

Abstract
Chronic inflammation is often associated with alcohol-related medical conditions. The key inducer of such inflammation, and also the best understood, is gut microflora-derived lipopolysaccharide (LPS). Alcohol can significantly increase the translocation of LPS from the gut. In healthy individuals, the adverse effects of LPS are kept in check by the actions and interactions of multiple organs. The liver plays a central role in detoxifying LPS and producing a balanced cytokine milieu. The central nervous system contributes to anti-inflammatory regulation through neuroimmunoendocrine actions. Chronic alcohol use impairs not only gut and liver functions, but also multi-organ interactions, leading to persistent systemic inflammation and ultimately, to organ damage. The study of these interactions may provide potential new targets for therapeutic intervention.
Keywords: Chronic alcohol use, Chronic inflammation, Lipopolysaccharides, Pro-inflammatory and anti-inflammatory cytokines, Kupffer cells, Monocytes, Tumor necrosis factor α, Interleukin-10, Neuroendocrine, Hypothalamo-pituitary-adrenal axis, Glucocorticoid
INTRODUCTION
Chronic inflammation is commonly associated with alcohol-related medical conditions. Accumulating evidence suggests that it acts as an etiological factor in the initiation and progression of many of these conditions. A significant number of illnesses of individuals with alcoholic liver diseases can be explained readily by a high level of circulating pro-inflammatory cytokines[1]. Humans with variants of pro- and anti-inflammatory cytokine genes show increased susceptibility to alcoholic liver disease[2]. Alcohol-mediated activation of inflammation signaling pathways can increase tumorigenesis in mice[3]. An alcohol-induced increase of tumor necrosis factor (TNF) α, a pro-inflammatory cytokine, in the mouse brain is closely linked to neurodegeneration[4]. In addition, mice in which the receptor gene for TNFα has been knocked out are resistant to alcohol-induced liver injury[5].
There are two broad sources of alcohol related inflammation inducers: those derived from alcohol damaged cells and those derived from gut microflora, specifically, lipopolysaccharide (LPS). Alcohol metabolism directly leads to the production of reactive oxygen species, known for their ability to stimulate activation of a key inflammation transcription factor nuclear factor-κB (NF-κB)[6]. Hypoxia, resulting from alcohol metabolism, is also known to induce the inflammatory response[7]. On the whole, information on such inflammation inducers and their contribution to alcohol related inflammatory conditions is still limited.
In comparison, LPS derived from gut microflora has been extensively studied as a key inducer of inflammation in alcohol-related conditions. Alcohol stimulates LPS translocation across the gut via a number of mechanisms, and alcoholics with liver diseases are known to have significantly elevated circulating LPS[8]. LPS is a major cell wall component of all Gram-negative bacteria and can mimic bacterial infection in causing an acute inflammatory response. Humans have evolved complex and coordinated defenses against LPS and its adverse effect, in which the gut, the liver, the brain, as well as innate immune cells within these organs, play crucial roles. Effects of LPS in combination with alcohol on individual organs have been reviewed extensively in the past, but less attention has been given to the communication and cooperation among these organs and their actions against LPS. In this review, our goals are to capture the latest understanding of (1) the effect of alcohol on LPS translocation and dissemination; (2) the actions of the liver and brain to manage the adverse effect of LPS; and (3) the impairment by alcohol use to the integrated host control of inflammation. We hope that such an approach will lead to a better understanding of the pathogenesis of alcohol-related tissue damage and disease.
INFLAMMATORY RESPONSE
Inflammation, mediated largely by the innate immune system, is the physiological response triggered by “danger” signals derived from tissue injury or infection[9,10]. In response to such signals, innate immune cells, such as macrophages, release pro-inflammatory cytokines and chemokines that stimulate neutrophil recruitment to the site of injury and increase vascular permeability to fluid and plasma proteins. Neutrophils, in turn, release reactive oxygen species and anti-microbial peptides and phagocytose dead and foreign material. The plasma proteins contribute proteases and opsonins to combat infection. The liver is an important innate immune organ that plays a major role in the inflammatory response by producing acute phase reactants, including C-reactive protein, mannose binding protein, complement factors, ferritin, serum amyloid A and P, and surfactant proteins[11,12]. All acute inflammation facets work together to eliminate the insult, prevent further damage, and promote tissue repair.
An inflammatory response is triggered by the binding of a microbial product or an endogenous product of tissue damage to pattern recognition receptors (PRRs) including toll-like receptors (TLRs). Upon ligand binding, PRRs activate intracellular signaling that ultimately leads to the expression of genes encoding pro-inflammatory cytokines and other effectors that promote, sustain and amplify the inflammatory response. However, inflammation is a double-edged sword, equipped to destroy invading pathogens, but also equally capable of damaging healthy tissue. Thus, the process also includes the synthesis of anti-inflammatory effectors to regulate the extent and duration of the inflammatory response. Neuroimmunoendocrine mechanisms coordinated in the central nervous system (CNS) also contribute to anti-inflammatory regulation. In contrast to acute inflammation, chronic inflammation in alcoholic liver disease is characterized by persistent expression of pro-inflammatory cytokines and persistent recruitment of monocytes and neutrophils[13,14]. These cytokines and immune effector cells maintain the damaging oxidative state, leading to sustained damage to host tissue.
INTERACTIONS OF GUT, LIVER AND BRAIN IN INFLAMMATION, AND ALCOHOL’S ROLE IN ORGAN DAMAGE
Translocation of gut microbial products
Gut microflora and translocation of microbial products: The human gastrointestinal tract is home to approximately 1014 microorganisms, consisting mostly of commensals. Besides providing the host nutritional and energy needs and shaping host mucosal and systemic immunity, the gut microflora are increasingly recognized as an important pathogenic factor in chronic inflammation-related diseases[15-18]. These physiological and pathological roles of gut microflora are mediated by two interactions: (1) inside the lumen of the gastrointestinal (GI) tract between microflora, or their products, and different types of host cells at the surface of the intestinal epithelium; and (2) outside the GI tract between microbial products that have translocated across the intestinal epithelial barrier and the host cells in the circulation or in different organs.
A causal role for a bacterial factor in human diseases in the absence of bacterial infection was first demonstrated in animal models of severe hemorrhagic shock-associated mortality and dietary hepatic injury[19,20]. In both cases, there was no apparent bacterial infection and sterilization of gut microflora with oral administration of non-absorbable antibiotics significantly improved the clinical outcomes. In addition, germfree animals were resistant to diet induced liver injury[21]. Search for a causative factor led to the findings that the diseased animals had increased circulating LPS or endotoxemia[22]. Oral administration of LPS resensitized the antibiotic treated animals to diet-induced liver injury[23]. Later studies showed that in healthy individuals LPS is readily detectable in the portal blood that flows from the GI tract to the liver[24]. More recently, evidence has emerged for translocation of microbial products and their potential pathogenic role in a variety of chronic diseases including chronic heart failure, spondyloarthritis, chronic fatigue syndrome, HIV/AIDS and AIDS-associated dementia, chronic hepatitis C, non-alcoholic steatohepatitis (as part of metabolic syndrome), and alcoholic liver disease[25-33].
Translocation of LPS across the gut epithelial barrier has been frequently ascribed to simple diffusion across epithelial cell membranes or leaking through tight junctions between adjacent cells[34]. Consistently, factors that reduce membrane permeability to various molecules and improve the function of tight junctions appear to reduce LPS translocation[35,36]. Tomita et al[37] have shown that a majority of LPS transport across colonic epithelial cells uses a receptor-mediated endocytosis system involving LPS binding proteins, CD14 and TLR4. A recent study demonstrated that LPS can also be translocated across the gut epithelium by its association with chylomicrons formed on the surface of the epithelium as a vehicle for fatty acid transport[38]. Whether these two mechanisms, receptor-mediated endocytosis and chylomicron association, represent most, if not all, of in vivo LPS transport across gut epithelial cell membranes is not yet known.
Enhancement of LPS translocation by alcohol: It has been well established in humans that heavy alcohol consumption is associated with an increase in gut permeability and LPS leakage, with or without liver disease. Abstinence for 2 wk or longer is necessary for increased gut permeability to return to a baseline level[39]. Acute heavy alcohol consumption is associated with a transient appearance of LPS in the circulation in normal human subjects[40]. Individuals with alcoholic fatty liver but not severe liver disease also show an elevated LPS level in plasma[8]. Studies of animal models show that acute alcohol feeding in mice increases LPS in plasma approximately five-fold within 30-90 min[41,42]. Daily binge feeding of alcohol in rats for 4 wk increases the plasma LPS level approximately 15-fold compared to control animals[43].
