

Abstract
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors that strongly influence molecular signalling in normal and cancer cells. Although increasing evidence suggests a role of PPARs in skin carcinogenesis, only expression of PPARγ has been investigated in human melanoma tissues. Activation of PPARα has been shown to inhibit the metastatic potential, whereas stimulation of PPARγ decreased melanoma cell proliferation. We show here that the third member of the PPAR family, PPARβ/δ is expressed in human melanoma samples. Specific pharmacological activation of PPARβ using GW0742 or GW501516 in low concentrations inhibits proliferation of human and murine melanoma cells. Inhibition of proliferation is accompanied by decreased expression of the Wilms’ tumour suppressor 1 (WT1), which is implicated in melanoma proliferation. We demonstrate that PPARβ directly represses WT1 as (1) PPARβ activation represses WT1 promoter activity; (2) in chromatin immunoprecipitation and electrophoretic mobility shift assays, we identified a binding element for PPARβ in the WT1 promoter; (3) deletion of this binding element abolishes repression by PPARβ and (4) the WT1 downstream molecules nestin and zyxin are down-regulated upon PPARβ activation. Our findings elucidate a novel mechanism of signalling by ligands of PPARβ, which leads to suppression of melanoma cell growth through direct repression of WT1.
Keywords: PPARβ, WT1, Melanoma, Proliferation, Transcriptional regulation, Tumour, Immunohistochemistry, Cancer cells, Skin, Cell line
Introduction
The incidence of malignant melanoma has been increasing steadily worldwide as a consequence of excessive exposure to sunlight [4, 14]. A sensitive skin phenotype with fair skin, tendency to burn, inability to tan, the presence of dysplastic nevi, family history of melanoma and immunosuppression are established risk factors [9, 35]. Despite advancements in early diagnosis and treatment of melanoma, morbidity and mortality do not decrease most likely because of poor understanding of the molecular mechanisms involved in skin repair, skin carcinogenesis and melanoma growth.
Increasing evidence suggests a role of peroxisome proliferator-activated receptors (PPARs) in skin formation, repair and skin carcinogenesis (reviewed in [28]). PPARs belong to the nuclear receptor superfamily of ligand-activated transcription factors [36]. They exist in three different isoforms termed PPARα, PPARβ/δ and PPARγ. All PPARs form heterodimers with retinoic X receptors, and, upon ligand binding, use the basal transcriptional machinery to regulate gene expression [24]. PPARs are expressed during normal skin development in the epidermis, hair follicles and sebaceous glands. In contrast to humans, where PPARs are also expressed in the adult epidermis, in rodents, PPARs are down-regulated in the epidermis after birth (reviewed in [28]). In skin repair, PPARα and PPARβ, but not PPARγ, expression is up-regulated in keratinocytes [27]. In this case, PPARβ is transcriptionally activated via the TNF-α pathway leading to survival, migration and differentiation of keratinocytes [38].
Several studies suggested that PPAR activation could interfere with skin carcinogenesis. Mice fed with a PPARα activator were more resistant to chemically induced carcinogenesis [40]. In line with this, PPARβ− and PPARγ-deficient mice were more susceptible against chronic application of chemical carcinogens [22, 31]. Less is known about the role of PPARs, especially PPARβ in melanocytes and melanoma. mRNA expression of all PPARs has been described in human melanocytes, and PPARα and γ activators were shown to inhibit cell proliferation and to stimulate melanin synthesis [21]. Expression of PPARα has been detected in melanoma cells [11, 16, 17] as well as in human melanoma samples (Wagner et al., unpublished observation). Pharmacological PPARα activation has been shown to inhibit the metastatic potential of melanoma cells, whereas no effect on proliferation could be observed [11, 16, 17]. In human melanoma samples, PPARγ expression was demonstrated, and PPARγ agonists were shown to inhibit the proliferation of human melanoma cell lines [29]. In addition, PPARβ mRNA expression had been reported in one melanoma cell line [15]. However, PPARβ expression in melanoma in vivo and its possible functional relevance have not been investigated yet.
Therefore, our study served the purposes to examine (1) PPARβ expression in melanoma in vivo, (2) to clarify the functional consequences of PPARβ activation in melanoma cells and (3) to elucidate possible molecular downstream pathways of PPARβ activation in melanoma cells.
