

Abstract
Hepatitis C virus (HCV) is one of the main etiological factors responsible for liver disease worldwide. It has been estimated that there are over 170 million people infected with HCV worldwide. Of these infected individuals, approximately 75% will go on to develop a life long necroinflammatory liver disease, which over decades, can result in serious complications, such as cirrhosis and hepatocellular carcinoma. Currently there is no effective vaccine and whilst antiviral therapies have been improved, they are still only effective in approximately 50% of individuals. HCV infection stands as a major cause of global morbidity and suffering, and places a significant burden on health systems. The second highest cause of liver disease in the western world is alcoholic liver disease. Frequently, HCV infected individuals consume alcohol, and the combined effect of HCV and alcohol consumption is deleterious for both liver disease and response to treatment. This review discusses the impact of alcohol metabolism on HCV replication and the negative impact on interferon (IFN)-α treatment, with a particular focus on how alcohol and HCV act synergistically to increase oxidative stress, ultimately leading to exacerbated liver disease and a reduction in the efficacy of IFN-α treatment. A better understanding of the complicated mechanisms at play in hepatocytes infected with HCV and metabolizing alcohol will hopefully provide better treatment options for chronic hepatitis C individuals that consume alcohol.
Keywords: Alcohol metabolism, Hepatitis C virus, Reactive oxygen species, Interferon signaling
INTRODUCTION
Hepatitis C virus (HCV) has emerged as a significant human pathogen worldwide and is now the most common cause of significant liver disease in many countries[1]. Although the disease is typically not severe in the acute phase, more than 70% of infected individuals develop a persistent infection that, over the course of 2-3 decades, can result in progressive hepatic fibrosis and loss of functioning hepatocyte mass[2,3]. A proportion of infected individuals will develop cirrhosis and eventually hepatocellular carcinoma (HCC)[4,5]. This progressive liver disease is thought to arise as a result of the chronic inflammatory response to clear HCV infected hepatocytes, resulting in an environment that is favorable for the fibrogenic process[6].
There are numerous clinical studies that suggest a strong epidemiological link between the consumption of alcohol and accelerated liver disease in HCV infected individuals[7-9]. The majority of alcohol metabolism takes place in hepatocytes, which is also the primary site of HCV replication, and thus it is logical that interactions between the two will occur, both at the clinical and molecular level. The majority of studies have concluded that HCV infected individuals that consume alcohol show a strong propensity for accelerated liver disease, and that alcohol metabolism and concurrent HCV infection act synergistically to facilitate this rapid disease progression[7-11]. In fact, excessive alcohol consumption is now a recognized co-factor in liver disease progression, and persons infected with HCV are recommended to limit their alcohol intake[12]. Despite strong epidemiological evidence, the molecular mechanisms by which alcohol consumption exacerbates chronic hepatitis C (CHC) remain unclear. Furthermore, the interactions between alcohol metabolism, HCV, and the host antiviral immune response are also unknown. Clearly, the relationship between alcohol and HCV is complex, and the mechanisms responsible for accelerated disease progression are most likely not related to a single factor, but are the result of alterations to hepatocyte homeostasis, production of cytokines, and modification of the immune system.
This review will focus on the role that alcohol metabolism has on HCV RNA replication and discuss the potential molecular mechanisms responsible, with a particular focus on oxidative stress. The effect of alcohol on interferon (IFN)-α action and its modulation of signal transduction pathways will also be discussed.
ALCOHOL CONSUMPTION ACCELERATES HCV RELATED LIVER DISEASE
One of the first reports documenting the role of alcohol consumption on CHC progression was published by Seeff et al[5], in which they reported that two thirds of HCV positive patients that died from end stage liver disease were chronic consumers of alcohol. Poynard et al[4] extended this study, showing that the consumption of 50 g/d of alcohol increased the rate at which fibrosis progresses in HCV infected individuals. They also identified three independent risk factors that were associated with increased rates of fibrosis: age greater than 40 years at time of infection, being male, and consuming more than 50 g of alcohol per day. At the virological level, Pessione et al[8] found a direct dose dependent correlation between increasing HCV RNA levels and increasing levels of alcohol consumption. The mechanism for this increase was not established, but it was postulated that the increase in HCV RNA could be due to a direct effect of alcohol increasing viral replication or through reduced clearance of the virus by the immune system. One of the most comprehensive studies investigating the effect of alcohol on HCV disease progression was performed by Corrao et al[9], in which they studied a large cohort of 417 patients. The most striking finding of this study was a comparison of the associated risk factors for developing cirrhosis between HCV infected patients that abused alcohol and those that abstained. The risk factor for developing cirrhosis in patients that were HCV positive but did not consume alcohol was 9, compared to a significantly higher risk factor of 147 for those patients that abused alcohol. This study added further weight to the hypothesis that HCV and alcohol metabolism synergistically contribute to exacerbated liver disease.
There have been a number of studies that have documented a clear link between excess alcohol consumption and an increased risk of HCC development. It has been suggested that HCV infected individuals that consume alcohol show a 100-fold increase in their risk of developing HCC[13-18]. Clearly, chronic consumption of alcohol in HCV infected individuals is a dangerous mix, with significant clinical implications.