Alcohol may increase LPS leakage from the gut by a variety of mechanisms. Recent studies have demonstrated that alcohol and/or acetaldehyde can directly alter gut permeability and leakiness by induction of inducible nitric oxide synthase (iNOS) and NF-κB signaling, which, in turn, modulates a differential expression of tight junction proteins that involves the action of miRNA-122[44-46]. In addition, chronic alcohol use can cause barrier structure defects[42,47]. In addition, alcohol can also alter gut integrity and permeability indirectly. First, the Zn2+ deficiency that is common in alcoholics also adversely affects gut epithelial integrity[48,49]. Second, in patients with alcoholic liver disease, increased circulating inflammatory cytokines and LPS may promote further increases in gut leakiness[50,51]. Third, alcohol use also alters the mucosal immune system, which, in turn, could have an adverse effect on the structure or function of the gut barrier. For example, alcohol feeding in Rhesus Macaques alters the number of CD4+ and the ratio of CD4+/CD8+ cells in the mucosal immune compartment[52]. Whether these changes affect gut barrier structure and function is yet to be explored. Finally, alcohol might also affect gut permeability via its stimulatory effect on neuroendocrine hormones, known to regulate gut permeability. Locally released corticotropin-releasing hormone can stimulate the degranulation of mast cells in the GI tract thus releasing mediators for “enhanced” gut permeability[53].
In addition to the adverse effect on gut barrier structure and permeability, chronic alcohol exposure also alters microflora content and composition. Alcoholics have an overgrowth of microflora compared to non-alcoholic individuals[54,55]. Such overgrowth includes LPS-producing Gram-negative bacteria and is expected to increase microbial products in the gut and thus promote their translocation. Chronic alcohol consumption also alters the composition of the microflora[54,56,57]. Consistent with these findings, probiotic or synbiotic treatments in alcoholic patients and animals with alcoholic liver disease restore gut permeability and reduce tissue injury[56,58-60].
Routes of LPS dissemination and their distinct responses: After passing through the gut epithelium, LPS in the interstitial fluid can enter the systemic circulation by two routes: the portal vein and the GI tract lymphatic vessels[61]. By the lymphatic route, lymph fluid from the GI tract moves through the ascending lymphatic vessel and the mesenteric lymph node and eventually releases LPS into the blood stream at the thoracic duct opening. By the portal vein route, LPS in the portal blood is delivered directly into the liver, where only a minor fraction that is not detoxified or excreted enters the systemic circulation via the hepatic central vein. In rats, under normal conditions, a vast majority of LPS absorbed from the GI tract is delivered to the liver via portal blood[62], possibly due to a faster flow rate of portal blood compared to that of lymph.
A recent study revealed, surprisingly, that dietary fatty acids can drive LPS dissemination via the lymphatic route[38]. Significant increases of lymphatic LPS dissemination have also been observed in experimental peritonitis and intestinal ischemic injury caused by hemorrhage or burn[62-64]. Although experimental data are lacking, increased portal hypertension and intestinal interstitial fluid volume associated with liver disease would be expected to drive more LPS into the lymphatics. Whether alcohol affects the routes of dissemination remains to be explored.
The lymphatic LPS dissemination, rather than the portal vein, plays a dominant role in determining the level of LPS in systemic circulation[61,62]. Upon entering the liver from the portal route most LPS is detoxified and only that escaping the process can enter circulation. However, the lymphatic route, with no significant detoxification organ in transit, will release most of the bioactive LPS into the circulation and thus make it available to different organs. Consequently, LPS released from the lymphatics is responsible for most of the LPS-induced inflammatory injury in many organs. Indeed, preference of the lymphatic LPS transport in the case of experimental peritonitis is associated with lung injury[65]. Interestingly, acute alcohol feeding causes a rapid rise of circulating LPS in healthy animals[41,42], implicating the involvement of the lymphatic route in alcohol use.
The most important host cell types that bind LPS and produce a large amount of inflammatory mediators are macrophages and monocytes. The liver contains the largest pool of tissue macrophages, Kupffer cells (KCs), whereas the spleen contains the largest reserve of monocytes[66]. In response to LPS, KCs produce a significant amount of both pro- and anti-inflammatory cytokines, whereas monocytes produce predominantly pro-inflammatory cytokines (see below). Therefore, the route of LPS dissemination will also influence the types of inflammatory mediators released into circulation, which would, in turn, affect the inflammatory state in an individual.
Liver plays a key role in controlling the impact of microbial products and bears its burden during pathogenesis
Dampening the inflammatory response by detoxification and induction of anti-inflammatory cytokines: Both clinical observation and experimental research suggest that the liver plays a critical role in LPS clearance and inactivation. Two of the best known mechanisms for dampening LPS-induced inflammation are: (1) the liver’s ability to detoxify (by both Kupffer cells and hepatocytes) and excrete (by hepatocytes) LPS; and (2) the Kupffer cell’s ability to generate a large amount of the anti-inflammatory cytokine, interleukin (IL)-10.
This role of liver in LPS detoxification (clearance and inactivation) has been best demonstrated by tracking the fate of labeled LPS introduced by i.v. injection. Most LPS is rapidly concentrated in the liver and either degraded in hepatocytes or excreted into the gut via the bile duct[67-69]. In a healthy liver, LPS rapidly loses its biological activity[22], and its uptake by the spleen increases when liver is diseased[69]. Thus, the liver plays a primary role in protecting cells from exposure to microbial products.
IL-10 is a major anti-inflammatory cytokine. Like TNFα, it is produced mainly by KCs in response to LPS. IL-10 has a pleiotropic inhibitory effect on innate and adaptive immune responses. In KCs and macrophages, IL-10 strongly inhibits LPS-induced production of cytokines, including TNFα, IL-1, IL6, and IL-10 itself[70,71]. IL-10 also inhibits the differentiation and maturation of pro-inflammatory Th1 T cells of the adaptive immune system[72,73].
Significantly, KCs are responsible for producing most infection- or LPS-induced IL-10 in circulation[74,75]. Two recent studies reveal that cell-cell interactions in the liver play an important role in KC’s ability to make IL-10. A liver endothelial cell-derived factor, hepatocyte growth factor (HGF), enhances the expression of the heme oxygenase-1 (HO-1) gene, which, in turn, stimulates IL-10 production in the KCs[76]. A study in a hepatocyte-monocyte coculture model also showed that LPS-TLR4 signaling in hepatocytes stimulates IL-10 production in monocytes and likely in KCs[77].
When liver function is compromised, more microbial products enter and/or stay in circulation[78]. LPS can accumulate in other organs, especially the spleen[79]. Consequently, the inflammatory response to the microbial products becomes amplified and prolonged. The impact of such circulating LPS on many organs might be significant, especially on the brain, and might exacerbate disease conditions in these organs.
Interconnections among microbial products, liver innate immune function, injury, and repair: Gut microflora and its products also play an important role in shaping hepatic innate immunity. Livers from germ-free animals have a significantly lower number of KCs, which also show reduced activity in response to LPS[80]. Translocation of microbial products including LPS from the gut is thus necessary for the host to maintain a sufficient number of KCs in the liver via proliferation or recruitment, and to stimulate and sustain (by priming) the maturation of these cells. Although additional definitive studies are needed, such primed KCs could play a number of important roles in the liver’s innate immune defense. First, by phagocytosis and by producing immune mediators, primed KCs are effectors for immune attack against infection in the liver and for clearing microbial products, damaged tissue and debris. In addition, such primed KCs are responsible for initiating the acute phase response (APR), another important innate immune function of liver, in which hepatocytes synthesize many accessory factors involved in combating infection by phagocytosis, leukocytosis and leukocyte infiltration[81]. Finally, primed KCs are likely to be competent in producing IL-10 to protect the host against an overzealous inflammatory response.