Here we show that PPARβ is expressed in human melanoma samples. Pharmacological PPARβ activation in human and murine melanoma cells at low doses inhibits cell proliferation without inducing apoptosis. This growth inhibition of melanoma cells is accompanied by a decrease in the expression of the Wilms’ tumour suppressor (WT1). Finally, we demonstrate that PPARβ directly binds to the WT1 promoter and represses its activity, therefore inhibiting the growth promoting effects of WT1 on melanoma cells.
Materials and methods
Cell culture
Human and mouse melanoma cell lines (A375, accession number CRL-1676, B16F0, accession number CRL-6322) were grown in Dulbecco’s modified eagle’s medium supplemented with 10% fetal calf serum, 100 IU/ml penicillin and 100 µg/ml streptomycin. Media and reagents were obtained from Invitrogen (Cergy Pontoise, France). A375 or B16F0 cells were maintained for 24 h in medium in the presence of GW0742 (Glaxo Smith Kline, Research Triangle Park, USA) or GW501516 (Alexis Biochemicals, Coger S.A., Paris, France) dissolved in dimethyl sulfoxide (DMSO) at concentrations of 100 or 500 nmol/l. Controls were treated with vehicle (DMSO) only.
Detection of cell proliferation
A375 and B16F0 cells were split into 96-well dishes, treated with GW0742, GW501516 or vehicle (DMSO). Additionally, B16F0 cells were treated with GW0742 in the presence or absence of a dominant negative PPARβ isoform (3) and A375 cells after transfection with WT1 expression constructs or PPARβ siRNA constructs. After 24 h, bromodeoxyuridine was added and the cells incubated for 3 h. Afterwards, cells were fixed and BrdU incorporation detected using a mouse monoclonal anti-BrdU antibody followed by incubation with a goat anti-mouse IgG peroxidase-coupled secondary antibody with TMB as peroxidase substrate and spectrophotometrical reading of the plates at 450 nm according to the manufacturer’s instructions (Millipore, Molsheim, France). Alternatively, 24 h after GW treatment, cells were methanol fixed and immunohistochemical detection of proliferating cell nuclear antigen (PCNA) was performed as described [44] with counterstaining of nuclei using 4',6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA).
Apoptosis assay
Apoptotic cells were detected by TdT-dUTP terminal nick-end labelling (TUNEL) staining 24 h after GW0742 treatment using the in situ cell death detection kit (Roche Molecular Biochemicals, Meylan, France) as described [44].
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis and Western blot
Total cell lysates were prepared, electrophoresed and blotted as described [47]. The following antibodies were used for immunodetection: polyclonal anti-WT1 antibody from rabbit (C-19, sc-846, Santa Cruz Biotechnology, Heidelberg, Germany; 1:500 dilution in phosphate-buffered saline (PBS), 2.5% Blotto, 0.05% Tween-20), polyclonal anti-PPARβ from rabbit (H-74, sc-7197, Santa Cruz Biotechnology, 1:500), monoclonal anti-nestin from mouse (MAB5326 and MAB353, Millipore, 1:500), polyclonal anti-zyxin antibody from rabbit (kind gift of M. Beckerle, 1:1,000), polyclonal anti-GAPDH from goat (L-20, sc-31915, Santa Cruz Biotechnology, 1:500) and polyclonal anti-actin from goat (C-11, sc-1615, Santa Cruz Biotechnology, 1:500) and peroxidase-coupled goat anti-rabbit secondary antibody (1:2,000, Vector Laboratories), peroxidase-coupled horse anti-goat secondary antibody (1:2,000, Vector Laboratories) and peroxidase-coupled horse anti-mouse secondary antibody (1:2,000, Vector Laboratories).
Quantitative RT-PCR
Reverse transcriptase polymerase chain reaction (RT-PCR) was performed with 2 µg of total RNA as described [47]. The following primers were used for PCR amplification: human WT1 (NCBI accession no. NM005238), 5′-GGACAAGCCTGTCATTCCTG-3′ (forward primer), 5′-AAGAAACTGCCATAGCTGGATT-3′ (reverse primer) and mouse wt1 (NCBI accession no. NM144783), 5′-CAGATGAACCTAGGAGCTACCTTAAA-3′ (forward primer), 5′-TGCCCTTCTGTCCATTTCA-3′ (reverse primer). Expression was normalised to the individual levels of the housekeeping gene GAPDH using the following primers: human GAPDH (NCBI accession no. NM002046), 5′-AGCTGTCCCACTTACAGATGC-3′ (forward primer), 5′-CCTTGAAGTCACACTGGTATGG-3′ (reverse primer) and mouse GAPDH (NCBI accession no. NM008084), 5′-ATTCAACGGCACAGTCAAGG-3′ (forward primer), 5′-TGGATGCAGGGATGATGTTC-3′ (reverse primer).
