

Abstract
This article reviews the evidence that ties the development of hepatocellular carcinoma (HCC) to the natural immune pro-inflammatory response to chronic liver disease, with a focus on the role of Toll-like receptor (TLR) signaling as the mechanism of liver stem cell/progenitor transformation to HCC. Two exemplary models of this phenomenon are reviewed in detail. One model applies chronic ethanol/lipopolysaccharide feeding to the activated TLR4 signaling pathway. The other applies chronic feeding of a carcinogenic drug, in which TLR2 and 4 signaling pathways are activated. In the drug-induced model, two major methyl donors, S-adenosylmethionine and betaine, prevent the upregulation of the TLR signaling pathways and abrogate the stem cell/progenitor proliferation response when fed with the carcinogenic drug. This observation supports a nutritional approach to liver cancer prevention and treatment. The observation that upregulation of the TLR signaling pathways leads to liver tumor formation gives evidence to the popular concept that the chronic pro-inflammatory response is an important mechanism of liver oncogenesis. It provides a nutritional approach, which could prevent HCC from developing in many chronic liver diseases.
Keywords: Toll-like receptor; Hepatocellular carcinoma; Methyl donors, Epigenetic processes; Inflammation; Alcohol; Drug toxicity; Lipopolysaccharides
INTRODUCTION
What is the mechanism that explains how alcohol abuse increases the risk of hepatocellular carcinoma (HCC) in hepatitis C, hepatitis B, diabetes[1], hemochromatosis[2], and α-1 antitrypsin deficiency[3]? Evidence is emerging that the synergism is due to an activation of a common pathway in which Toll-like receptor (TLR) signaling induces pro-inflammatory cytokine production through nuclear factor (NF)-κB activation and growth factors through activation of activator protein 1 (AP-1). A model of chronic ethanol feeding has been used as follows. Transgenic mice with hepatitis C non-structural 5A (NS5A), the NS5A protein of hepatitis C virus (HCV), were fed ethanol chronically. This induced HCC formation and resembled clinical alcohol abuse and hepatitis C progression to HCC[4].
Likewise, an experimental model of chronic drug toxicity, which induces HCC, resembles clinical drug-induced HCC[5]. These experimental models are reviewed to help understand how the TLR signaling pathway leads to HCC formation, as a mechanism that is shared by the clinical and experimental models.
TLR SIGNALING PATHWAY IN CHRONIC LIVER DISEASE
There is emerging evidence that upregulation of the TLR signaling pathway occurs in many chronic liver diseases. Many different cell types in the liver express TLRs[6]. Kupffer cells express TLR2, 3, 4 and 9. Hepatocytes express TLR2, 3, 4 and 5. Stellate cells express TLR4 and 9. Biliary epithelium expresses TLR2, 3, 4 and 5. Sinusoidal endothelium expresses TLR4. TLR-mediated signals result in hepatitis B, hepatitis C, alcoholic liver disease, non-alcoholic liver diseases, primary biliary cirrhosis, primary sclerosing cholangitis, hepatic fibrosis, ischemic-reperfusion injury and liver allograft rejection[6].
HEPATITIS B
TLR2 is downregulated in hepatocytes, Kupffer cells and peripheral blood monocytes in hepatitis-B-antigen-positive patients[6]. In hepatitis-B-e-antigen-negative patients, TLR2 expression and tumor necrosis factor (TNF)-α production is upregulated and probably mediated by pre-core proteins[6].
HEPATITIS C
The HCV core and NS3 protein activate TLR2/TLR1 and TLR2/TLR6 on monocytes to produce inflammatory cytokines[6]. Patients with hepatitis C lack tolerance to lipopolysaccharide (LPS). Marked upregulation of TLR2 and TLR4 have been observed in patients with chronic hepatitis C and were detected in hepatocytes, Kupffer cells, and peripheral monocytes. TLR2-mediated activation by hepatitis C may contribute to the pro-inflammatory cytokine activation[7].