By playing a central role in detoxifying microbial products and supplying mediators for immune defense, the liver also uniquely subjects itself to non-specific inflammatory damage. The pro-inflammatory cytokines TNFα, IL-1, and IL-6, produced by KCs in response to LPS and endogenous inflammatory inducers (e.g. from apoptotic cells), are critical for inducing secondary cytokines and chemokines, and the acute phase proteins, which, in turn, play roles in other vascular events associated with inflammation, including infiltration of neutrophils. Neutrophils are cytotoxic due to the release of superoxides (via the respiratory burst) and proteolytic enzymes that can cause cell injury. Activation of the liver APR and chemokine production, for example, due to injury in the brain, can cause liver injury[82]. The same cytokines also play important roles in liver tissue regeneration and repair[83]. Therefore, it is important to identify and recognize those conditions under which this balance of injury and regeneration is disrupted and a pathological course ensues.
Alcohol’s synergistic interaction with LPS in amplifying liver and systemic inflammatory conditions: The liver is the first internal organ after the GI tract to be exposed to alcohol and the principal organ for its metabolism. Simultaneously, the liver is exposed to increased alcohol concentrations and LPS translocated from the gut microflora. Alcohol and its metabolism can modulate the cellular response to LPS in the liver and independently generate endogenous inflammatory inducers. Thus, both alcohol and LPS act simultaneously to influence the inflammatory response in the liver and other organs.
There are four broad mechanisms by which alcohol can modulate inflammatory conditions in the liver and circulation. First, chronic alcohol exposure can amplify inflammation in the liver via a short term sensitizing/priming of KCs. For example, daily alcohol feeding for up to 8 wk sensitizes KCs in their response to LPS. KC sensitization depends on the increased LPS translocation by alcohol feeding[84]. The sensitized KCs produce an increased amount of the pro-inflammatory cytokine TNFα that contributes to liver inflammation and injury[5]. Consistent with these observations, depletion of KCs can prevent early liver injury associated with alcohol exposure[85].
Second, long term alcohol exposure impairs the liver’s ability to produce pro-inflammatory, and more significantly, anti-inflammatory cytokines. Circulating monocytes in alcoholics show reduced IL-10 production in response to LPS[86,87]. Similarly, production of plasma IL-10 in mice is depressed after 7 wk of alcohol exposure[88]. Although it remains to be confirmed in this setting, KC dysfunction may be responsible for the reduced plasma IL-10 because depletion of them abolishes circulating IL-10[83]. The reduced IL-10 production in alcoholics coincides with an increased production of plasma TNFα by circulating monocytes[89]. In supporting the anti-inflammatory function of IL-10, knockout of IL-10 and macrophage/neutrophil specific knockout of STAT3, a key IL-10 downstream target, drastically increased alcohol-induced liver inflammation in mice[90] (Bin Gao, personal communication). In addition, reduced circulating IL-10, in combination with increased IL-6, might also play a direct role in the appearance of pro-inflammatory Th17 cells in circulation and in the liver[91]. Thus, chronic alcohol exposure creates an imbalance in the cytokine milieu favoring systemic inflammatory conditions that can cause cell injury in various tissues.
Third, chronic alcohol consumption can independently stimulate inflammation via the formation of endogenous inflammation inducers: (1) Alcohol metabolism generates highly reactive byproducts, including reactive oxygen species and acetaldehyde[92,93]. These reactive byproducts modify proteins and lipids to form adducts that are processed as foreign antigens by the host immune system. Antibodies and lymphocytes against these products are commonly found in patients with alcoholic liver diseases[94,95]. A recent study has shown that alcoholics with liver disease have increased levels of the pro-inflammatory T-cell, Th17, both in circulation and in the liver[91], which might be related to a cytokine imbalance caused by chronic alcohol, as in the case of IL-10 knockout mice[96]; (2) Alcohol metabolism can promote necrosis or apoptosis of hepatocytes that can independently induce inflammation. Alcohol metabolism, especially by pericentral hepatocytes, can lead to mitochondrial damage and hypoxia, events that cause cells to undergo necrosis and apoptosis[97-99]. Intracellular components derived from these dead cells can also act as endogenous “danger signals” to activate the inflammatory response[9]; and (3) Alcohol metabolism also leads to dysregulation of fatty acid metabolism, which invariably contributes to the accumulation of fatty acids in hepatocytes. Fat accumulation in cultured cells can induce the synthesis of IL-8, a pro-inflammatory chemokine important for neutrophil recruitment[100]. Though the relative contribution to systemic inflammation remains unknown, the above alcohol-involved mechanisms are expected to significantly affect the inflammatory state within the liver.
Finally, chronic alcohol use impairs the liver’s ability to detoxify LPS and other microbial products. Circulating LPS increases significantly even in those alcoholics with no clinical liver disease other than fatty liver[8]. Thus, it is likely that fat accumulation in hepatocytes can significantly reduce their ability to detoxify LPS. Other alcohol-induced effects, such as reactive oxygen species formation, decrease in antioxidants, hypoxia, reduction of ATP production, and mitochondrial defects can cause cell injury or death. Such liver damage is expected to further diminish the liver’s ability to detoxify LPS. Consequently, more LPS will escape from the liver and stay in the circulation, which in turn increases systemic inflammatory conditions and injury among different organs. This conclusion is supported by the observations that in alcoholic hepatitis patients there is a significant increase of multi-organ failure[101]. Interestingly, the rise of circulating LPS and increased production of pro-inflammatory cytokines also raises the possibility that circulating LPS (monocyte response), not liver LPS (KC response), plays a more important role in liver and other organ injury[87,101,102].
Brain participates in the regulation of peripheral inflammation and also bears its burden
Inflammation induces brain damage: Any event leading to an increased pro-inflammatory cytokine presence in the brain creates the potential for oxidative conditions, which can result in neuronal damage. In cases of CNS trauma or infection, further escalation of the cytotoxic conditions occurs when leukocytes infiltrate the brain. While necessary to stem the infection, leukocytes in the brain are also believed to contribute to neurodegeneration in CNS disorders such as Alzheimer’s disease and multiple sclerosis. Surprisingly, even when the injury initiating event is CNS inflammation, chemokines originating in the liver are responsible for mobilizing neutrophils from the bone marrow into circulation, making them available for infiltration into the brain. Although the mechanism by which CNS inflammation is signaled to the liver has not been determined, there is evidence that liver KCs are the source of the chemokines that mobilize the neutrophils[103,104].
Peripheral LPS injection, as a model of bacterial infection in rodents, leads to elevation of the pro-inflammatory cytokine TNFα in the serum and the CNS, even though LPS does not cross the blood brain barrier. Remarkably, TNFα remains elevated in the brain for months after exposure to LPS, thus exerting a prolonged detrimental effect on the brain[105].
The central pro-inflammatory cytokines induced by peripherally-injected LPS, in particular IL-6, inhibit neurogenesis in the hippocampus through blockade of the differentiation of neural progenitor cells into neurons[106]. The TLR signaling pathway can also be activated by currently unidentified endogenous ligands and is believed to contribute to inflammation-mediated cell damage in neurodegenerative disorders such as Alzheimer’s disease[107-109].
Alcohol-related inflammation causes brain damage: Even in the absence of specific neurological or hepatic complications, excessive drinking can lead to regional structural brain damage and cognitive dysfunction[110], neuronal death and inhibition of neurogenesis[111], resulting in a reduction in brain volume, including white matter[112].
Similar to the CNS response to peripheral LPS, TNFα, and the pro-inflammatory chemokine MCP-1 are induced in the brains of mice treated intragastrically with alcohol[4]. Separately, it has been proposed that alcohol causes neurodegeneration by reducing the activity of the pro-neuronal survival transcription factor CREB and by activating the inflammation-promoting transcription factor NF-κB[111]. Alcohol can also inhibit neuronal differentiation by disrupting activity-dependent neuroprotective protein signaling[113].
Neuroimmune sensing and regulation of peripheral inflammation: The brain plays a vital role in coordinating defense against systemic infection or injury and in restoring homeostasis to safeguard the integrity of the entire organism. Physiologically important CNS responses include fever, hyperalgesia, and other sickness behaviors, as well as activation of homeostatic stress responses [the hypothalamo-pituitary-adrenal (HPA) axis and the sympathetic nervous system (SNS)]. The presence of systemic infection or injury is rapidly signaled to the brain, both by neural routes and by circulating pro-inflammatory cytokines[114].