Transient transfection experiments
To investigate the effect of PPARβ expression on WT1 promoter activity, a 767-bp fragment of the WT1 promoter in the pGl2 basic luciferase expression vector was co-transfected with PPARβ constructs. A375 and B16F0 cells were transfected at 60–80% confluency using Fugene 6 reagent (Roche Molecular Biochemicals) or Lipofectamine 2000 (Invitrogen), respectively. About 0.3 µg of the reporter constructs together with 0.1 µg of a cytomegalovirus (CMV)-driven β-galactosidase plasmid, and 1.6 µg of the expression construct encoding PPARβ were transiently co-transfected and assayed for luciferase- and β-galactosidase activity as described in detail elsewhere [47]. Alternatively, the WT1 promoter construct [42] was co-transfected only with the β-galactosidase reporter plasmid and the cells cultured for 48 h in the presence of 200 nM GW0742 or vehicle. The putative PPAR responsive element was deleted from the WT1 promoter construct using the Quik Change II site directed mutagenesis kit (Stratagene, Agilent Technologies, Massy, France) with the following oligonucleotides 5′-CCCCGCAGCTAGCCTGGACATGGGAG-3′ (forward, reverse primer in the corresponding antisense orientation). This deletion construct was again co-transfected with the PPARβ expression construct. To obtain transient over-expression of WT1, A375 cells were transfected with plasmids encoding either the WT1(-KTS) or the WT1(+KTS) splice variant or a combination of both isoforms (50:50% ratio). The empty expression vector (pCB6+) served as negative control. To down-regulate PPARβ expression, siRNA constructs directed against human PPARβ (sc-36305-SH, Santa Cruz Biotechnology) were transfected. Subsequently, GW0742 or vehicle (DMSO) was added to the cultures for a period of 24 h before Western blot or BrdU incorporation-based proliferation analysis.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay was performed on B16F0 cells using manufacturer’s instructions (Millipore). Antibodies (3 µg each) against acetylated histone 3 (rabbit polyclonal antibody, 06-599, Millipore) and PPARβ (rabbit polyclonal antibody H-74, sc-7197, Santa Cruz Biotechnology) were used. Normal rabbit serum served as a negative control and a 1:5 and a 1:10 dilution of the input sample as positive control. The histone H3 antibody was used to check for preservation of nucleosomes at the genomic locus. Following immunoprecipitation, the purified DNA was eluted in 30 µl UltraPure DNase, RNase-free water (Sigma, Saint-Quentin Fallavier, France). For amplification of purified DNA fragments by PCR, 1 µl of the diluted input DNA or the immunoprecipitated DNA’s were mixed with primers, DNase-free water and Red Taq Ready mix (Sigma). The following primers were used: WT1 promoter, 5′-CGCAGCTAGCCTCTAGAATT-3′ (forward), 5′-GCCGTCTAGGTAAGTAATGA-3′ (reverse); 3′UTR, 5′-TTCAAGGTGTCTAGAAAGTC-3′ (forward), 5′-TTACATTAGCAGGCACATAC-3′ (reverse). PCR products were electrophoresed on a 2% agarose gel yielding DNA fragments of 215 and 196 bp, respectively.
Electrophoretic mobility shift assays
The putative PPAR responsive element from the WT1 promoter contained the following sequence: 5′-TAGCCTCTAGAATTCTGGACATGGGA-3′. The PPAR responsive element from the acyl-CoA oxidase gene (5′-CCCGAACGTGACCTTTGTCCTGGTCC-3′) served as positive control. Annealed oligonucleotides were 32P-end labelled in a T4 polynucleotide kinase reaction (New England Biolabs, Ozyme, Saint Quentin Yvelines, France). PPARβ and RxRα proteins were generated from full-length cDNAs in pSG5 vector (Stratagene) using the coupled TNT in-vitro-transcription-translation system (Promega, Charbonnières-les Bains, France). For supershift assays, the same antibodies as for the ChIP experiments were used. DNA binding reactions were performed on ice for 30 min with approximately 20 ng of proteins in 15 µl of a 1× reaction buffer containing 10 mM Tris–HCl, pH 7.5, 50 mM KCl, 50 mM NaCl, 1 mM MgCl2, 1 mM EDTA, 5 mM DTT, 5% glycerol and 0.025 mg/ml denatured herring sperm DNA. For supershift experiments, the reaction mixes were pre-incubated for 45 min with the PPARβ antibodies mentioned above prior to addition of the labelled oligonucleotides.
