line | OGB2 | 12 | 1.05 ± 0.36 | 6.05 ± 4.74 |
| Cy1-I | Control polymorphism | OGB1 | 5 | 0.81 ± 0.32 | 4.52 ± 1.73 |
| Mutant | |||||
| Cy2-I | Mild deficit | OGB1 | 21 | 0.92 ± 0.50 | 5.73 ± 2.66 |
| Cy3-I | Severe deficit | OGB1 | 11 | 1.06 ± 0.56 | 6.04 ± 1.81 |
Figure 2.

Ca2+ entry through NMDA and voltage-gated Ca2+ channels. Ca2+ fluorescence in ES-I cells in response to a 3 pulse train of glutamate picospritz pulses (0.33 Hz), normalized to the initial control response. Pharmacology measures were only taken for cells which had a constant control response (<10% variation over 3 repeats). The pharmacological agents were bath applied, as described in the ‘Material and methods’ section.
Subtle Ca2+ kinetic phenotype in cells with mitochondrial mutations
In all neuronal lines, the Ca2+ response to a single brief application of glutamate was variable from cell to cell (Fig. 3), suggestive of heterogeneity within the populations. The Ca2+ dye acts as a weak buffer, and this can influence the decay kinetics (Neher and Augustine, 1992; Sabatini et al., 2002), as evidenced by the fact that extending the dye loading time caused about a 25% increase in baseline fluorescence, and for the transients to be significantly slower [τdecay for short loading (30mins) = 3.08 ± 0.20 s (avg ± SEM, n = 21); long loading (>1 h) = 4.38 ± 0.25 s (n = 17); ts = 4.16, P < 0.001] (Supplementary Fig. S3). Notably though, when the loading protocol was standardized, the various cell lines showed no significant difference in the fluorescence kinetics in response to a single glutamate stimulus (Fig. 3, Table 1).
Figure 3.

Mitochondrial mutations do not affect the Ca2+ transient in response to isolated, brief glutamate applications. Sample traces of OGB1 fluorescence in response to a single brief glutamate application in a control cell line (ES-I) (A) and the cell line with severe mitochondrial mutation (Cy3-I) (B). (C) Pooled average responses in all four cell lines examined (n is the number of different cells for each line).
Since fatigue is a well-established feature of mitochondrial disease, we therefore examined how repeated stimuli affected the time course of the Ca2+ transients. To do this, we first imaged the response to a single puff of glutamate, and then after a pause for recovery of 2–5 mins, we delivered a train of glutamate puffs (same field of view and location of puffer pipette). We limited our analysis to cells that showed <5% variation in the response to the initial glutamate puff (Fig. 4), and a consistent pattern of change of responses to all subsequent puffs in the train [whether this be steady (e.g. Fig. 4A), incremental (e.g. Fig. 4B) or decremental signals]. The rationale for these tight selection criteria was to isolate consistent progressive changes induced by repeated stimuli.
Figure 4.

Progressive distortion of the Ca2+ transient during a train of glutamate pulses, in cells with severe mitochondrial dysfunction. (A) Overlays of the Ca2+ fluorescence in response to a single glutamate application and to a short train of applications in the parental cell line, ES-I. The single application (‘singlet’) response is duplicated and time shifted (black trace) to overlay the final response in the train. The final response in the train typically shows the same decay kinetics in control cell lines. These analyses were only performed for cells which showed identical initial responses (<2% variability). (B) Similar traces for Cy3-I, the cell line with a severe mitochondrial deficit. Note the marked deviation of the decay of the train relative to that of the singlet. (C) A ‘residual’ trace is calculated simply by subtracting the singlet response from the final response in the train. The example shows the derivation for the traces in panel B (Cy3-I line).
A striking feature of the control cell lines (ES-I and Cy1-I) was that consecutive transients in the train routinely matched the initial transient very well (Fig. 4A). The slow decay of the transients meant that the subsequent stimuli occur before the fluorescence returned to baseline, and yet these cells did not show simple summation of events, but rather the response appeared to be capped (cells which were distant from the glutamate electrode did sometimes show summation, albeit from small and variable amplitude, initial events).