The very early effectors of the immune response also communicate to the CNS that homeostasis has been threatened. The CNS, in turn, responds by activating the HPA axis and the SNS, the actions of which are largely anti-inflammatory, to keep inflammation in check. The anti-inflammatory signals of the HPA and SNS supplement intrinsic immune anti-inflammatory mechanisms to restrict the extent and duration of inflammation.
Signals reaching the paraventricular nucleus of the hypothalamus lead to the activation of the HPA axis, resulting in the synthesis of corticotropin releasing hormone by the hypothalamus, which in turn activates the pituitary gland to produce adrenocorticotropic hormone, which subsequently induces synthesis and release of the glucocorticoid cortisol from the adrenal cortex. Glucocorticoids inhibit the production of pro-inflammatory cytokines and promote the release of anti-inflammatory cytokines including IL-10 following binding to glucocorticoid receptors on leukocytes[115-119].
In parallel, the SNS is also activated by peripheral inflammation. SNS activation leads to peripheral release of epinephrine and norepinephrine, which also have anti-inflammatory effects at the systemic level. These catecholamines act through specific receptors expressed on innate immune cells to suppress the pro-inflammatory response and promote anti-inflammatory cytokine synthesis[120]. Thus, the glucocorticoids and neuroendocrine catecholamines produced by the HPA axis and SNS response work in concert with the anti-inflammatory cytokines produced by immune cells to contain inflammation. Since an unchecked pro-inflammatory response will damage healthy tissue due to the highly oxidative and cytotoxic milieu, the counter-regulatory mechanisms are essential to maintain tissue integrity.
Alcohol interferes with neuroimmune response and regulation: Alcohol ingestion is recognized as a stressful event by the HPA axis; a single dose of alcohol resulting in a blood alcohol level of 100 mg percent activates the HPA axis, leading to elevated cortisol levels[121]. Chronic exposure to alcohol increases the tolerance of the HPA axis to alcohol, such that serum cortisol after alcohol consumption is lower than in non-alcohol-dependent controls[121]. In addition, chronic alcohol also leads to a blunting of the HPA axis in response to external stressors other than alcohol. Tolerance to alcohol by the neuroendocrine system seems to begin to develop prior to full blown alcohol dependence and may correlate with the amount of alcohol consumed[122]. In addition, alcohol-induced complications can uncouple the HPA axis and the SNS responses to stress. Patients with alcoholic cirrhosis have increased basal sympathetic tone, as measured by neuropeptide Y production, compared to healthy controls while displaying reduced serum cortisol levels compared to controls[123]. The absence of cortisol due to the blunting of the stress responses allows inflammation to persist beyond the point where it is beneficial. Furthermore, in the context of alcoholic cirrhosis, the uncoupling of the HPA axis and the SNS has harmful hemodynamic consequences[123]. The potential role of neuroendocrine factors to mediate the effects of acute alcohol on inflammation has been investigated[124]. However, whether the blunting of the HPA axis directly mediates the effect of chronic alcohol consumption on the inflammatory state remains to be determined.
Since the low-grade chronic inflammation associated with chronic alcohol consumption is present even in the absence of any obvious infection, it is possible that LPS leakage from the gut and poor clearance by the liver may be underlying factors. In view of the tolerance that humans and animals develop to the stimulation of the HPA axis by either alcohol[125-127] or LPS[128], LPS will not be able to sufficiently activate the HPA axis and subsequent glucocorticoid release. The net outcome is that the alcohol-dependent individual would have a reduced capacity to suppress LPS-induced inflammation.
CONCLUSION
Heavy alcohol consumption contributes to systemic inflammation by interfering with the body’s natural defenses against the influx of gut microbiota and its products. Chronic alcohol use impairs the balance of microflora in the gut, the gut barrier function, the liver’s ability to detoxify bacterial products and to generate a balanced cytokine milieu, and the brain’s ability to regulate inflammation in the periphery. When these defenses are impaired, systemic inflammation ensues. The sustained inflammation has the potential to damage host tissues, beyond the local injury to the gut and the liver, including the brain. It also has the potential to alter adaptive immunity that can leads to lymphocyte-mediated inflammation and tissue injury.
As detailed in this review and Figure 1, many potential sites for alteration of gut and liver function and the complex network of neuroimmunoendocrine interactions by alcohol remain to be explored. Both acute and chronic alcohol consumption can cause devastating morbidity and mortality in the drinking population. Research into the effects of alcohol on gut leakiness and LPS translocation, mucosal and liver immunity, and CNS-liver interactions are important to the understanding of alcohol’s potential to disrupt host inflammation control mechanisms and consequent damage to all tissues in the body. Delineating the mechanism by which alcohol disrupts homeostatic control of inflammation may provide insights into effective preventative or therapeutic measures.
Figure 1.
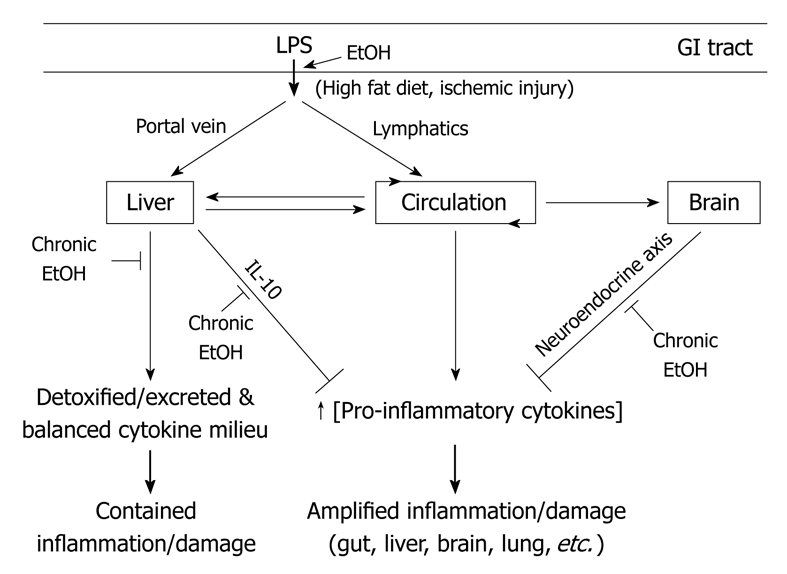
Gut microflora derived lipopolysaccharide (LPS)-dissemination, detoxification, and multi-organ host response and effects of alcohol use. A number of key events in LPS translocation and host response are depicted. First, upon translocation across the gut barrier, LPS enters circulation via two different routes: the portal vein or lymphatic vessels. LPS in the portal vein is directly delivered to the liver and most of it is detoxified and excreted, whereas most LPS in lymphatics are released into the circulation at the thoracic duct opening. Between the two routes, portal dissemination is dominant under normal conditions whereas a few known conditions including fatty diet and ischemic injury stimulate lymphatic dissemination. Second, a healthy liver plays a key role in the detoxification of LPS in the portal vein and systemic circulation but also generates a balanced cytokine milieu that lead to a contained inflammation and limited damage. Third, LPS in circulation is accessible to many organs and plays a major role in multi-organ damage, especially when LPS detoxification in liver is compromised. Lastly, LPS and its immune mediators (pro-inflammatory cytokines) can activate the neuroendocrine response in the central nervous system (CNS) that leads to the activation of the hypothalamo-pituitary-adrenal (HPA) axis and then the synthesis of cortisol, which down-regulates inflammatory responses in the periphery. A number of key sites under alcohol influence are depicted. First, alcohol enhances LPS translocation across the gut barrier by increasing microflora content and impairing gut barrier function. Second, alcohol stimulates LPS dissemination via lymphatics, evidenced by a rapid rise in circulating LPS in healthy animals fed with acute alcohol. The potential use of the portal route remains to be determined. Third, chronic alcohol use not only adversely affects the liver’s ability to detoxify LPS but also affects the liver’s ability to synthesize a key anti-inflammatory cytokine, interleukin (IL)-10. Lastly, chronic alcohol reduces the synthesis of neuroendocrine effectors that regulate systemic inflammation. Together, chronic alcohol impairs the balance of microflora in the gut, gut barrier function, the liver’s ability to detoxify bacterial products and to generate a balanced cytokine milieu, and the brain’s ability to regulate inflammation in the periphery. GI: Gastrointestinal.