Tissue samples and immunohistology
The study adheres to the principles of the Declaration of Helsinki and to title 45, US code of Federal Regulations, Part 46, Protection of human subjects. PPARβ immunohistochemical expression was evaluated in normal skin samples (n = 5) and tissue specimens of cutaneous malignant melanomas (seven superficial spreading melanomas, five nodular melanomas and one subcutaneous melanoma metastasis), which were obtained from patients who had undergone surgery at the University of Nice or the University of Florence Medical School (Table 1). Tissues were fixed in 10% buffered formalin and paraffin-embedded. Paraffin sections were dewaxed in xylene, hydrated in ethanol series and washed in phosphate-buffered saline. An antigen retrieval method using a pressure cooker was performed before immunohistochemical staining [33]. Antigen detection was performed using the EnVision + Dual Link System-HRP from Dako (Trappes, France) according to the manufacturer’s instructions using Vector VIP substrate (Vector VIP substrate kit, SK-4600, Vector Laboratories).
Table 1.
Summary of tumour lesions used for the investigation of PPARβ expression
| No. | Sex | Age | Location | Histological type | Ulceration | Clark’s level | Breslow thickness (mm) |
|---|---|---|---|---|---|---|---|
| 1 | m | 82 | Shoulder | NM | Present | IV | 2 |
| 2 | m | 61 | Back | NM | Present | IV | 7.5 |
| 3 | f | 65 | Arm | NM | Absent | IV | 5 |
| 4 | f | 77 | Arm | NM | Absent | III | 1.2 |
| 5 | f | 70 | Back | NM | Absent | IV | 3.6 |
| 6 | m | 77 | Thigh | SSM | Present | IV | 1.3 |
| 7 | m | 87 | Shoulder | SSM | Present | IV | 2.7 |
| 8 | f | 56 | Trunk | SSM | Present | IV | 5.5 |
| 9 | m | 52 | Back | SSM | Absent | IV | 2.1 |
| 10 | m | 70 | Abdomen | SSM | Present | IV | 3 |
| 11 | m | 69 | Back | SSM | Absent | IV | 1.7 |
| 12 | f | 47 | Leg | SSM | Absent | IV | 1.45 |
| 13 | f | 74 | Leg | Metastasis | Absent | NA | NA |
To predict the likelihood of metastatic spread at the time of surgery, Breslow’s tumour thickness, ulceration and Clark’s level are the currently most accepted prognostic factors [2]
NM nodular melanoma, SSM superficial spreading melanoma, NA not applicable
Polyclonal anti-PPARβ from rabbit (H-74, sc-7197, Santa Cruz Biotechnology) or monoclonal anti-PPARβ from mouse (MAB 3892, Millipore) were used in a dilution of 1:100 and 1:500 for the latter in PBS, 0.1% bovine serum albumin and 0.1% Triton X-100. The primary antibody was replaced by normal serum in the negative controls. As an additional positive control to the PPARβ-positive keratinocytes in the skin samples, paraffin sections of human colon samples were equally processed, using DAB as a substrate. Sections were counterstained with hematoxylin (Sigma) and analysed by four independent investigators, three of them experienced dermatopathologists.
For immunofluorescence double-labelling, methanol-fixed A375 cells or paraffin sections were incubated with the polyclonal PPARβ antibody and a monoclonal anti-WT1 antibody from mouse in a 1:100 dilution (clone 6F-H2, MAB 4234, Millipore). Antigens were visualised using Cy2 and Cy3 coupled secondary antibodies in a 1:150 dilution (Jackson Immuno-Research, Suffolk, UK). Slides were viewed under an epifluorescence microscope (DMLB, Leica, Wetzlar, Germany) connected to a digital camera (Spot RT Slider, Diagnostic Instruments, Livingston, Scotland) with the Spot software (Universal Imaging, Downingtown, PA, USA).