Importantly, this apparent cap was not caused by saturation of the dye, as shown by several experiments (Supplementary Figs S2 and S3). In short, the uniformity of successive fluorescence signals in the train was also seen using OGB2 (Supplementary Fig. S2A), a variant of OGB1 with a much higher KD (Haughland, 2005). Moreover, increasing the OGB1 buffer concentration, by extending the loading time (>1 h loading), did not alter the relative decay rates after singlets and trains (i.e. it slowed the decays after both patterns of stimulation equivalently; Supplementary Fig. S3). These results also show that the apparent reproducibility of responses was not influenced by the basal occupancy of the dye, since this will be different for OGB1 and OGB2, or if the dye concentration is increased. Furthermore, a rapid application of the calcium ionophore, A23187, caused the fluorescence to greatly exceed that produced by glutamatergic stimuli (Supplementary Fig. S4; Supplementary Movies 4 and 5). The OGB1 fluorescence signal could also be increased beyond the apparent cap, by applying the uncoupling agent, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP; data not shown), indicating a possible role for mitochondria in capping the Ca2+ transient. Further details of these control experiments are provided in the supplementary figure legends. Collectively, they show that the striking feature of the normal cybrid population, in which repeated stimuli produce identical Ca2+ transients, is likely to reflect accurately the pattern of cytosolic Ca2+.
In contrast with the singlet responses, there were marked differences between the various cell lines in the responses to trains of glutamate puffs (Figs 4 and 5). Cell lines with impaired mitochondrial function (Cy2-I and Cy3-I) showed a progressive deviation of the response to successive stimuli (Fig. 4B). To analyse this further, we derived ‘residual’ traces (Fig. 4C), which represent the difference between the decays of the nth transient in the train and the time-shifted, singlet response. These residuals showed a maximal deviation of the train decay from the singlet decay at around 8–10 s after the stimulus for cell lines with both mild (Cy2-I) and severe (Cy3-I) mitochondrial dysfunction. The deviation of decays was extremely protracted for Cy3-I (severe mutants), being significantly different 50 s after the final stimulus (Fig. 5B). In other words, in cells with severe mitochondrial dysfunction, the cytosolic Ca2+ following a train of stimuli remains significantly above resting levels for tens of seconds after the stimulus ends. The deficit appears to be progressive, with a steady incremental deviation for each successive stimulus in a train (Fig. 5C).
Figure 5.

Cell lines with mutant mitochondria show altered Ca2+ handling in response to repeated glutamate applications. (A) Average ‘residuals’ for the four different cell lines to show the alteration in decay kinetics when a cell with impaired mitochondrial function is repeatedly stimulated (n is the number of different cells for each line). (B) Statistical comparisons at the two time points indicated by the arrow heads in A. (C) The progressive change in kinetics in a mutated cell line, as measured 8 s after each successive stimulus in a train of ten pulses. Data are shown only for the ES-I (parental control) and for the Cy3-I (severe mutant) lines (error bars are the SEM of the residual measures for each cell; parental, n = 9; severe, n = 7).
Discussion
We present here data that show a progressive Ca2+-handling deficit in neurons with pathogenic mtDNA mutations. To show this, we used cybrid cell lines, which allow a direct comparison of the effects on the cell biology of different mitochondrial genotypes since the somatic genotype is constant. We mimicked synaptic activation with very brief (tens of milliseconds) applications of physiological concentrations of glutamate, and restricted our analyses to cells which showed highly stable (<5% difference) Ca2+ responses to the initial stimulus in a train. By making this restriction, we revealed a striking feature of Ca2+ handling in the control cell lines, which was that the initial event was exactly reproduced following each stimulus in a train of stimuli. In contrast, cells with mtDNA mutations showed normal responses to a single glutamate application, but the response to subsequent applications became increasingly distorted. We suggest that this represents a novel form of cellular fatigue in cells with impaired oxidative phosphorylation.