ALCOHOL AND HCV INDUCE OXIDATIVE STRESS
Reactive oxygen species (ROS) are defined as small highly reactive oxygen-containing molecules that cause oxidative stress when the rate at which they are produced is greater than the rate at which they are removed, leading to a disturbance in the pro-oxidant/anti-oxidant balance. Cytochrome P450-2E1 (CYP2E1) metabolism of alcohol stimulates the microsomal production of ROS, such as superoxide anions (O2.-), hydroxyl radicals (OH-), 1-hydroxyethyl radicals (CH3C.HOH), lipid hydroperoxides (LOOH), and (when iron levels increase due to alcohol metabolism) the production of ferryl radicals[19]. Oxidative stress can potentially lead to cellular damage that can play a role in a variety of pathological conditions[20]. The generation of hepatic oxidative stress in CHC is now well established, and most likely a consequence of HCV proteins disrupting mitochondrial and hepatocyte organelle function, in addition to the inflammatory response directed towards HCV infected hepatocytes. Under normal conditions, oxidative stress exists in a state of equilibrium with cellular antioxidants that scavenge ROS and prevent cellular injury. However, when cellular antioxidant mechanisms are overwhelmed through chronic oxidative stress or disease processes, oxidative stress production continues unchecked, with pathological consequences. This chronic exposure of the liver to oxidative stress in CHC has significant clinical implications, as it is well documented that oxidative stress is a mediator of hepatic inflammation, fibrosis, and the development of HCC[21].
One of the most significant mediators of oxidative stress in the liver is the metabolism of alcohol by the enzyme CYP2E1. Metabolism of alcohol can also occur via the other main alcohol metabolizing enzyme alcohol dehydrogenase (ADH). Whilst ADH-mediated metabolism of alcohol does produce ROS, CYP2E1-mediated metabolism of alcohol produces levels of ROS that greatly exceed that of the ROS produced by ADH. Alcohol metabolism not only directly produces ROS, but it also creates an environment that is favorable for oxidative stress. It is becoming increasingly clear that oxidative stress plays a prominent role in the pathogenesis of alcohol-induced liver disease[22] and CHC[21]. It is therefore not difficult to envisage the potentially explosive situation where oxidative stress produced by HCV and alcohol leads to a synergistic exacerbation of liver disease.
HCV INFECTION INDUCES A STATE OF OXIDATIVE STRESS
While clinical studies have suggested that markers of oxidative stress are increased in CHC, it was the development of mice transgenic for the HCV core protein that clearly demonstrated that HCV directly induces a state of oxidative stress[23]. Mice expressing either the HCV core or the complete HCV polyprotein developed pathologies consistent with those observed in human HCV infection[23,24], such as steatosis and development of HCC. Prior to HCC development, the HCV core-expressing mice showed a marked increase in lipid peroxidation and activation of the anti-oxidant system, suggesting that the expression of HCV core is sufficient to induce oxidative stress in the mouse liver and initiate HCC through DNA damage and modulation of signaling cascades[23]. It was subsequently shown in vitro that HCV core expression results in increased generation of ROS and expression of antioxidant enzymes[25-27]. Mechanistically, it was shown that this increase in oxidative stress was due to interactions between HCV core and destabilisation of the mitochondrial electron transport chain and that this was further enhanced in the presence of alcohol[28,29].
In addition to the core protein, the HCV nonstructural protein non-structural 5A (NS5A) has also been demonstrated to increase cellular ROS, albeit through a different mechanism to that of the HCV core. HCV NS5A localizes to the endoplasmic reticulum (ER) and lipid droplets, and is part of the HCV replication complex that results in the formation of altered cytoplasmic membrane structures, known as the membranous web. It has been postulated that this change in the membrane structure results in ER stress and the unfolded protein response, leading to the release of ER Ca2+ stores and resulting in the formation of oxidative stress[30]. Expression of ectopic NS5A results in oxidative stress, and NS5A-induced transcriptional activation can be blocked by the treatment of cells with the free radical scavenges pyrrolidine-2,4-dicarboxylate acid and N-acetyl-cysteine (NAC)[31], suggesting that NS5A induces a state of oxidative stress in the cells. However, these studies should be interrupted with caution, as they are reliant on ectopic over-expression of HCV proteins in the absence of the complete repertoire of HCV proteins and RNA replication. However, Huh-7 cells harboring the HCV replicon do induce a state of oxidative stress[32,33]. Thus, it is logical to hypothesize that HCV replication and alcohol metabolism lead to a synergistic increase in hepatic oxidative stress that contributes to accelerated liver disease.
ALCOHOL MODULATES HCV REPLICATION
As previously outlined, there is clinical evidence to suggest that alcohol metabolism increases HCV replication and modulates the host response to HCV[4,7,8]. While the exact molecular mechanisms are unclear, there have been a number of postulated mechanisms, such as (1) an alcohol-induced increase in HCV RNA replication; (2) enhancement of HCV quasispecies complexity; (3) modulation of the immune system; and (4) synergistic increase in ROS. However, pinpointing the precise mechanism of how alcohol and HCV interact in the laboratory has been hampered by the lack of a small animal model of HCV pathogenesis and, until recently, the inability to culture the virus. However, the recent development of a fully permissive cell culture system for HCV has been a significant advancement for the study of HCV biology[34]. This is further compounded by the fact that hepatocyte-derived cell lines (including the Huh-7 cell line that is permissive for HCV replication) do not express the alcohol metabolizing enzymes ADH and CYP2E1 in culture. However, numerous studies have been conducted using non-CYP2E1/ADH hepatocyte derived cell lines to determine the impact of alcohol on HCV replication, often with conflicting conclusions. To overcome this limitation, hepatocyte derived cell lines have been engineered to express CYP2E1[19], including Huh-7 cell lines that support HCV replication and metabolize alcohol[32]. These cells provide a useful tool to study the molecular interactions between alcohol metabolism and HCV.