ALCOHOL LIVER DISEASE
Alcohol ingestion decreases the intestinal mucosal barrier to LPS, which activates TLR4 on Kupffer cells, which leads to pro-inflammatory cytokine production[8]. Chronic alcohol consumption activates other TLRs, such as TLR1, 2 and 6-9, which further increases the TNF-α response to LPS in mice[6]. Human monocytes exposed to ethanol for 7 d develop hypersensitivity to LPS through decreased IRAK-M expression and activation of NF-κB and extracellular signal-regulated kinase (ERK)[9]. IRAK-M is part of the cascade that activates NF-κB and mitogen-activated protein kinase, initiated by TLR4 signaling.
CIRRHOSIS
TLR4 expression is downregulated in monocytes from patients with Child-Pugh C cirrhosis, which indicates LPS tolerance, because the expression returns to normal after antibiotic therapy[6].
HCC
Mice deficient in TLR4 and MyD88 develop fewer and smaller liver tumors after treatment with a chemical carcinogen, which implies that TLR4-MyD88 signaling is involved in the development of liver tumors[6]. This suggests that chronic inflammation that results from hepatitis C and TLR-induced increase in NF-κB activation and TNFα production promotes tumor growth[10].
ROLE OF TLR4 SIGNALING IN ETHANOL-FACILITATED TUMOR FORMATION IN THE NS5A TRANSGENIC MOUSE MODEL OF HCC CARCINOGENESIS
Ethanol feeding upregulates the TLR4-dependent, MyD88-independent signaling pathway in mice[11], presumably by increasing endotoxemia due to gut leakage of LPS. TLR-4 upregulation in Kupffer and stellate cells occurs in mice fed ethanol for 1 mo using the intragastric tube feeding model[12].
Machida et al[4] have taken advantage of upregulation of the TLR4 signaling pathway induced by ethanol, to accelerate HCC formation in NS5A transgenic (NS5A Tg) mice. NS5A Tg mouse livers already overexpressed TLR4, but ethanol and LPS feeding increased the TLR4 signaling due to the endotoxemia caused by ethanol. LPS challenge killed 30% of the NS5A Tg mice, but not the wild-type mice or the NS5A Tg/TLR4-/- mice. The NS5A Tg mice overreacted to LPS, with the development of massive hemorrhagic, necrosis, and inflammation. The TAK1-TRAF6 and TAK1-IRAK1 interactions in NS5A Tg mice given LPS were enhanced, but not enhanced in the wild-type or TLR-/- mice. Phosphorylation of Jun N-terminal kinase (JNK) and IκBα, two further downstream components of TLR4 signaling, was increased by LPS.
To determine whether TLR upregulation in the NS5A Tg mice was due to overexpression by Kupffer cells or hepatocytes, the Kupffer cells were depleted by injection of liposome-encapsulated clodronate 2 d before LPS injection. This reduced TLR4 activation by 35% in the NS5A Tg mice, and did not reduce the mortality, whereas, in wild-type mice, it reduced the TLR4 activation by 70% to 80%.
Machida et al[4] have tested human liver from patients with hepatitis C, including those who abused alcohol. NS5A was increased in both groups of hepatitis C patients. NS5A expression in the liver from hepatitis C patients was about one-third of the level seen in the NS5A Tg mice. TLR4, p-JNK, and p-1KBα were upregulated in the hepatitis C livers, which confirmed the clinical importance of TLR4 activation in NS5A Tg mice.
The question was: were the NS5A Tg mice more susceptible to alcohol-induced liver damage? The NS5A Tg mice were fed ethanol for 4 wk by intragastric tube together with wild-type, TLR4-/- and TLR4-/- NS5A Tg mice. The NS5A Tg mice fed ethanol developed higher alanine aminotransferase levels, and spotty liver necrosis with inflammatory cell infiltration. These liver injury markers did not occur in the TLR4-/-/NS5A Tg mice fed ethanol, which confirmed the pathogenic role of TLR4 in this mouse model. LPS levels in the ethanol-fed mice were equally elevated. The liver injury, induced by ethanol feeding, was abrogated by feeding the antibiotics polymyxin B and neomycin, when ethanol-feeding-induced injury was augmented by feeding LPS weekly and ethanol daily. These results indicate the importance of endotoxin in activated TLR4 signaling in alcohol-fed NS5A Tg mice. Lipid peroxidation was increased in the alcohol-fed NS5A Tg mice, compared to the wild-type mice fed ethanol.