Acknowledgments
The authors are grateful to Dr. Bin Gao, Dr. Gary Murray, Dr. Svetlana Radaeva, and members of the Division of Metabolism and Health Effects, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, for their critical reading of the manuscript.
Footnotes
Peer reviewer: Shinji Shimoda, MD, PhD, Medicine and Biosystemic Science, Kyushu University Graduate School of Medical Sciences, 3-1-1 Maidashi, Higashi-Ku, Fukuoka 812-8582, Japan
S- Editor Tian L L- Editor O’Neill M E- Editor Zheng XM
References
- 1.McClain CJ, Barve S, Deaciuc I, Kugelmas M, Hill D. Cytokines in alcoholic liver disease. Semin Liver Dis. 1999;19:205–219. doi: 10.1055/s-2007-1007110. [DOI] [PubMed] [Google Scholar]
- 2.Wilfred de Alwis NM, Day CP. Genetics of alcoholic liver disease and nonalcoholic fatty liver disease. Semin Liver Dis. 2007;27:44–54. doi: 10.1055/s-2006-960170. [DOI] [PubMed] [Google Scholar]
- 3.Machida K, Tsukamoto H, Mkrtchyan H, Duan L, Dynnyk A, Liu HM, Asahina K, Govindarajan S, Ray R, Ou JH, et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl Acad Sci USA. 2009;106:1548–1553. doi: 10.1073/pnas.0807390106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflammation. 2008;5:10. doi: 10.1186/1742-2094-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, Thurman RG. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942–952. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- 6.Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 7.Oliver KM, Taylor CT, Cummins EP. Hypoxia. Regulation of NFkappaB signalling during inflammation: the role of hydroxylases. Arthritis Res Ther. 2009;11:215. doi: 10.1186/ar2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukui H, Brauner B, Bode JC, Bode C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J Hepatol. 1991;12:162–169. doi: 10.1016/0168-8278(91)90933-3. [DOI] [PubMed] [Google Scholar]
- 9.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 10.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 11.Baumann H, Gauldie J. The acute phase response. Immunol Today. 1994;15:74–80. doi: 10.1016/0167-5699(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 12.Gao B, Jeong WI, Tian Z. Liver: An organ with predominant innate immunity. Hepatology. 2008;47:729–736. doi: 10.1002/hep.22034. [DOI] [PubMed] [Google Scholar]
- 13.Lee PY, Li Y, Kumagai Y, Xu Y, Weinstein JS, Kellner ES, Nacionales DC, Butfiloski EJ, van Rooijen N, Akira S, et al. Type I interferon modulates monocyte recruitment and maturation in chronic inflammation. Am J Pathol. 2009;175:2023–2033. doi: 10.2353/ajpath.2009.090328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brunt PW, Kew MC, Scheuer PJ, Sherlock S. Studies in alcoholic liver disease in Britain. I. Clinical and pathological patterns related to natural history. Gut. 1974;15:52–58. doi: 10.1136/gut.15.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol. 2008;8:411–420. doi: 10.1038/nri2316. [DOI] [PubMed] [Google Scholar]
- 16.Kelly D, Conway S, Aminov R. Commensal gut bacteria: mechanisms of immune modulation. Trends Immunol. 2005;26:326–333. doi: 10.1016/j.it.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 17.Othman M, Agüero R, Lin HC. Alterations in intestinal microbial flora and human disease. Curr Opin Gastroenterol. 2008;24:11–16. doi: 10.1097/MOG.0b013e3282f2b0d7. [DOI] [PubMed] [Google Scholar]
- 18.Mai V, Draganov PV. Recent advances and remaining gaps in our knowledge of associations between gut microbiota and human health. World J Gastroenterol. 2009;15:81–85. doi: 10.3748/wjg.15.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fine J, Frank H, Schweinburg F, Jacob S, Gordon T. The bacterial factor in traumatic shock. Ann N Y Acad Sci. 1952;55:429–445. doi: 10.1111/j.1749-6632.1952.tb26558.x. [DOI] [PubMed] [Google Scholar]
- 20.Gyorgy P. Antibiotics and liver injury. Ann N Y Acad Sci. 1954;57:925–931. doi: 10.1111/j.1749-6632.1954.tb36471.x. [DOI] [PubMed] [Google Scholar]
- 21.Luckey TD, Reyniers JA, Gyorgy P, Forbes M. Germfree animals and liver necrosis. Ann N Y Acad Sci. 1954;57:932–935. doi: 10.1111/j.1749-6632.1954.tb36472.x. [DOI] [PubMed] [Google Scholar]
- 22.Ravin HA, Rowley D, Jenkins C, Fine J. On the absorption of bacterial endotoxin from the gastro-intestinal tract of the normal and shocked animal. J Exp Med. 1960;112:783–792. doi: 10.1084/jem.112.5.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broitman SA, Gottlieb LS, Zamcheck N. Influence of neomycin and ingested endotoxin in the pathogenesis of choline deficiency cirrhosis in the adult rat. J Exp Med. 1964;119:633–642. doi: 10.1084/jem.119.4.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacob AI, Goldberg PK, Bloom N, Degenshein GA, Kozinn PJ. Endotoxin and bacteria in portal blood. Gastroenterology. 1977;72:1268–1270. [PubMed] [Google Scholar]
- 25.Sandek A, Anker SD, von Haehling S. The gut and intestinal bacteria in chronic heart failure. Curr Drug Metab. 2009;10:22–28. doi: 10.2174/138920009787048374. [DOI] [PubMed] [Google Scholar]
- 26.Vaarala O. Leaking gut in type 1 diabetes. Curr Opin Gastroenterol. 2008;24:701–706. doi: 10.1097/MOG.0b013e32830e6d98. [DOI] [PubMed] [Google Scholar]
- 27.Jacques P, Elewaut D. Joint expedition: linking gut inflammation to arthritis. Mucosal Immunol. 2008;1:364–371. doi: 10.1038/mi.2008.24. [DOI] [PubMed] [Google Scholar]
- 28.Caradonna L, Mastronardi ML, Magrone T, Cozzolongo R, Cuppone R, Manghisi OG, Caccavo D, Pellegrino NM, Amoroso A, Jirillo E, et al. Biological and clinical significance of endotoxemia in the course of hepatitis C virus infection. Curr Pharm Des. 2002;8:995–1005. doi: 10.2174/1381612024606983. [DOI] [PubMed] [Google Scholar]
- 29.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 30.Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, Zaman T, Stone D, Mefford M, Morgello S, et al. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One. 2008;3:e2516. doi: 10.1371/journal.pone.0002516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maes M, Mihaylova I, Leunis JC. Increased serum IgA and IgM against LPS of enterobacteria in chronic fatigue syndrome (CFS): indication for the involvement of gram-negative enterobacteria in the etiology of CFS and for the presence of an increased gut-intestinal permeability. J Affect Disord. 2007;99:237–240. doi: 10.1016/j.jad.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 32.Cani PD, Delzenne NM. The role of the gut microbiota in energy metabolism and metabolic disease. Curr Pharm Des. 2009;15:1546–1558. doi: 10.2174/138161209788168164. [DOI] [PubMed] [Google Scholar]
- 33.Bode C, Bode JC. Activation of the innate immune system and alcoholic liver disease: effects of ethanol per se or enhanced intestinal translocation of bacterial toxins induced by ethanol? Alcohol Clin Exp Res. 2005;29:166S–171S. doi: 10.1097/01.alc.0000189280.19073.28. [DOI] [PubMed] [Google Scholar]
- 34.Keshavarzian A, Farhadi A, Forsyth CB, Rangan J, Jakate S, Shaikh M, Banan A, Fields JZ. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J Hepatol. 2009;50:538–547. doi: 10.1016/j.jhep.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keshavarzian A, Choudhary S, Holmes EW, Yong S, Banan A, Jakate S, Fields JZ. Preventing gut leakiness by oats supplementation ameliorates alcohol-induced liver damage in rats. J Pharmacol Exp Ther. 2001;299:442–448. [PubMed] [Google Scholar]