Ca2+ handling in wild-type neurons
The electrophysiological responses to glutamate are reasonably stable when stimuli are presented at 0.1 Hz, and in control cells, this is reflected in the normalized pattern of Ca2+ transient. The stability of the Ca2+ transient has some noteworthy features. The long decay means that, if stimulated at 0.1 Hz, the Ca2+ level is still markedly raised at the start of the next transient. As such, one might expect summation of events, and indeed this does occur if the initial transient is small, as happens far from the glutamate pipette. Other cells, typically close to the glutamate pipette, show large amplitude and very stable initial responses, and in these cells there is no summation; rather, the amplitudes of successive peaks are constant, and since the decay is also stable, the entire shape of the event is reproduced.
The conventional view of Ca2+ cycling in neurons is that mitochondria rapidly absorb most of the Ca2+ which enters the cells when cytosolic Ca2+ concentration exceeds a threshold value of around 0.8 μM (Nicholls, 1978). This threshold for mitochondrial uptake is a powerful means of producing a cytosolic Ca2+ cap because as the cytosolic Ca2+ levels rise and once the threshold for mitochondrial uptake is surpassed, there is a great acceleration in the rate of Ca2+ clearance from the cytosol. This will produce a relatively stable capping effect over a range of Ca2+ influx parameters. Mitochondrial uptake is dependent on the large electrochemical potential generated across the inner mitochondrial membrane by the electron transfer chain. Ca2+ entry rapidly dissipates the electrochemical gradient, and thus diminishes the driving force for Ca2+ uptake, but also stimulates the recovery process by activating various mitochondrial metabolic pathways. The mitochondria thus greatly curtail the initial cytosolic transient, but then Ca2+ is returned to the cytosol over a much longer period as part of the process of recycling the cation to the extracellular space (Fierro et al., 1998).
Altered Ca2+-handling phenotype associated with mitochondrial mutations
In both mutant cell lines, the response to an isolated glutamate application was no different from controls for all measures (peak ΔF/F, 10–90% rise time, decay time constants). Our experimental protocol did not permit an estimate of baseline cytosolic Ca2+ levels (which requires ratiometric imaging), but the similarity of the ‘singlet’ response to control responses strongly suggests that baseline Ca2+ regulation in the mutant cells was ostensibly normal. In contrast to the normal cell lines, however, the responses to successive stimuli were not maintained; there was a progressive small increase in the peak fluorescence, peak on peak, and a marked slowing of the decay. Moreover, the Ca2+-handling deficit is more severe in those neurons showing the greatest reduction in oxidative phosphorylation capacity, consistent with a direct causal link between the genetic deficit and the Ca2+-handling phenotype.
It is worth considering these changes in terms of the single compartment model of Ca2+ fluxes proposed by Neher and Augustine (1992). In their formulation, the time constant of the fluorescence decay, τ, can be written as a function of the buffering capacities (Κ) of the exogenous buffer (namely the Ca2+ dye; suffix ‘e’) and endogenous buffer (suffix ‘b’), and the plasmalemmal clearance rate, γ, as follows:
to produce the requisite change during the train of stimuli, there would have to be either an acute increase in the buffering capacities of the cell (endogenous or exogenous buffering), or an acute reduction in the clearance rate. It is difficult to envisage though, how a mitochondrial mutation may allow an acute increase in buffering; if anything, the opposite might be expected. Nor is it likely that other endogenous buffering systems could be liberated over this acute time course, although we cannot rule this out (a chronic increase in Ca2+ buffering capacity might be expected as an adaptive response, although the ‘normal’ decays of the singlet Ca2+ transients provide no evidence of such a change). Far more likely is that the reduced oxidative phosphorylation in these cells causes a deficit in ATP, and thereby compromises the clearance of Ca2+ through Ca2+-ATPases. A reduced rate of clearance would also be expected to cause the small upward shift in the apparent Ca2+ cap seen in these neurons. Note though, that this increase in the peak is still less than expected from simple summation for events with a time constant, 6 s (expected increase ∼20%) indicative that the cap still exists in these cells, albeit in a compromised state.