The NS5A Tg mice and the same three control groups, wild-type, TLR4-/- and TLR4-/- NS5A Tg mice, were fed the Lieber deCarli diet that contained ethanol for 12 mo to induce liver tumor formation. Liver tumors developed only in the ethanol-fed NS5A Tg mice. TLR4 signaling induced by NS5A and alcohol feeding was documented by showing an increased TAK1 interaction with TRAF6 and by elevation of pJNK and pIκB, which occurred only in the ethanol-fed mice. It was concluded that alcohol and NS5A synergistically induced liver tumors by enhancing the expression of TLR signaling.
To determine the molecular mechanisms that cause synergism between alcohol feeding and NS5A Tg mice, microarray analysis of livers was performed[4]. The non-tumor areas of the NS5A Tg mice fed ethanol were compared with wild-type controls. Of the 83 genes that were changed > 4-fold, for example Nanog, TLR4 and interferon (IFN)-α4 were upregulated. By immunofluorescent antibody staining, Nanog was colocalized with the stem cell markers CD133 and CD49f, which suggested that cancer stem cells were present in the non-tumor livers of the NS5A Tg ethanol-fed mice. The TLR4-/- NS5A Tg mice fed ethanol did not develop Nanog-positive stem cells, which implied that the mechanism of stem cell and tumor induction was dependent on TLR4 signaling. To support this idea, NS5A Tg Huh7 cells treated with LPS upregulated the expression of Nanog, and this was reduced in TLR4 knockdown cells using shRNA. Overexpression of TLR4 in Huh7 cells increased the expression of Nanog in the presence of LPS[4]. LPS increased the reporter activity in NS5A transduced Huh7 cells. The results indicate that the promoter of the Nanog gene is directly activated by TLR4 and that Nanog is a novel downstream gene of TLR4 signaling.
Further studies indicated that Nanog mediated the TLR-dependent liver tumor formation by hepatic progenitor cells[4], using the hepatic progenitor cell transplant model. Hepatoblasts were isolated from p53-deficient embryonic liver infected with a retroviral vector that expressed TLR4 or Nanog along with green fluorescent protein (GFP). The cells were transplanted into wild-type mice via the spleen. These mice were given three injections of CCl4 to stimulate the establishment of the progenitor cells. After this, the mice were repeatedly injected with LPS several times a week for 25 wk. Liver tumors formed after 48-52 d, as judged by GFP imaging in vivo and by autopsy. Direct Nanog transduction in hepatic progenitor cells also produced tumors, but less than when LPS was injected subcutaneously into nude mice and GFP imaging was performed over 88 d. The TLR4-transduced mice began to form tumors 40 d after LPS treatment. These cells were p53-/-[4]. Silencing Nanog expression by Nanog shRNA delayed tumor formation. It was concluded that Nanog increases HCC formation by the TLR4-transduced progenitor cells, but Nanog alone does not confer the full oncogenic potential[4].