There are conflicting reports surrounding the role of alcohol metabolism on HCV replication in vitro, which most likely reflects the different model systems used in different laboratories. Using HCV replicon cell lines that constitutively express CYP2E1 (replicon cells constitutively replicate HCV RNA under the control of an antibiotic selection marker, but do not produce infection virus particles), it was shown that physiological concentrations of alcohol (0-100 mmol/L) resulted in a CYP2E1-dependent increase in HCV RNA levels, 72 h following alcohol stimulation[32]. This alcohol-induced increase in replication was blocked in the presence of the anti-oxidant NAC, strongly suggesting that oxidative stress plays a central role in this alcohol-induced effect. Furthermore, consistent with the data suggesting that HCV induces a state of oxidative stress alone, NAC reduced HCV replication by 50% in both HCV replicon cell lines and Huh-7 cells infected with HCV cell culture derived virus (JFH-1) (Beard MR, personal communication). Therefore, it is conceivable that HCV uses oxidative stress to its replicative advantage. Moreover, when the infectious HCV JFH-1 virus was used to infect CYP2E1 expressing Huh-7 cells, treatment with alcohol also resulted in a significant increase in HCV (JFH-1) replication (Beard MR, personal communication). Collectively, these results suggest that CYP2E1-mediated metabolism of alcohol results in an increase of HCV replication, at least in vitro, and are consistent with the clinical observations described earlier. In contrast, CYP2E1-independent, alcohol-induced increases in HCV replication have been reported using the HCV replicon system[35,36]. Conversely, it was shown that a single dose of acute ethanol exposure inhibits HCV replication[37]. There are several methodological issues that explain these apparent discrepancies. Firstly, Huh-7 cells are different between laboratories and it is possible that low basal levels of CYP2E1/ADH exist. Secondly, there are significant differences in these studies in regards to experimental design that could account for the differences noted. For example, significant differences may occur depending on whether experimental conditions mimic acute or chronic exposure to ethanol. Acute alcohol metabolism results in a rapid increase in cellular ROS and it is possible that this burst of oxidative stress can inhibit replication[38,39]. This correlates with unpublished findings in our laboratory, where the treatment of HCV replicon cells and HCVcc (JFH-1) infected Huh-7 cells with H202, to induce an acute burst of oxidative stress, results in a decrease in HCV replication, although chronic exposure to alcohol increases HCV replication. We have also shown in our laboratory that there is a bi-phasic effect of alcohol metabolism on HCV replication, suggesting that perhaps moderate levels of oxidative stress stimulate replication whereas more pronounced levels of oxidative stress could repress replication[32].
The molecular mechanism whereby ROS modulates HCV replication is unknown. However, ROS have the ability to act as potent second messengers and activate cellular transcription factors, such as STAT3, nuclear factor κB, NF-AT, and AP-1[31,40]. Interestingly, it has been reported that ROS induced by HCV replication in vitro can activate the transcription factor STAT3, which can, in turn, lead to the stimulation of STAT3-dependent genes that are capable of creating a cellular environment favourable for HCV replication[41]. Our laboratory has also shown this HCV/ROS increase in STAT3 activation. Expression of a constitutively active STAT3 molecule increases HCV replication, while conversely, specific inhibitors of STAT3 (AG490 and STA-21) cause significant decreases in replication (Beard MR, unpublished results). Clearly, the role of STAT3 and STAT3-dependent gene expression warrants further investigation.
However, not all studies implicate ROS in the increase in HCV replication following alcohol stimulation. Seronello et al[42] suggested that metabolism of alcohol modulates host lipid metabolism, thus, potentiating HCV replication. Obviously, the interactions between HCV and alcohol metabolism is a complex and multi-factorial process (Figure 1), and further investigations are required to ascertain the role of the alcohol-induced modulation of HCV replication.
Figure 1.

Proposed model of hepatitis C virus (HCV)/alcohol interactions in hepatocytes. Metabolism of alcohol by cytochrome P450-2E1 (CYP2E1) and HCV replication leads to a synergistic induction of oxidative stress in the cell. Oxidative stress inhibits interferon (IFN)-α signalling and also leads to increased rates of inflammation, cirrhosis, and hepatocellular carcinoma (HCC). NS5A: Non-structural 5A; NF-κB: Nuclear factor κB; ROS: Reactive oxygen species.