Thus, our data suggest that baseline Ca2+ regulation is apparently well maintained, but there is a rapid loss of the ability to regulate cytosolic Ca2+ concentration when the system is strained, which may have long-lasting consequences. This observation would fit with previous studies, which show that even in the presence of a mitochondrial respiratory chain defect, ATP levels within the cell and the mitochondrial membrane potential are maintained under resting conditions (von Kleist-Retzow et al., 2007). ATP can be generated by glycolysis, and this ATP may also be used to maintain the mitochondrial membrane potential which is seen as essential for maintaining cell viability (Campanella et al., 2008; Abramov et al., 2010). With repetitive stimulation, the ability to maintain ATP levels will be impaired (Taylor et al., 1994) without effective oxidative phosphorylation and thus be likely to result in altered Ca2+ regulation.
The consequences of Ca2+ dysregulation
The consequences of this altered Ca2+ regulation are likely to be manifold. Ca2+ occupies a pivotal role coordinating many different molecular cascades in all cell types, over many different timescales (Berridge et al., 2003). In neurons, fluxes in cytosolic Ca2+ concentration coordinate transmitter release, long-term potentiation and long-term depression, dendritic spine remodelling, developmental remodelling of dendritic and axonal branches, regulation of gene expression and entry into different cell death pathways. It is believed that the exact temporal and spatial extent of the transient Ca2+ elevation explains how this single ionic species can coordinate these disparate pathways. For instance, both long-term potentiation and long-term depression have been shown to require Ca2+ entry through NMDA receptors at a dendritic spine, but the exact timing of the entry relative to the timing of a post-synaptic action potential and the associated Ca2+ elevation in the dendrite adjacent to the spine determines which of these two processes occurs. If the kinetics of the Ca2+ transient are altered by dysfunctional mitochondria, then the specificity of the Ca2+ signal will be affected, leading to the activation of either the wrong molecular cascade, or of multiple cascades in multiple spatial locations. These mtDNA mutations may also affect mitochondrial movement and distribution around the cell, with further consequences for local Ca2+ regulation (Kang et al., 2008). The pathological distortion of the Ca2+ signal may be expected to affect brain function, even without ostensible neuronal loss. This functional pathology will of course be further exacerbated by neuronal cell death caused by dysregulation.
These findings are relevant to the clinical features of mtDNA disease. In these patients, there is often development of symptoms related to neurodegeneration later in life, which is supportive of the relatively subtle changes seen in Ca2+ dysregulation (McFarland et al., 2002). In addition, there is often deterioration in patients following epileptic seizures (Okumura et al., 2008), a situation in which intense neuronal activity is associated with extreme cytosolic Ca2+ loading (Trevelyan et al., 2006). This raises issues in terms of management of patients with mitochondrial disease, especially at times of seizures. Aborting the seizure as quickly as possible is clearly crucial, but we should consider the possibility of interventions that might also limit toxicity due to high intracellular Ca2+.
In conclusion, we show here for the first time, a change in Ca2+ handling in neurons that arises as a direct consequence of mutations in the mitochondrial genome. Although we are still far from achieving a complete understanding of the consequences of such mutations, our results give insight into many facets of mitochondrial disease by showing that neuronal dysfunction may only be apparent with repetitive stimulation, and that a key feature is a progressive distortion of Ca2+ signalling with widespread consequences for neuronal development, communication and cell death.
Funding
The Wellcome Trust, Wolfson Foundation Marriott Foundation and Newcastle Centre for Brain Ageing and Vitality (supported by Biotechnology and Biological Sciences Research Council, Engineering and Physical Sciences Research Council, Economic and Social Research Council and Medical Research Council). Australian National Health and Medical Research Council (NHMRC) CJ Martin Postdoctoral Fellowship (to D.M.K.). A.J.T currently holds an Epilepsy Research UK Fellowship.
Supplementary material
Supplementary material is available at Brain online.