- 36.Lambert JC, Zhou Z, Wang L, Song Z, McClain CJ, Kang YJ. Prevention of alterations in intestinal permeability is involved in zinc inhibition of acute ethanol-induced liver damage in mice. J Pharmacol Exp Ther. 2003;305:880–886. doi: 10.1124/jpet.102.047852. [DOI] [PubMed] [Google Scholar]
- 37.Tomita M, Ohkubo R, Hayashi M. Lipopolysaccharide transport system across colonic epithelial cells in normal and infective rat. Drug Metab Pharmacokinet. 2004;19:33–40. doi: 10.2133/dmpk.19.33. [DOI] [PubMed] [Google Scholar]
- 38.Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res. 2009;50:90–97. doi: 10.1194/jlr.M800156-JLR200. [DOI] [PubMed] [Google Scholar]
- 39.Bjarnason I, Peters TJ, Wise RJ. The leaky gut of alcoholism: possible route of entry for toxic compounds. Lancet. 1984;1:179–182. doi: 10.1016/s0140-6736(84)92109-3. [DOI] [PubMed] [Google Scholar]
- 40.Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J Hepatol. 1987;4:8–14. doi: 10.1016/s0168-8278(87)80003-x. [DOI] [PubMed] [Google Scholar]
- 41.Rivera CA, Bradford BU, Seabra V, Thurman RG. Role of endotoxin in the hypermetabolic state after acute ethanol exposure. Am J Physiol. 1998;275:G1252–G1258. doi: 10.1152/ajpgi.1998.275.6.G1252. [DOI] [PubMed] [Google Scholar]
- 42.Zhou Z, Wang L, Song Z, Lambert JC, McClain CJ, Kang YJ. A critical involvement of oxidative stress in acute alcohol-induced hepatic TNF-alpha production. Am J Pathol. 2003;163:1137–1146. doi: 10.1016/s0002-9440(10)63473-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Enomoto N, Yamashina S, Kono H, Schemmer P, Rivera CA, Enomoto A, Nishiura T, Nishimura T, Brenner DA, Thurman RG. Development of a new, simple rat model of early alcohol-induced liver injury based on sensitization of Kupffer cells. Hepatology. 1999;29:1680–1689. doi: 10.1002/hep.510290633. [DOI] [PubMed] [Google Scholar]
- 44.Ferrier L, Bérard F, Debrauwer L, Chabo C, Langella P, Buéno L, Fioramonti J. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol. 2006;168:1148–1154. doi: 10.2353/ajpath.2006.050617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology. 2009;50:638–644. doi: 10.1002/hep.23009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang Y, Banan A, Forsyth CB, Fields JZ, Lau CK, Zhang LJ, Keshavarzian A. Effect of alcohol on miR-212 expression in intestinal epithelial cells and its potential role in alcoholic liver disease. Alcohol Clin Exp Res. 2008;32:355–364. doi: 10.1111/j.1530-0277.2007.00584.x. [DOI] [PubMed] [Google Scholar]
- 47.Bode C, Bode JC. Effect of alcohol consumption on the gut. Best Pract Res Clin Gastroenterol. 2003;17:575–592. doi: 10.1016/s1521-6918(03)00034-9. [DOI] [PubMed] [Google Scholar]
- 48.McClain CJ, Antonow DR, Cohen DA, Shedlofsky SI. Zinc metabolism in alcoholic liver disease. Alcohol Clin Exp Res. 1986;10:582–589. doi: 10.1111/j.1530-0277.1986.tb05149.x. [DOI] [PubMed] [Google Scholar]
- 49.Lambert JC, Zhou Z, Wang L, Song Z, McClain CJ, Kang YJ. Preservation of intestinal structural integrity by zinc is independent of metallothionein in alcohol-intoxicated mice. Am J Pathol. 2004;164:1959–1966. doi: 10.1016/S0002-9440(10)63756-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hietbrink F, Besselink MG, Renooij W, de Smet MB, Draisma A, van der Hoeven H, Pickkers P. Systemic inflammation increases intestinal permeability during experimental human endotoxemia. Shock. 2009;32:374–378. doi: 10.1097/SHK.0b013e3181a2bcd6. [DOI] [PubMed] [Google Scholar]
- 51.Emmanuel DG, Madsen KL, Churchill TA, Dunn SM, Ametaj BN. Acidosis and lipopolysaccharide from Escherichia coli B:055 cause hyperpermeability of rumen and colon tissues. J Dairy Sci. 2007;90:5552–5557. doi: 10.3168/jds.2007-0257. [DOI] [PubMed] [Google Scholar]
- 52.Poonia B, Nelson S, Bagby GJ, Veazey RS. Intestinal lymphocyte subsets and turnover are affected by chronic alcohol consumption: implications for SIV/HIV infection. J Acquir Immune Defic Syndr. 2006;41:537–547. doi: 10.1097/01.qai.0000209907.43244.ee. [DOI] [PubMed] [Google Scholar]
- 53.Söderholm JD, Perdue MH. Stress and gastrointestinal tract. II. Stress and intestinal barrier function. Am J Physiol Gastrointest Liver Physiol. 2001;280:G7–G13. doi: 10.1152/ajpgi.2001.280.1.G7. [DOI] [PubMed] [Google Scholar]
- 54.Bode JC, Bode C, Heidelbach R, Dürr HK, Martini GA. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology. 1984;31:30–34. [PubMed] [Google Scholar]
- 55.Hauge T, Persson J, Danielsson D. Mucosal bacterial growth in the upper gastrointestinal tract in alcoholics (heavy drinkers) Digestion. 1997;58:591–595. doi: 10.1159/000201507. [DOI] [PubMed] [Google Scholar]
- 56.Kirpich IA, Solovieva NV, Leikhter SN, Shidakova NA, Lebedeva OV, Sidorov PI, Bazhukova TA, Soloviev AG, Barve SS, McClain CJ, et al. Probiotics restore bowel flora and improve liver enzymes in human alcohol-induced liver injury: a pilot study. Alcohol. 2008;42:675–682. doi: 10.1016/j.alcohol.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mutlu E, Keshavarzian A, Engen P, Forsyth CB, Sikaroodi M, Gillevet P. Intestinal dysbiosis: a possible mechanism of alcohol-induced endotoxemia and alcoholic steatohepatitis in rats. Alcohol Clin Exp Res. 2009;33:1836–1846. doi: 10.1111/j.1530-0277.2009.01022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stadlbauer V, Mookerjee RP, Hodges S, Wright GA, Davies NA, Jalan R. Effect of probiotic treatment on deranged neutrophil function and cytokine responses in patients with compensated alcoholic cirrhosis. J Hepatol. 2008;48:945–951. doi: 10.1016/j.jhep.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 59.Marotta F, Barreto R, Wu CC, Naito Y, Gelosa F, Lorenzetti A, Yoshioka M, Fesce E. Experimental acute alcohol pancreatitis-related liver damage and endotoxemia: synbiotics but not metronidazole have a protective effect. Chin J Dig Dis. 2005;6:193–197. doi: 10.1111/j.1443-9573.2005.00230.x. [DOI] [PubMed] [Google Scholar]
- 60.Forsyth CB, Farhadi A, Jakate SM, Tang Y, Shaikh M, Keshavarzian A. Lactobacillus GG treatment ameliorates alcohol-induced intestinal oxidative stress, gut leakiness, and liver injury in a rat model of alcoholic steatohepatitis. Alcohol. 2009;43:163–172. doi: 10.1016/j.alcohol.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Azuma K, Akiyama M, Ebata T, Totsuka M, Hayasaka H. Endogenous endotoxin absorption and the role of intestinal lymphatics. Jpn J Surg. 1983;13:535–539. doi: 10.1007/BF02469499. [DOI] [PubMed] [Google Scholar]
- 62.Olofsson P, Nylander G, Olsson P. Endotoxin: routes of transport in experimental peritonitis. Am J Surg. 1986;151:443–446. doi: 10.1016/0002-9610(86)90098-x. [DOI] [PubMed] [Google Scholar]
- 63.Olofsson P, Nylander G, Olsson P. Endotoxin-transport routes and kinetics in intestinal ischemia. Acta Chir Scand. 1985;151:635–639. [PubMed] [Google Scholar]
- 64.Gosain A, Gamelli RL. Role of the gastrointestinal tract in burn sepsis. J Burn Care Rehabil. 2005;26:85–91. doi: 10.1097/01.bcr.0000150212.21651.79. [DOI] [PubMed] [Google Scholar]