EFFICACY OF IFN-α IN THE PRESENCE OF ALCOHOL METABOLISM
IFN-α2b/Ribavirin combination therapy is the current and only treatment strategy for CHC. Whilst the exact mode of IFN-α action is not well understood, IFN-α therapy is thought to result in the induction of interferon-stimulated genes (ISGs), many of which have antiviral activity. The binding of IFN to its cellular receptor results in rapid autophosphorylation and activation of receptor-associated Janus activated kinases (JAKs), TYK2 and JAK1, and activation of the JAK/STAT signaling cascade[43]. TYK2 and JAK1 phosphorylate tyrosine residues on the cytoplasmic tail of the receptor, this provides docking sites for the signal transducer and activator of transcription (STATs), which are latent cytoplasmic transcription factors that transduce signals from the cell surface to the nucleus, where they directly regulate transcription. STAT1 is phosphorylated at tyrosine residue 701 (Y-701) and STAT2 at tyrosine residue 690 (Y-690). Once phosphorylated, STAT1 and STAT2 then form active heterodimers and translocate to the nucleus where they directly activate transcription via the multi component transcription factor ISG factor 3, which binds to the interferon-stimulated response element (ISRE), a cis-acting DNA element found in the promoters of the majority of type I IFN genes. Within the nucleus, STAT-1 is further phosphorylated on serine residue 727, resulting in an increase in the transcriptional activation ability of this complex. The end step of this signaling cascade culminates in the transcriptional activation of hundreds of ISGs. These produce anti-viral proteins capable of limiting HCV replication in hepatocytes and modulating the immune response (Figure 2). As mentioned previously, it is a well established clinical observation that alcohol consumption reduces the efficacy of IFN treatment[7]. The molecular basis that underlies the reduced anti-viral capacity of IFN-α in the presence of alcohol metabolism remains to be established; however, evidence suggests that alcohol might directly inhibit the actions of IFN-α in patients at the signaling level.
Figure 2.
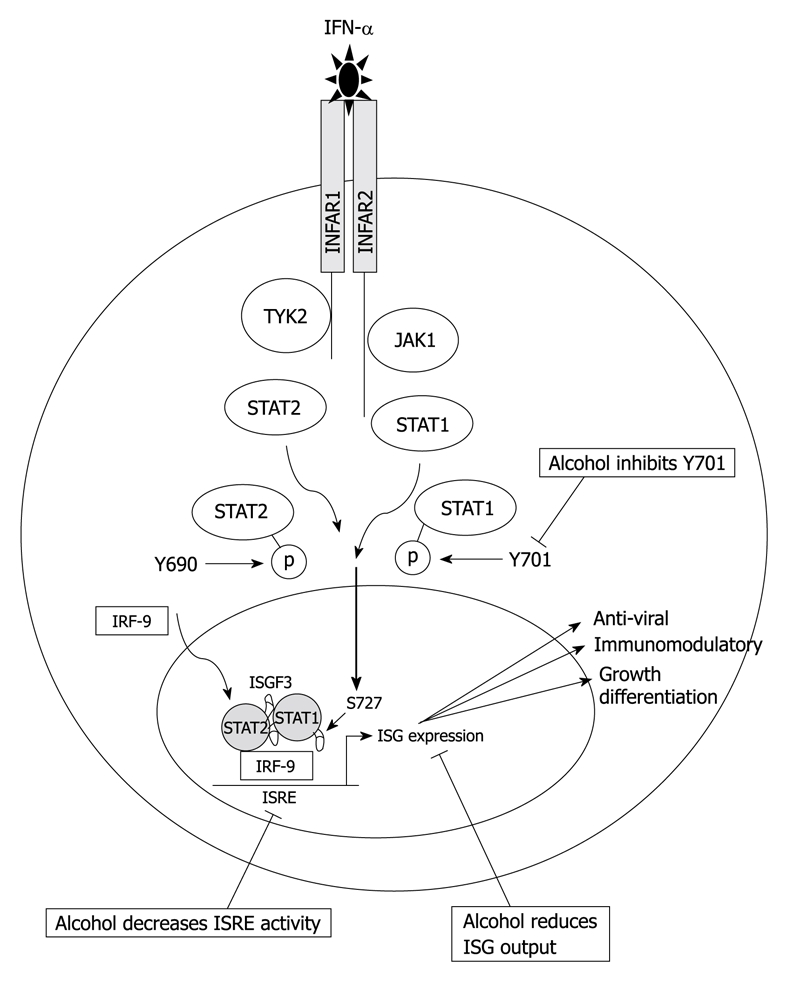
IFN-α signal transduction. Binding of IFN-α to its cognate receptor on the cell surface results in activation of the Janus activated kinase (JAK)/STAT signaling pathway culminating in the production of interferon-stimulated genes (ISGs), many of which have anti-viral properties that act to limit HCV infection. Alcohol inhibits Y701, decreases interferon-stimulated response element (ISRE) activity, and subsequently dampens the ISG response.