Supplementary Material
Glossary
Abbreviations
- mtDNA
mitochondrial DNA
- NMDA
N-methyl-d-aspartic acid
- OGB
Oregon Green 488 BAPTA
References
- Abramov AY, Smulders-Srinivasan TK, Kirby DM, Acin-Perez R, Énriquez JA, Lightowlers RN, et al. Mechanism of neurodegeneration of neurons with mitochondrial DNA mutations. Brain. 2010 doi: 10.1093/brain/awq015. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain G, Kitchens D, Yao M, Huettner JE, Gottlieb DI. Embryonic stem cells express neuronal properties in vitro. Dev Biol. 1995;168:342–57. doi: 10.1006/dbio.1995.1085. [DOI] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–7. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Bender A, Schwarzkopf RM, McMillan A, Krishnan KJ, Rieder G, Neumann M, et al. Dopaminergic midbrain neurons are the prime target for mitochondrial DNA deletions. J Neurol. 2008;255:1231–5. doi: 10.1007/s00415-008-0892-9. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–29. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- Brini M, Pinton P, King MP, Davidson M, Schon EA, Rizzuto R. A calcium signaling defect in the pathogenesis of a mitochondrial DNA inherited oxidative phosphorylation deficiency. Nat Med. 1999;5:951–4. doi: 10.1038/11396. [DOI] [PubMed] [Google Scholar]
- Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR, Abramov AY, et al. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 2008;8:13–25. doi: 10.1016/j.cmet.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Carafoli E, Rossi CS, Lehninger AL. Cation and anion balance during active accumulation of Ca++ and Mg++ by isolated mitochondria. J Biol Chem. 1964;239:3055–61. [PubMed] [Google Scholar]
- Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, et al. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–52. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- Crompton M, Moser R, Ludi H, Carafoli E. The interrelations between the transport of sodium and calcium in mitochondria of various mammalian tissues. Eur J Biochem. 1978;82:25–31. doi: 10.1111/j.1432-1033.1978.tb11993.x. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Davidzon G. Mitochondrial DNA and disease. Ann Med. 2005;37:222–32. doi: 10.1080/07853890510007368. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet. 2008;83:254–60. doi: 10.1016/j.ajhg.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Waymire KG, Narula N, Li P, Rocher C, Coskun PE, et al. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science. 2008;319:958–62. doi: 10.1126/science.1147786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro L, DiPolo R, Llano I. Intracellular calcium clearance in Purkinje cell somata from rat cerebellar slices. J Physiol. 1998;510 (Pt 2):499–512. doi: 10.1111/j.1469-7793.1998.499bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch MD, Bakowski D, Parekh AB. Store-operated Ca2+ entry depends on mitochondrial Ca2+ uptake. EMBO J. 2002;21:6744–54. doi: 10.1093/emboj/cdf675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–14. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Haughland RP. The Handbook: A guide to fluorescent probes and labelling techniques. USA: Invitrogen Corporation; 2005. [Google Scholar]
- Hille B. Ionic channels of excitable membranes. Sunderland, MA: Sinauer Associates; 2002. [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, et al. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–48. doi: 10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby DM, Rennie KJ, Smulders-Srinivasan TK, Acin-Perez R, Whittington M, Enriquez JA, et al. Transmitochondrial embryonic stem cells containing pathogenic mtDNA mutations are compromised in neuronal differentiation. Cell Prolif. 2009;42:413–24. doi: 10.1111/j.1365-2184.2009.00612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–20. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- McFarland R, Taylor RW, Turnbull DM. The neurology of mitochondrial DNA disease. Lancet Neurol. 2002;1:343–51. doi: 10.1016/s1474-4422(02)00159-x. [DOI] [PubMed] [Google Scholar]
- McFarland R, Taylor RW, Turnbull DM. Mitochondrial disease–its impact, etiology, and pathology. Curr Top Dev Biol. 2007;77:113–55. doi: 10.1016/S0070-2153(06)77005-3. [DOI] [PubMed] [Google Scholar]