If Nanog activation is not the whole story, what else is required to transform progenitor cells into HCC-promoting cells? It turned out that TLR4-dependent, Nanog-expressing HCC stem cells exhibited defective transforming growth factor (TGF)-β signaling[13]. The TGF-β signaling pathway inhibits liver cell regeneration. It has been shown that the TGF-β defective pathway in mice insufficient for β2 spectrin (β2SP) leads to spontaneous development of HCC. Cancer stem cells from alcohol-fed NS5A Tg mice were examined for growth in soft agar. Their lentiviral cDNA library was created and tested for transformation of the oval cell line and screening for oncogenic genes. GFP-labeled cancer stem cells were injected into nude mice that were repeatedly injected with LPS after the initial stem cell injection, and tumor formation was followed by GFP imaging. The interaction of the TGF-β pathway with the TLR4-dependent oncogenic activity was studied. The cancer stem cells had upregulation of Nanog and sex determining region Y-box 2 (Sox-2). Tumors were formed in the nude mice, and knockdown of Nanog prevented tumor formation. The cancer stem cells were defective in the expression of TGF-β1 and β2SP. The TLR4 response element promoter activity under TLR4 activation by LPS E2F1 was induced by LPS in the cancer stem cells. E2F1 is a transcriptional activator for Nanog and is inhibited by TGF-β. The authors conclude that heightened TLR4 activation in the Nanog-positive cancer stem cells is associated with and interactive with the defective TGF-β tumor suppressor pathway for oncogene activity of Nanog-positive cancer stem cells[13].
ROLE OF TLR4/2 SIGNALING PATHWAY IN CHEMICAL-CARCINOGEN-INDUCED LIVER TUMOR MODEL
Oliva et al[5] have developed a chemically-induced mouse model of liver tumor formation, which recently has been shown to be associated with TLR-4/2 activation[14]. In this model of experimental carcinogenesis, mice were fed diethyl 1,4-dehydro-2,3,6-trimethyl-3,5-pyridine decarboxylate (DDC). 0.1% DDC was fed ad libitum in the diet for 10 wk, at which time a large percentage of hepatocytes had become transformed into stem cell/progenitors. These cells stained positive for stem cell markers (UbD, OV-6, GSTP), and formed Mallory-Denk bodies. When the drug was withdrawn, these stem cell/progenitors persisted in small numbers scattered throughout the liver lobules. When the drug was reintroduced after 1 mo withdrawal, the stem cell/progenitors proliferated with a growth advantage over the intervening normal hepatocytes[5,15-17], which indicated that the stem cell/progenitors had been epigenetically changed[5,18-20]. The replication of stem cell/progenitors was prevented by feeding S-adenosylmethionine (SAMe) or betaine as methyl donors[5,19,20]. The replication of the stem cell/progenitors was stimulated by refeeding DDC or other liver toxins. In primary liver cultures, stem cell/progenitor formation was associated with NF-κB and AP-1 activation, as well as phosphorylation of p38 JNK and ERK[17,21-25]. All of this could be the result of increased TLR4/2 (Figure 1), according to signaling microarray analysis data mining of livers of control mice compared with mice refed DDC for 7 d and those fed DDC with SAMe (4 g/kg body weight[14]. TLR2 and TLR4 were both upregulated and this was prevented by SAMe. RT-PCR confirmed the upregulation of TLR2/4 expression, and this was prevented by SAMe. Levels of MyD88 adapter protein were increased, but the TLR2/4-TR1F-1RF3 pathway (MyD88-independent pathway) was not upregulated, which differs from the case when ethanol is fed[11]. TRAF6 tended to be upregulated and this was prevented by SAMe feeding. TRAF6 is down stream from the MyD88/IRAK complex. TRAF6 leads to activation of NF-κB and cytokine upregulation, and indirectly to the activation of p38, JNK and ERK, which initiates growth of hepatocytes (Figure 1). With an increase in NF-κB activation, TNFα (R21 and 12a) and IFN-γ (21 and 2) receptor expression was upregulated, coupled with the proliferation of stem cell/progenitors when DDC was refed, and SAMe prevented these changes[14,27].
Figure 1.