In combination with the effect of alcohol on HCV replication and accelerated disease progression, alcohol metabolism also decreases the efficacy of IFN-α. Patients who consume alcohol do not respond effectively to IFN-α therapy and for this reason alcohol consumption is contraindicated during IFN-α treatment. There have been numerous studies confirming that alcoholics do not respond well to IFN-α therapy. Mochida et al[44] showed that less than 10% of alcoholic patients responded to IFN-α therapy. Loguercio et al[45] showed a direct relationship between alcohol consumption and response to treatment, with the numbers of patients achieving a sustained virological response decreasing as alcohol consumption increased. Safdar et al[7] recently published their findings that alcohol abuse decreases response to IFN treatment in HCV patients and therefore, it is recommended that HCV infected patients abstain from alcohol consumption whilst on treatment. A number of studies have shown that alcohol metabolism directly interferes with IFN signaling, indicating that the consumption of alcohol could directly inhibit IFN-α treatment in patients. However, there has been some recent evidence indicating that the high non-compliance rate of alcoholics adhering to treatment programs could account for the reduced response rates of alcoholic patients in the literature[46]. Whilst this is certainly a contributing factor, it does not detract from the strong in vitro data showing that alcohol has a direct inhibitory effect on IFN-α signaling. These studies have implications not only for IFN-α treatment, but also for the activity of endogenously produced type I and II IFN’s (IFN-α, β, and γ, respectively) that might result in a weakened host response to HCV infection.
ALCOHOL AND IFN-α SIGNALING
There are a number of reports investigating the effects of HCV on IFN-α signaling, suggesting that HCV negatively impacts on IFN-α signaling[47-51]; however, there are a limited number of studies investigating the combined effects of HCV and alcohol metabolism on IFN-α signaling. Insights into the effect of alcohol metabolism on IFN-α signaling can be gleaned from the study conducted by Osna et al[52], in which the effects of alcohol metabolism by ADH and CYP2E1 on IFN-γ signal transduction were investigated. This study documented a decrease in STAT1 tyrosine phosphorylation in the presence of alcohol metabolism, suggesting that alcohol can effectively dampen the IFN signaling cascade. The most comprehensive study investigating the effects of alcohol and HCV replication on IFN signaling was conducted by Plumlee et al[37]. This study showed that acute treatment of HCV genomic replicon cells with ethanol lead to the inhibition of the anti-HCV effects of IFN and interestingly, caused a decrease in STAT1 tyrosine phosphorylation, but induced STAT1 serine phosphorylation. This study also showed induction of ISRE promoter activity in the presence of alcohol, which is somewhat contradictory to their finding that IFN signaling was abrogated. It has also been documented that the oxidative stress generated via the treatment of Huh-7 cells with H202, disrupted the JAK-STAT signaling pathway specifically, by blocking STAT1, STAT2, JAK1, and TYK2 tyrosine phosphorylation[53]. More recently it has been shown that alcohol metabolism decreases the efficacy of IFN-α in vitro in genomic replicon cells that express CYP2E1[32]. In this study, alcohol metabolism preferentially inhibited STAT1 tyrosine phosphorylation at residue 701 (Y701), the critical residue required for STAT1 heterodimerisation with STAT2[32]. These findings suggest that a decrease in STAT1-Y701 phosphorylation due to alcohol metabolism would decrease downstream ISG expression and, in part, explain the reduced efficacy of IFN-α in HCV positive patients that consume alcohol. Consistent with this finding, a decrease in ISRE activity in the presence of alcohol metabolism was also observe[32] and a number of anti-viral ISGs have been shown to be downregulated by alcohol metabolism (Beard MR, unpublished findings). Collectively, these studies suggest that alcohol can specifically inhibit IFN-α at the molecular level.
Clearly, the factors at play in the alcohol-induced suppression of IFN signaling in the background of HCV replication are multifactorial and complicated in an infected patient. It is also possible that alcohol can inhibit other components of the cellular innate immune response, such as components of the cellular recognition of viral pathogen-associated molecular patterns, and by activation of the toll-like-receptor-3 and the retinoic acid-induced gene-1 pathways. Another possibility is that alcohol might modulate SOCS-1 and -3, which are part of a negative feedback loop that acts to suppress IFN-signaling. It has been shown using a concanavalin-A model of hepatitis in mice that activated STAT3 plays an important role in inducing SOCS-3[54]. Interestingly, as outlined previously, we have been able to demonstrate that both alcohol metabolism and HCV replication are capable of activating STAT3, and it appears that this increase is due to oxidative stress.
CONCLUSION
In summary, it is well established clinically that chronic alcohol consumption in the setting of CHC is a dangerous combination leading to exacerbated liver disease. While the factors that lead to increased liver disease are complex, it seems that a common thread throughout many investigations is the generation of oxidative stress by both alcohol metabolism and HCV replication. Thus, it is a logical conclusion that HCV and alcohol metabolism act synergistically to accelerate disease progression via oxidative stress. In an attempt to understand these processes we have developed a model to portray these interactions (Figure 1). Evidence suggests that this oxidative stress synergy impacts on HCV replication and the anti-viral action of IFN-α, although we are waiting for the development of a small animal model to confirm these in vitro observations. Defining the molecular mechanisms and complex interactions between HCV and alcohol will further our understanding of the pathogenic role alcohol plays in CHC development and hopefully lead to novel therapeutic strategies and patient management.