- 65.Watkins AC, Caputo FJ, Badami C, Barlos D, Xu da Z, Lu Q, Feketeova E, Deitch EA. Mesenteric lymph duct ligation attenuates lung injury and neutrophil activation after intraperitoneal injection of endotoxin in rats. J Trauma. 2008;64:126–130. doi: 10.1097/TA.0b013e3181574a8a. [DOI] [PubMed] [Google Scholar]
- 66.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Braude AI, Carey FJ, Zalesky M. Studies with radioactive endotoxin. II. Correlation of physiologic effects with distribution of radioactivity in rabbits injected with radioactive sodium chromate. J Clin Invest. 1955;34:858–866. doi: 10.1172/JCI103141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mimura Y, Sakisaka S, Harada M, Sata M, Tanikawa K. Role of hepatocytes in direct clearance of lipopolysaccharide in rats. Gastroenterology. 1995;109:1969–1976. doi: 10.1016/0016-5085(95)90765-3. [DOI] [PubMed] [Google Scholar]
- 69.Nakatani Y, Fukui H, Kitano H, Nagamoto I, Tsujimoto T, Kuriyama S, Kikuchi E, Hoppou K, Tsujii T. Endotoxin clearance and its relation to hepatic and renal disturbances in rats with liver cirrhosis. Liver. 2001;21:64–70. doi: 10.1034/j.1600-0676.2001.210110.x. [DOI] [PubMed] [Google Scholar]
- 70.Knolle P, Schlaak J, Uhrig A, Kempf P, Meyer zum Büschenfelde KH, Gerken G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J Hepatol. 1995;22:226–229. doi: 10.1016/0168-8278(95)80433-1. [DOI] [PubMed] [Google Scholar]
- 71.Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147:3815–3822. [PubMed] [Google Scholar]
- 72.Fiorentino DF, Zlotnik A, Vieira P, Mosmann TR, Howard M, Moore KW, O'Garra A. IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J Immunol. 1991;146:3444–3451. [PubMed] [Google Scholar]
- 73.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 74.Emmanuilidis K, Weighardt H, Maier S, Gerauer K, Fleischmann T, Zheng XX, Hancock WW, Holzmann B, Heidecke CD. Critical role of Kupffer cell-derived IL-10 for host defense in septic peritonitis. J Immunol. 2001;167:3919–3927. doi: 10.4049/jimmunol.167.7.3919. [DOI] [PubMed] [Google Scholar]
- 75.Kurachi K, Suzuki S, Sakaguchi T, Yokoi Y, Konno H, Baba S, Nakamura S. Kupffer cells modulate splenic interleukin-10 production in endotoxin-induced liver injury after partial hepatectomy. J Hepatol. 2003;38:193–199. doi: 10.1016/s0168-8278(02)00354-9. [DOI] [PubMed] [Google Scholar]
- 76.Kamimoto M, Mizuno S, Nakamura T. Reciprocal regulation of IL-6 and IL-10 balance by HGF via recruitment of heme oxygenase-1 in macrophages for attenuation of liver injury in a mouse model of endotoxemia. Int J Mol Med. 2009;24:161–170. doi: 10.3892/ijmm_00000219. [DOI] [PubMed] [Google Scholar]
- 77.Petrasek J, Nath B, Dolganiuc A, Hritz I, Kodys K, Catalano D, Kurt-Jones EA, Mandrekar P, Szabo G. Hepatocyte-specific Irf3 signaling protects from alcohol-induced liver injury in mice through regulation of monocyte/macrophage-derived cytokines via type-I interferons. Hepatology. 2009;50 Suppl 4:A869. [Google Scholar]
- 78.Tachiyama G, Sakon M, Kambayashi J, Iijima S, Tsujinaka T, Mori T. Endogenous endotoxemia in patients with liver cirrhosis--a quantitative analysis of endotoxin in portal and peripheral blood. Jpn J Surg. 1988;18:403–408. doi: 10.1007/BF02471464. [DOI] [PubMed] [Google Scholar]
- 79.Freudenberg M, Galanos C. Metabolic fate of endotoxin in rat. Adv Exp Med Biol. 1990;256:499–509. doi: 10.1007/978-1-4757-5140-6_44. [DOI] [PubMed] [Google Scholar]
- 80.Billiar TR, Maddaus MA, West MA, Dunn DL, Simmons RL. The role of intestinal flora on the interactions between nonparenchymal cells and hepatocytes in coculture. J Surg Res. 1988;44:397–403. doi: 10.1016/0022-4804(88)90182-5. [DOI] [PubMed] [Google Scholar]
- 81.Prins HA, Meijer C, Boelens PG, Diks J, Holtz R, Masson S, Daveau M, Meijer S, Scotté M, van Leeuwen PA. Kupffer cell-depleted rats have a diminished acute-phase response following major liver resection. Shock. 2004;21:561–565. doi: 10.1097/01.shk.0000126649.96850.36. [DOI] [PubMed] [Google Scholar]
- 82.Campbell SJ, Hughes PM, Iredale JP, Wilcockson DC, Waters S, Docagne F, Perry VH, Anthony DC. CINC-1 is an acute-phase protein induced by focal brain injury causing leukocyte mobilization and liver injury. FASEB J. 2003;17:1168–1170. doi: 10.1096/fj.02-0757fje. [DOI] [PubMed] [Google Scholar]
- 83.Rai RM, Loffreda S, Karp CL, Yang SQ, Lin HZ, Diehl AM. Kupffer cell depletion abolishes induction of interleukin-10 and permits sustained overexpression of tumor necrosis factor alpha messenger RNA in the regenerating rat liver. Hepatology. 1997;25:889–895. doi: 10.1002/hep.510250417. [DOI] [PubMed] [Google Scholar]
- 84.Enomoto N, Ikejima K, Yamashina S, Hirose M, Shimizu H, Kitamura T, Takei Y, Sato And N, Thurman RG. Kupffer cell sensitization by alcohol involves increased permeability to gut-derived endotoxin. Alcohol Clin Exp Res. 2001;25:51S–54S. doi: 10.1097/00000374-200106001-00012. [DOI] [PubMed] [Google Scholar]
- 85.Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology. 1994;20:453–460. [PubMed] [Google Scholar]
- 86.Le Moine O, Marchant A, De Groote D, Azar C, Goldman M, Devière J. Role of defective monocyte interleukin-10 release in tumor necrosis factor-alpha overproduction in alcoholics cirrhosis. Hepatology. 1995;22:1436–1439. [PubMed] [Google Scholar]
- 87.McClain CJ, Hill DB, Song Z, Deaciuc I, Barve S. Monocyte activation in alcoholic liver disease. Alcohol. 2002;27:53–61. doi: 10.1016/s0741-8329(02)00212-4. [DOI] [PubMed] [Google Scholar]
- 88.Hill DB, D'Souza NB, Lee EY, Burikhanov R, Deaciuc IV, de Villiers WJ. A role for interleukin-10 in alcohol-induced liver sensitization to bacterial lipopolysaccharide. Alcohol Clin Exp Res. 2002;26:74–82. [PubMed] [Google Scholar]
- 89.McClain CJ, Cohen DA. Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology. 1989;9:349–351. doi: 10.1002/hep.1840090302. [DOI] [PubMed] [Google Scholar]
- 90.Horiguchi N, Wang L, Mukhopadhyay P, Park O, Jeong WI, Lafdil F, Osei-Hyiaman D, Moh A, Fu XY, Pacher P, et al. Cell type-dependent pro- and anti-inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology. 2008;134:1148–1158. doi: 10.1053/j.gastro.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lemmers A, Moreno C, Gustot T, Maréchal R, Degré D, Demetter P, de Nadai P, Geerts A, Quertinmont E, Vercruysse V, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 2009;49:646–657. doi: 10.1002/hep.22680. [DOI] [PubMed] [Google Scholar]
- 92.Hoek JB, Pastorino JG. Ethanol, oxidative stress, and cytokine-induced liver cell injury. Alcohol. 2002;27:63–68. doi: 10.1016/s0741-8329(02)00215-x. [DOI] [PubMed] [Google Scholar]
- 93.Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology. 2006;43:S63–S74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- 94.Duryee MJ, Klassen LW, Thiele GM. Immunological response in alcoholic liver disease. World J Gastroenterol. 2007;13:4938–4946. doi: 10.3748/wjg.v13.i37.4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Batey RG, Wang J. Molecular pathogenesis of T lymphocyte-induced liver injury in alcoholic hepatitis. Front Biosci. 2002;7:d1662–d1675. doi: 10.2741/A870. [DOI] [PubMed] [Google Scholar]