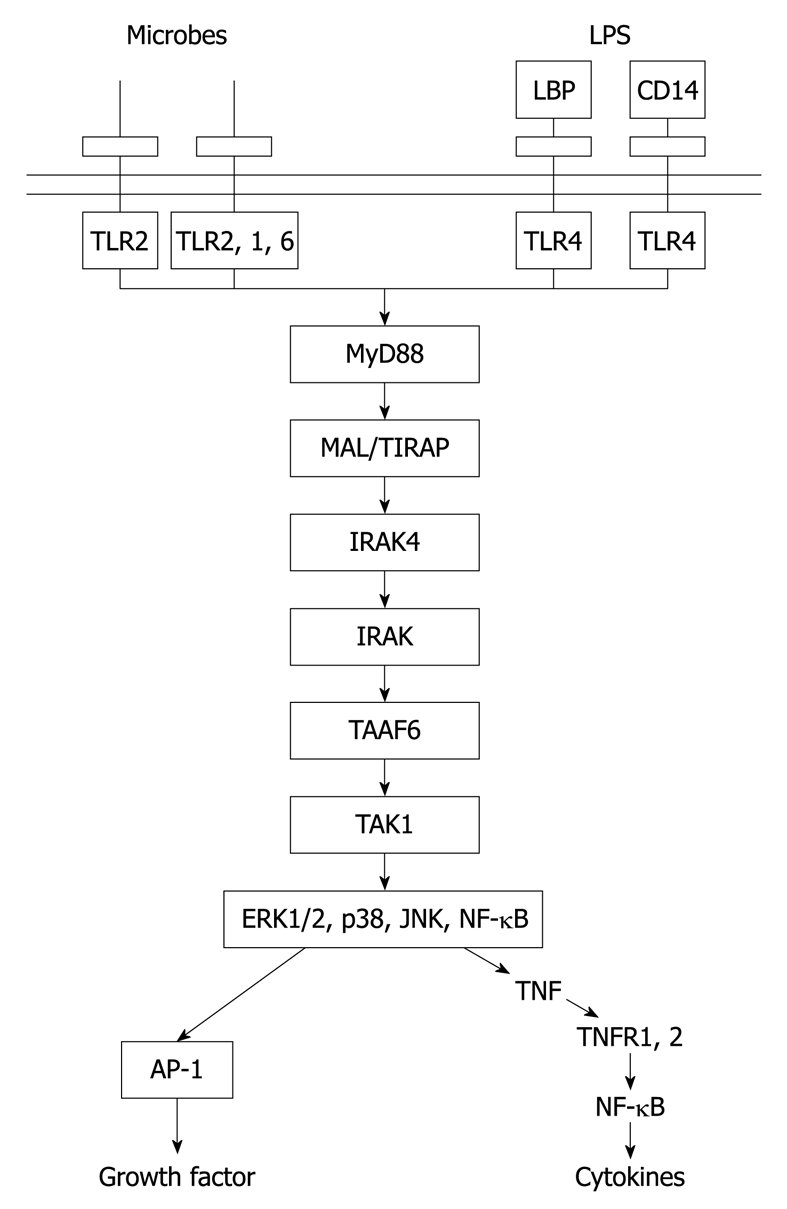
Main relationship between Toll-like receptor (TLR)2 and 4, their adaptors, protein kinases, which are linked to them and downstream signaling effects[26]. LPS: Lipopolysaccharide; LBP: LPS binding protein; CD14: Cluster of differentiation 14; ERK: Extracellular signal-regulated kinase; JNK: Jun N-terminal kinase; NF-κB: Nuclear factor κB; AP-1: Activator protein 1; TNF: Tumor necrosis factor.
Other signaling pathways such as STAT3, Wnt NOTCH, hedgehog and TGFβ, which are involved in stem cell renewal, differentiation and survival may also be involved in stem cell/progenitor transition to cancer stem cells found in HCC[28].
CONCLUSION
There are increasing numbers of examples of the TLR signaling pathway in which it plays a key role in activating stem cell/progenitor proliferation and conversion to cancer-stem-cell-based liver tumor formation. Two examples were given in detail, one driven by chronic ethanol and LPS stem cell/progenitor feeding, and the other by chronic drug feeding. These two examples provide proof of the principle that TLR signaling pathway activation is required for stem cell/progenitor activation and transformation, and supports the concept that inflammation leads to cancer formation. Gene knockout of TLR4 expression prevented tumor formation in the first example and SAMe prevented all the steps leading to pro-inflammatory activation, including TLR signaling in the second example, therefore, it is likely that blocking TLR signaling would prevent tumor formation prophylactically by feeding SAMe.