Footnotes
Peer reviewer: Dr. Claudia Zwingmann, PhD, Professor, Department of Medicine, University of Montreal, Centre de Recherche, 264 Rene-Levesque Est, Montreal, QC, H2X 1P1, Canada
S- Editor Tian L L- Editor Stewart GJ E- Editor Zheng XM
References
- 1.Dore GJ, Law M, MacDonald M, Kaldor JM. Epidemiology of hepatitis C virus infection in Australia. J Clin Virol. 2003;26:171–184. doi: 10.1016/s1386-6532(02)00116-6. [DOI] [PubMed] [Google Scholar]
- 2.Thomas DL, Astemborski J, Rai RM, Anania FA, Schaeffer M, Galai N, Nolt K, Nelson KE, Strathdee SA, Johnson L, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA. 2000;284:450–456. doi: 10.1001/jama.284.4.450. [DOI] [PubMed] [Google Scholar]
- 3.Harris HE, Ramsay ME, Andrews N, Eldridge KP. Clinical course of hepatitis C virus during the first decade of infection: cohort study. BMJ. 2002;324:450–453. doi: 10.1136/bmj.324.7335.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet. 1997;349:825–832. doi: 10.1016/s0140-6736(96)07642-8. [DOI] [PubMed] [Google Scholar]
- 5.Seeff LB, Buskell-Bales Z, Wright EC, Durako SJ, Alter HJ, Iber FL, Hollinger FB, Gitnick G, Knodell RG, Perrillo RP. Long-term mortality after transfusion-associated non-A, non-B hepatitis. The National Heart, Lung, and Blood Institute Study Group. N Engl J Med. 1992;327:1906–1911. doi: 10.1056/NEJM199212313272703. [DOI] [PubMed] [Google Scholar]
- 6.Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol. 2006;1:23–61. doi: 10.1146/annurev.pathol.1.110304.100230. [DOI] [PubMed] [Google Scholar]
- 7.Safdar K, Schiff ER. Alcohol and hepatitis C. Semin Liver Dis. 2004;24:305–315. doi: 10.1055/s-2004-832942. [DOI] [PubMed] [Google Scholar]
- 8.Pessione F, Degos F, Marcellin P, Duchatelle V, Njapoum C, Martinot-Peignoux M, Degott C, Valla D, Erlinger S, Rueff B. Effect of alcohol consumption on serum hepatitis C virus RNA and histological lesions in chronic hepatitis C. Hepatology. 1998;27:1717–1722. doi: 10.1002/hep.510270635. [DOI] [PubMed] [Google Scholar]
- 9.Corrao G, Aricò S. Independent and combined action of hepatitis C virus infection and alcohol consumption on the risk of symptomatic liver cirrhosis. Hepatology. 1998;27:914–919. doi: 10.1002/hep.510270404. [DOI] [PubMed] [Google Scholar]
- 10.Wiley TE, McCarthy M, Breidi L, McCarthy M, Layden TJ. Impact of alcohol on the histological and clinical progression of hepatitis C infection. Hepatology. 1998;28:805–809. doi: 10.1002/hep.510280330. [DOI] [PubMed] [Google Scholar]
- 11.Bellentani S, Pozzato G, Saccoccio G, Crovatto M, Crocè LS, Mazzoran L, Masutti F, Cristianini G, Tiribelli C. Clinical course and risk factors of hepatitis C virus related liver disease in the general population: report from the Dionysos study. Gut. 1999;44:874–880. doi: 10.1136/gut.44.6.874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters MG, Terrault NA. Alcohol use and hepatitis C. Hepatology. 2002;36:S220–S225. doi: 10.1053/jhep.2002.36811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tagger A, Donato F, Ribero ML, Chiesa R, Portera G, Gelatti U, Albertini A, Fasola M, Boffetta P, Nardi G. Case-control study on hepatitis C virus (HCV) as a risk factor for hepatocellular carcinoma: the role of HCV genotypes and the synergism with hepatitis B virus and alcohol. Brescia HCC Study. Int J Cancer. 1999;81:695–699. doi: 10.1002/(sici)1097-0215(19990531)81:5<695::aid-ijc4>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 14.Aizawa Y, Shibamoto Y, Takagi I, Zeniya M, Toda G. Analysis of factors affecting the appearance of hepatocellular carcinoma in patients with chronic hepatitis C. A long term follow-up study after histologic diagnosis. Cancer. 2000;89:53–59. [PubMed] [Google Scholar]
- 15.Matsuda Y, Amuro Y, Higashino K, Hada T, Yamamoto T, Fujikura M, Yamaguchi K, Shimomura S, Iijima H, Nakano T. Relation between markers for viral hepatitis and clinical features of Japanese patients with hepatocellular carcinoma: possible role of alcohol in promoting carcinogenesis. Hepatogastroenterology. 1995;42:151–154. [PubMed] [Google Scholar]
- 16.Kuwana K, Ichida T, Kamimura T, Ohkoshi S, Ogata N, Harada T, Endoh K, Asakura H. Risk factors and the effect of interferon therapy in the development of hepatocellular carcinoma: a multivariate analysis in 343 patients. J Gastroenterol Hepatol. 1997;12:149–155. doi: 10.1111/j.1440-1746.1997.tb00398.x. [DOI] [PubMed] [Google Scholar]
- 17.Khan KN, Yatsuhashi H. Effect of alcohol consumption on the progression of hepatitis C virus infection and risk of hepatocellular carcinoma in Japanese patients. Alcohol Alcohol. 2000;35:286–295. doi: 10.1093/alcalc/35.3.286. [DOI] [PubMed] [Google Scholar]