- 96.McKinstry KK, Strutt TM, Buck A, Curtis JD, Dibble JP, Huston G, Tighe M, Hamada H, Sell S, Dutton RW, et al. IL-10 deficiency unleashes an influenza-specific Th17 response and enhances survival against high-dose challenge. J Immunol. 2009;182:7353–7363. doi: 10.4049/jimmunol.0900657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Casey CA, Nanji A, Cederbaum AI, Adachi M, Takahashi T. Alcoholic liver disease and apoptosis. Alcohol Clin Exp Res. 2001;25:49S–53S. doi: 10.1097/00000374-200105051-00009. [DOI] [PubMed] [Google Scholar]
- 98.Thurman RG, Bradford BU, Iimuro Y, Frankenberg MV, Knecht KT, Connor HD, Adachi Y, Wall C, Arteel GE, Raleigh JA, et al. Mechanisms of alcohol-induced hepatotoxicity: studies in rats. Front Biosci. 1999;4:e42–e46. doi: 10.2741/A478. [DOI] [PubMed] [Google Scholar]
- 99.Greijer AE, van der Wall E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J Clin Pathol. 2004;57:1009–1014. doi: 10.1136/jcp.2003.015032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Joshi-Barve S, Barve SS, Amancherla K, Gobejishvili L, Hill D, Cave M, Hote P, McClain CJ. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology. 2007;46:823–830. doi: 10.1002/hep.21752. [DOI] [PubMed] [Google Scholar]
- 101.Fukui H. Relation of endotoxin, endotoxin binding proteins and macrophages to severe alcoholic liver injury and multiple organ failure. Alcohol Clin Exp Res. 2005;29:172S–179S. doi: 10.1097/01.alc.0000189278.30237.e9. [DOI] [PubMed] [Google Scholar]
- 102.Tanaka S, Kumashiro R, Tanikawa K. Role of the spleen in endotoxin-induced hepatic injury in chronic alcohol-fed rats. Liver. 1992;12:306–310. doi: 10.1111/j.1600-0676.1992.tb00578.x. [DOI] [PubMed] [Google Scholar]
- 103.Wilcockson DC, Campbell SJ, Anthony DC, Perry VH. The systemic and local acute phase response following acute brain injury. J Cereb Blood Flow Metab. 2002;22:318–326. doi: 10.1097/00004647-200203000-00009. [DOI] [PubMed] [Google Scholar]
- 104.Campbell SJ, Zahid I, Losey P, Law S, Jiang Y, Bilgen M, van Rooijen N, Morsali D, Davis AE, Anthony DC. Liver Kupffer cells control the magnitude of the inflammatory response in the injured brain and spinal cord. Neuropharmacology. 2008;55:780–787. doi: 10.1016/j.neuropharm.2008.06.074. [DOI] [PubMed] [Google Scholar]
- 105.Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- 107.Balistreri CR, Grimaldi MP, Chiappelli M, Licastro F, Castiglia L, Listì F, Vasto S, Lio D, Caruso C, Candore G. Association between the polymorphisms of TLR4 and CD14 genes and Alzheimer's disease. Curr Pharm Des. 2008;14:2672–2677. doi: 10.2174/138161208786264089. [DOI] [PubMed] [Google Scholar]
- 108.Tang SC, Lathia JD, Selvaraj PK, Jo DG, Mughal MR, Cheng A, Siler DA, Markesbery WR, Arumugam TV, Mattson MP. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp Neurol. 2008;213:114–121. doi: 10.1016/j.expneurol.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV. Toll-like receptors in neurodegeneration. Brain Res Rev. 2009;59:278–292. doi: 10.1016/j.brainresrev.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Harper C, Matsumoto I. Ethanol and brain damage. Curr Opin Pharmacol. 2005;5:73–78. doi: 10.1016/j.coph.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 111.Crews FT, Nixon K. Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol. 2009;44:115–127. doi: 10.1093/alcalc/agn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Harper C. The neuropathology of alcohol-specific brain damage, or does alcohol damage the brain? J Neuropathol Exp Neurol. 1998;57:101–110. doi: 10.1097/00005072-199802000-00001. [DOI] [PubMed] [Google Scholar]
- 113.Chen S, Charness ME. Ethanol inhibits neuronal differentiation by disrupting activity-dependent neuroprotective protein signaling. Proc Natl Acad Sci USA. 2008;105:19962–19967. doi: 10.1073/pnas.0807758105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Quan N, Banks WA. Brain-immune communication pathways. Brain Behav Immun. 2007;21:727–735. doi: 10.1016/j.bbi.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 115.Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Scheinman RI, Gualberto A, Jewell CM, Cidlowski JA, Baldwin AS Jr. Characterization of mechanisms involved in transrepression of NF-kappa B by activated glucocorticoid receptors. Mol Cell Biol. 1995;15:943–953. doi: 10.1128/mcb.15.2.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Miyamasu M, Misaki Y, Izumi S, Takaishi T, Morita Y, Nakamura H, Matsushima K, Kasahara T, Hirai K. Glucocorticoids inhibit chemokine generation by human eosinophils. J Allergy Clin Immunol. 1998;101:75–83. doi: 10.1016/S0091-6749(98)70196-4. [DOI] [PubMed] [Google Scholar]
- 118.Sewell WA, Scurr LL, Orphanides H, Kinder S, Ludowyke RI. Induction of interleukin-4 and interleukin-5 expression in mast cells is inhibited by glucocorticoids. Clin Diagn Lab Immunol. 1998;5:18–23. doi: 10.1128/cdli.5.1.18-23.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Richards DF, Fernandez M, Caulfield J, Hawrylowicz CM. Glucocorticoids drive human CD8(+) T cell differentiation towards a phenotype with high IL-10 and reduced IL-4, IL-5 and IL-13 production. Eur J Immunol. 2000;30:2344–2354. doi: 10.1002/1521-4141(2000)30:8<2344::AID-IMMU2344>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 120.Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 121.Spencer RL, Hutchison KE. Alcohol, aging, and the stress response. Alcohol Res Health. 1999;23:272–283. [PMC free article] [PubMed] [Google Scholar]
- 122.Richardson HN, Lee SY, O'Dell LE, Koob GF, Rivier CL. Alcohol self-administration acutely stimulates the hypothalamic-pituitary-adrenal axis, but alcohol dependence leads to a dampened neuroendocrine state. Eur J Neurosci. 2008;28:1641–1653. doi: 10.1111/j.1460-9568.2008.06455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wiest R, Moleda L, Zietz B, Hellerbrand C, Schölmerich J, Straub R. Uncoupling of sympathetic nervous system and hypothalamic-pituitary-adrenal axis in cirrhosis. J Gastroenterol Hepatol. 2008;23:1901–1908. doi: 10.1111/j.1440-1746.2008.05456.x. [DOI] [PubMed] [Google Scholar]
- 124.Glover M, Cheng B, Fan R, Pruett S. The role of stress mediators in modulation of cytokine production by ethanol. Toxicol Appl Pharmacol. 2009;239:98–105. doi: 10.1016/j.taap.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rasmussen DD, Boldt BM, Bryant CA, Mitton DR, Larsen SA, Wilkinson CW. Chronic daily ethanol and withdrawal: 1. Long-term changes in the hypothalamo-pituitary-adrenal axis. Alcohol Clin Exp Res. 2000;24:1836–1849. [PubMed] [Google Scholar]
- 126.Wand GS, Dobs AS. Alterations in the hypothalamic-pituitary-adrenal axis in actively drinking alcoholics. J Clin Endocrinol Metab. 1991;72:1290–1295. doi: 10.1210/jcem-72-6-1290. [DOI] [PubMed] [Google Scholar]
- 127.Dai X, Thavundayil J, Santella S, Gianoulakis C. Response of the HPA-axis to alcohol and stress as a function of alcohol dependence and family history of alcoholism. Psychoneuroendocrinology. 2007;32:293–305. doi: 10.1016/j.psyneuen.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 128.Rivier C. Acute interactions between cytokines and alcohol on ACTH and corticosterone secretion in the rat. Alcohol Clin Exp Res. 1993;17:946–950. doi: 10.1111/j.1530-0277.1993.tb05646.x. [DOI] [PubMed] [Google Scholar]