Acknowledgments
The authors thank Adriana Flores for typing the manuscript.
Footnotes
Supported by NIH/NIAAA 8116 and Alcohol Center Grant on Liver and Pancreas P50-011999, Morphology Core
Peer reviewer: Dr. Pankaj K Singh, Peptide Biology Laboratories, Salk Insitute, 10010 North Torrey Pines Road, La Jolla, CA 92037, United States
S- Editor Tian L L- Editor Kerr C E- Editor Zheng XM
References
- 1.Hassan MM, Hwang LY, Hatten CJ, Swaim M, Li D, Abbruzzese JL, Beasley P, Patt YZ. Risk factors for hepatocellular carcinoma: synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology. 2002;36:1206–1213. doi: 10.1053/jhep.2002.36780. [DOI] [PubMed] [Google Scholar]
- 2.Britton RS, Bacon BR. Hereditary hemochromatosis and alcohol: a fibrogenic cocktail. Gastroenterology. 2002;122:563–565. doi: 10.1053/gast.2002.31652. [DOI] [PubMed] [Google Scholar]
- 3.Kage M, Kage M, Liew CT, Xu Y, Peters RL. Alpha-1-antitrypsin deficiency in adults. Acta Pathol Jpn. 1986;36:1139–1148. doi: 10.1111/j.1440-1827.1986.tb02835.x. [DOI] [PubMed] [Google Scholar]
- 4.Machida K, Tsukamoto H, Mkrtchyan H, Duan L, Dynnyk A, Liu HM, Asahina K, Govindarajan S, Ray R, Ou JH, et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl Acad Sci USA. 2009;106:1548–1553. doi: 10.1073/pnas.0807390106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oliva J, Bardag-Gorce F, French BA, Li J, McPhaul L, Amidi F, Dedes J, Habibi A, Nguyen S, French SW. Fat10 is an epigenetic marker for liver preneoplasia in a drug-primed mouse model of tumorigenesis. Exp Mol Pathol. 2008;84:102–112. doi: 10.1016/j.yexmp.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Testro AG, Visvanathan K. Toll-like receptors and their role in gastrointestinal disease. J Gastroenterol Hepatol. 2009;24:943–954. doi: 10.1111/j.1440-1746.2009.05854.x. [DOI] [PubMed] [Google Scholar]
- 7.Szabo G, Dolganiuc A, Mandrekar P. Pattern recognition receptors: a contemporary view on liver diseases. Hepatology. 2006;44:287–298. doi: 10.1002/hep.21308. [DOI] [PubMed] [Google Scholar]
- 8.Wheeler MD. Endotoxin and Kupffer cell activation in alcoholic liver disease. Alcohol Res Health. 2003;27:300–306. [PMC free article] [PubMed] [Google Scholar]
- 9.Mandrekar P, Bala S, Catalano D, Kodys K, Szabo G. The opposite effects of acute and chronic alcohol on lipopolysaccharide-induced inflammation are linked to IRAK-M in human monocytes. J Immunol. 2009;183:1320–1327. doi: 10.4049/jimmunol.0803206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 11.Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, Kurt-Jones E, Szabo G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology. 2008;48:1224–1231. doi: 10.1002/hep.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seki E, Inokuchi S, Tsukamoto H, Brenner DA. Kupffer cells and stellate cells promote alcohol-mediated liver injury through TLR4. Hepatology. 2009:50 Suppl; A871. [Google Scholar]
- 13.Machida K, Liu J, Jeong H, Mishra L, Tsukamoto H. TLR4-dependent Nanog + cancer stem cells exhibit defective TGF-beta signaling. Hepatology. 2009;50 Suppl:A852. [Google Scholar]