- 18.Donato F, Tagger A, Gelatti U, Parrinello G, Boffetta P, Albertini A, Decarli A, Trevisi P, Ribero ML, Martelli C, et al. Alcohol and hepatocellular carcinoma: the effect of lifetime intake and hepatitis virus infections in men and women. Am J Epidemiol. 2002;155:323–331. doi: 10.1093/aje/155.4.323. [DOI] [PubMed] [Google Scholar]
- 19.Cederbaum AI, Wu D, Mari M, Bai J. CYP2E1-dependent toxicity and oxidative stress in HepG2 cells. Free Radic Biol Med. 2001;31:1539–1543. doi: 10.1016/s0891-5849(01)00743-2. [DOI] [PubMed] [Google Scholar]
- 20.Caro AA, Cederbaum AI. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu Rev Pharmacol Toxicol. 2004;44:27–42. doi: 10.1146/annurev.pharmtox.44.101802.121704. [DOI] [PubMed] [Google Scholar]
- 21.Beard MR, Jones BE. Hepatitis C virus and oxidative stress: a dangerous liaison. Future Virol. 2006;1:223–232. [Google Scholar]
- 22.Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology. 2006;43:S63–S74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- 23.Moriya K, Nakagawa K, Santa T, Shintani Y, Fujie H, Miyoshi H, Tsutsumi T, Miyazawa T, Ishibashi K, Horie T, et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 2001;61:4365–4370. [PubMed] [Google Scholar]
- 24.Lerat H, Honda M, Beard MR, Loesch K, Sun J, Yang Y, Okuda M, Gosert R, Xiao SY, Weinman SA, et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology. 2002;122:352–365. doi: 10.1053/gast.2002.31001. [DOI] [PubMed] [Google Scholar]
- 25.Li K, Prow T, Lemon SM, Beard MR. Cellular response to conditional expression of hepatitis C virus core protein in Huh7 cultured human hepatoma cells. Hepatology. 2002;35:1237–1246. doi: 10.1053/jhep.2002.32968. [DOI] [PubMed] [Google Scholar]
- 26.Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM, Weinman SA. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology. 2002;122:366–375. doi: 10.1053/gast.2002.30983. [DOI] [PubMed] [Google Scholar]
- 27.Abdalla MY, Ahmad IM, Spitz DR, Schmidt WN, Britigan BE. Hepatitis C virus-core and non structural proteins lead to different effects on cellular antioxidant defenses. J Med Virol. 2005;76:489–497. doi: 10.1002/jmv.20388. [DOI] [PubMed] [Google Scholar]
- 28.Otani K, Korenaga M, Beard MR, Li K, Qian T, Showalter LA, Singh AK, Wang T, Weinman SA. Hepatitis C virus core protein, cytochrome P450 2E1, and alcohol produce combined mitochondrial injury and cytotoxicity in hepatoma cells. Gastroenterology. 2005;128:96–107. doi: 10.1053/j.gastro.2004.10.045. [DOI] [PubMed] [Google Scholar]
- 29.Korenaga M, Okuda M, Otani K, Wang T, Li Y, Weinman SA. Mitochondrial dysfunction in hepatitis C. J Clin Gastroenterol. 2005;39:S162–S166. doi: 10.1097/01.mcg.0000155517.02468.46. [DOI] [PubMed] [Google Scholar]
- 30.Tardif KD, Waris G, Siddiqui A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 2005;13:159–163. doi: 10.1016/j.tim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 31.Gong G, Waris G, Tanveer R, Siddiqui A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc Natl Acad Sci USA. 2001;98:9599–9604. doi: 10.1073/pnas.171311298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCartney EM, Semendric L, Helbig KJ, Hinze S, Jones B, Weinman SA, Beard MR. Alcohol metabolism increases the replication of hepatitis C virus and attenuates the antiviral action of interferon. J Infect Dis. 2008;198:1766–1775. doi: 10.1086/593216. [DOI] [PubMed] [Google Scholar]
- 33.Qadri I, Iwahashi M, Capasso JM, Hopken MW, Flores S, Schaack J, Simon FR. Induced oxidative stress and activated expression of manganese superoxide dismutase during hepatitis C virus replication: role of JNK, p38 MAPK and AP-1. Biochem J. 2004;378:919–928. doi: 10.1042/BJ20031587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang T, Li Y, Lai JP, Douglas SD, Metzger DS, O'Brien CP, Ho WZ. Alcohol potentiates hepatitis C virus replicon expression. Hepatology. 2003;38:57–65. doi: 10.1053/jhep.2003.50295. [DOI] [PubMed] [Google Scholar]
- 36.Trujillo-Murillo K, Alvarez-Martínez O, Garza-Rodríguez L, Martínez-Rodríguez H, Bosques-Padilla F, Ramos-Jiménez J, Barrera-Saldaña H, Rincón-Sánchez AR, Rivas-Estilla AM. Additive effect of ethanol and HCV subgenomic replicon expression on COX-2 protein levels and activity. J Viral Hepat. 2007;14:608–617. doi: 10.1111/j.1365-2893.2006.00837.x. [DOI] [PubMed] [Google Scholar]