- Mitchell P, Moyle J. Chemiosmotic hypothesis of oxidative phosphorylation. Nature. 1967;213:137–9. doi: 10.1038/213137a0. [DOI] [PubMed] [Google Scholar]
- Moudy AM, Handran SD, Goldberg MP, Ruffin N, Karl I, Kranz-Eble P, et al. Abnormal calcium homeostasis and mitochondrial polarization in a human encephalomyopathy. Proc Natl Acad Sci USA. 1995;92:729–33. doi: 10.1073/pnas.92.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Augustine GJ. Calcium gradients and buffers in bovine chromaffin cells. J Physiol. 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. The regulation of extramitochondrial free calcium ion concentration by rat liver mitochondria. Biochem J. 1978;176:463–74. doi: 10.1042/bj1760463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature. 2000;408:584–8. doi: 10.1038/35046067. [DOI] [PubMed] [Google Scholar]
- Okumura A, Kidokoro H, Itomi K, Maruyama K, Kubota T, Kondo Y, et al. Subacute encephalopathy: clinical features, laboratory data, neuroimaging, and outcomes. Pediatr Neurol. 2008;38:111–7. doi: 10.1016/j.pediatrneurol.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Parekh AB. Store-operated Ca2+ entry: dynamic interplay between endoplasmic reticulum, mitochondria and plasma membrane. J Physiol. 2003;547:333–48. doi: 10.1113/jphysiol.2002.034140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB. Mitochondrial regulation of store-operated CRAC channels. Cell Calcium. 2008;44:6–13. doi: 10.1016/j.ceca.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca(2+) ions in dendritic spines. Neuron. 2002;33:439–52. doi: 10.1016/s0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, Taylor RW, et al. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63:35–9. doi: 10.1002/ana.21217. [DOI] [PubMed] [Google Scholar]
- Sparaco M, Bonilla E, DiMauro S, Powers JM. Neuropathology of mitochondrial encephalomyopathies due to mitochondrial DNA defects. J Neuropathol Exp Neurol. 1993;52:1–10. doi: 10.1097/00005072-199301000-00001. [DOI] [PubMed] [Google Scholar]
- Taylor DJ, Kemp GJ, Radda GK. Bioenergetics of skeletal muscle in mitochondrial myopathy. J Neurol Sci. 1994;127:198–206. doi: 10.1016/0022-510x(94)90073-6. [DOI] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D, Watson BO, Yuste R. Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J Neurosci. 2006;26:12447–55. doi: 10.1523/JNEUROSCI.2787-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasington FD, Murphy JV. Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J Biol Chem. 1962;237:2670–7. [PubMed] [Google Scholar]
- Visch HJ, Koopman WJ, Zeegers D, van Emst-de Vries SE, van Kuppeveld FJ, van den Heuvel LW, et al. Ca2+-mobilizing agonists increase mitochondrial ATP production to accelerate cytosolic Ca2+ removal: aberrations in human complex I deficiency. Am J Physiol Cell Physiol. 2006;291:C308–16. doi: 10.1152/ajpcell.00561.2005. [DOI] [PubMed] [Google Scholar]
- Visch HJ, Rutter GA, Koopman WJ, Koenderink JB, Verkaart S, de Groot T, et al. Inhibition of mitochondrial Na+-Ca2+ exchange restores agonist-induced ATP production and Ca2+ handling in human complex I deficiency. J Biol Chem. 2004;279:40328–36. doi: 10.1074/jbc.M408068200. [DOI] [PubMed] [Google Scholar]
- von Kleist-Retzow JC, Hornig-Do HT, Schauen M, Eckertz S, Dinh TA, Stassen F, et al. Impaired mitochondrial Ca2+ homeostasis in respiratory chain-deficient cells but efficient compensation of energetic disadvantage by enhanced anaerobic glycolysis due to low ATP steady state levels. Exp Cell Res. 2007;313:3076–89. doi: 10.1016/j.yexcr.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci. 1994;14:348–56. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems PH, Valsecchi F, Distelmaier F, Verkaart S, Visch HJ, Smeitink JA, et al. Mitochondrial Ca2+ homeostasis in human NADH:ubiquinone oxidoreductase deficiency. Cell Calcium. 2008;44:123–33. doi: 10.1016/j.ceca.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Ying QL, Stavridis M, Griffiths D, Li M, Smith A. Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat Biotechnol. 2003;21:183–6. doi: 10.1038/nbt780. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.