- 14.Bardag-Gorce F, Oliva J, Li J, French BA, French SW. SAMe blocks the up regulation of Toll-like receptors signaling in Mallory-Denk body forming hepatocytes. Hepatology. 2009;50 Suppl:A867. doi: 10.1016/j.yexmp.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nan L, Bardag-Gorce F, Wu Y, Li J, French BA, French SW. Mallory body forming cells express the preneoplastic hepatocyte phenotype. Exp Mol Pathol. 2006;80:109–118. doi: 10.1016/j.yexmp.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Roomi MW, Gaal K, Yuan QX, French BA, Fu P, Bardag-Gorce F, French SW. Preneoplastic liver cell foci expansion induced by thioacetamide toxicity in drug-primed mice. Exp Mol Pathol. 2006;81:8–14. doi: 10.1016/j.yexmp.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 17.Nagao Y, Yuan QX, Wan YJY, Barbara A. French BA, French SW. Pathogenesis of mallory body formation: studies using the drug-primed mouse model. Hepatol Res. 1998;13:42–54. [Google Scholar]
- 18.Bardag-Gorce F, Oliva J, Villegas J, Fraley S, Amidi F, Li J, Dedes J, French B, French SW. Epigenetic mechanisms regulate Mallory Denk body formation in the livers of drug-primed mice. Exp Mol Pathol. 2008;84:113–121. doi: 10.1016/j.yexmp.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, Bardag-Gorce F, Dedes J, French BA, Amidi F, Oliva J, French SW. S-adenosylmethionine prevents Mallory Denk body formation in drug-primed mice by inhibiting the epigenetic memory. Hepatology. 2008;47:613–624. doi: 10.1002/hep.22029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oliva J, Bardag-Gorce F, Li J, French BA, Nguyen SK, Lu SC, French SW. Betaine prevents Mallory-Denk body formation in drug-primed mice by epigenetic mechanisms. Exp Mol Pathol. 2009;86:77–86. doi: 10.1016/j.yexmp.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nan L, Wu Y, Bardag-Gorce F, Li J, French BA, Wilson LT, French SW. The p105/50 NF-kappaB pathway is essential for Mallory body formation. Exp Mol Pathol. 2005;78:198–206. doi: 10.1016/j.yexmp.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Nagao Y, French BA, Cai Y, French SW, Wan YJ. Inhibition of PPAR alpha/RXR alpha-mediated direct hyperplasia pathways during griseofulvin-induced hepatocarcinogenesis. J Cell Biochem. 1998;69:189–200. doi: 10.1002/(sici)1097-4644(19980501)69:2<189::aid-jcb9>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 23.Nan L, Dedes J, French BA, Bardag-Gorce F, Li J, Wu Y, French SW. Mallory body (cytokeratin aggresomes) formation is prevented in vitro by p38 inhibitor. Exp Mol Pathol. 2006;80:228–240. doi: 10.1016/j.yexmp.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 24.Wu Y, Nan L, Bardag-Gorce F, Li J, French BA, Wilson LT, Dedes J, French SW. The role of laminin-integrin signaling in triggering MB formation. An in vivo and in vitro study. Exp Mol Pathol. 2005;79:1–8. doi: 10.1016/j.yexmp.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Yuan QX, Nagao Y, French BA, Wan YJ, French SW. Dexamethasone enhances mallory body formation in drug-primed mouse liver. Exp Mol Pathol. 2000;69:202–210. doi: 10.1006/exmp.2000.2320. [DOI] [PubMed] [Google Scholar]
- 26.Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- 27.Oliva J, Bardag-Gorce F, Li J, French BA, French SW. Mallory Denk body formation is associated with an increase of the immunoproteasome and a decrease of the 26S proteasome. FASEB J. 2009:24 Suppl; A219. [Google Scholar]
- 28.Yao Z, Mishra L. Cancer stem cells and hepatocellular carcinoma. Cancer Biol Ther. 2009;8:1691–1698. doi: 10.4161/cbt.8.18.9843. [DOI] [PMC free article] [PubMed] [Google Scholar]