- 37.Plumlee CR, Lazaro CA, Fausto N, Polyak SJ. Effect of ethanol on innate antiviral pathways and HCV replication in human liver cells. Virol J. 2005;2:89. doi: 10.1186/1743-422X-2-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi J, Lee KJ, Zheng Y, Yamaga AK, Lai MM, Ou JH. Reactive oxygen species suppress hepatitis C virus RNA replication in human hepatoma cells. Hepatology. 2004;39:81–89. doi: 10.1002/hep.20001. [DOI] [PubMed] [Google Scholar]
- 39.Choi J, Forman HJ, Ou JH, Lai MM, Seronello S, Nandipati A. Redox modulation of the hepatitis C virus replication complex is calcium dependent. Free Radic Biol Med. 2006;41:1488–1498. doi: 10.1016/j.freeradbiomed.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 40.Carballo M, Conde M, El Bekay R, Martín-Nieto J, Camacho MJ, Monteseirín J, Conde J, Bedoya FJ, Sobrino F. Oxidative stress triggers STAT3 tyrosine phosphorylation and nuclear translocation in human lymphocytes. J Biol Chem. 1999;274:17580–17586. doi: 10.1074/jbc.274.25.17580. [DOI] [PubMed] [Google Scholar]
- 41.Waris G, Turkson J, Hassanein T, Siddiqui A. Hepatitis C virus (HCV) constitutively activates STAT-3 via oxidative stress: role of STAT-3 in HCV replication. J Virol. 2005;79:1569–1580. doi: 10.1128/JVI.79.3.1569-1580.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Seronello S, Ito C, Wakita T, Choi J. Ethanol enhances hepatitis C virus replication through lipid metabolism and elevated NADH/NAD+ J Biol Chem. 2010;285:845–854. doi: 10.1074/jbc.M109.045740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silvennoinen O, Ihle JN, Schlessinger J, Levy DE. Interferon-induced nuclear signalling by Jak protein tyrosine kinases. Nature. 1993;366:583–585. doi: 10.1038/366583a0. [DOI] [PubMed] [Google Scholar]
- 44.Mochida S, Ohnishi K, Matsuo S, Kakihara K, Fujiwara K. Effect of alcohol intake on the efficacy of interferon therapy in patients with chronic hepatitis C as evaluated by multivariate logistic regression analysis. Alcohol Clin Exp Res. 1996;20:A371–A377. [PubMed] [Google Scholar]
- 45.Loguercio C, Di Pierro M, Di Marino MP, Federico A, Disalvo D, Crafa E, Tuccillo C, Baldi F, del VecchioBlanco C. Drinking habits of subjects with hepatitis C virus-related chronic liver disease: prevalence and effect on clinical, virological and pathological aspects. Alcohol Alcohol. 2000;35:296–301. doi: 10.1093/alcalc/35.3.296. [DOI] [PubMed] [Google Scholar]
- 46.Anand BS, Currie S, Dieperink E, Bini EJ, Shen H, Ho SB, Wright T. Alcohol use and treatment of hepatitis C virus: results of a national multicenter study. Gastroenterology. 2006;130:1607–1616. doi: 10.1053/j.gastro.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 47.Lin W, Kim SS, Yeung E, Kamegaya Y, Blackard JT, Kim KA, Holtzman MJ, Chung RT. Hepatitis C virus core protein blocks interferon signaling by interaction with the STAT1 SH2 domain. J Virol. 2006;80:9226–9235. doi: 10.1128/JVI.00459-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melén K, Fagerlund R, Nyqvist M, Keskinen P, Julkunen I. Expression of hepatitis C virus core protein inhibits interferon-induced nuclear import of STATs. J Med Virol. 2004;73:536–547. doi: 10.1002/jmv.20123. [DOI] [PubMed] [Google Scholar]
- 49.Heim MH, Moradpour D, Blum HE. Expression of hepatitis C virus proteins inhibits signal transduction through the Jak-STAT pathway. J Virol. 1999;73:8469–8475. doi: 10.1128/jvi.73.10.8469-8475.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blindenbacher A, Duong FH, Hunziker L, Stutvoet ST, Wang X, Terracciano L, Moradpour D, Blum HE, Alonzi T, Tripodi M, et al. Expression of hepatitis c virus proteins inhibits interferon alpha signaling in the liver of transgenic mice. Gastroenterology. 2003;124:1465–1475. doi: 10.1016/s0016-5085(03)00290-7. [DOI] [PubMed] [Google Scholar]
- 51.Lin W, Choe WH, Hiasa Y, Kamegaya Y, Blackard JT, Schmidt EV, Chung RT. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology. 2005;128:1034–1041. doi: 10.1053/j.gastro.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 52.Osna NA, Clemens DL, Donohue TM Jr. Ethanol metabolism alters interferon gamma signaling in recombinant HepG2 cells. Hepatology. 2005;42:1109–1117. doi: 10.1002/hep.20909. [DOI] [PubMed] [Google Scholar]
- 53.Di Bona D, Cippitelli M, Fionda C, Cammà C, Licata A, Santoni A, Craxì A. Oxidative stress inhibits IFN-alpha-induced antiviral gene expression by blocking the JAK-STAT pathway. J Hepatol. 2006;45:271–279. doi: 10.1016/j.jhep.2006.01.037. [DOI] [PubMed] [Google Scholar]
- 54.Hong F, Jaruga B, Kim WH, Radaeva S, El-Assal ON, Tian Z, Nguyen VA, Gao B. Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS. J Clin Invest. 2002;110:1503–1513. doi: 10.1172/JCI15841. [DOI] [PMC free article] [PubMed] [Google Scholar]