Statistics
Data are expressed as means ± SEM. ANOVA with the Bonferroni test as post hoc test was used vs. control. Differences between two groups were tested using the Mann–Whitney test for non-parametric samples. A p value less than 0.05 was considered statistically significant.
Results
PPARβ expression in normal skin and melanoma
Several reports focussed on PPARβ expression and function in keratinocytes whereas to our knowledge, PPARβ expression in melanocytes and melanoma in vivo has not been investigated yet. In normal human skin samples, epidermal keratinocytes, melanocytes, adipocytes, hair follicles, eccrine and sebaceous glands as well as vascular endothelial cells showed mostly nuclear immunoreactivity for PPARβ (Fig. 1a). In all melanoma samples tested (n = 13), PPARβ expression could be observed (Fig. 1b–d). This PPARβ expression was heterogeneous, with both, a nuclear and granular cytoplasmic pattern. Interestingly, in nodular melanomas and in the melanoma metastasis, we observed an overall heterogeneous expression pattern within the tumour lesion, whereas, in all superficial spreading melanomas tested, less PPARβ expression could be observed. In superficial spreading melanomas, however, PPARβ expression was mostly confined to the deeper invasive front, which might suggest that PPARβ could be connected to invasion or proliferation of melanoma cells. No staining could be observed when the first antibody was replaced with normal serum (Fig. 1e). In addition to the internal positive control of PPARβ reactive keratinocytes, human colon sections were stained and depicted the described expression [20] of PPARβ in epithelial, mesenchymal and crypt cells (Fig. 1f).
Fig. 1.
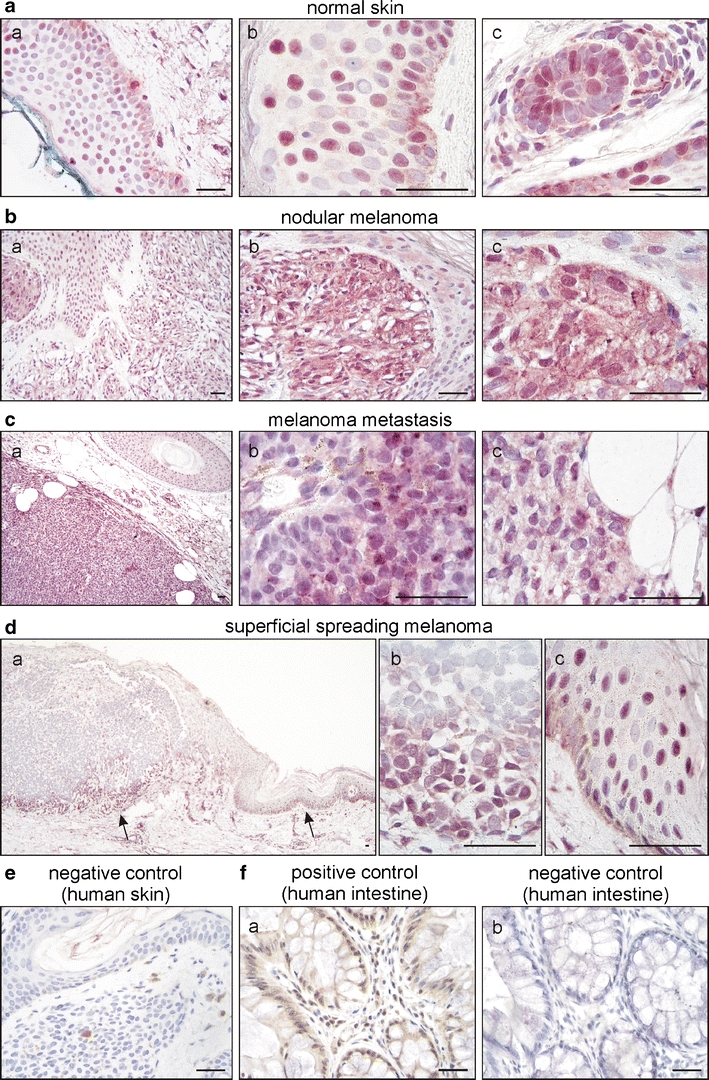
PPARβ expression in normal skin, primary and metastatic melanoma. Representative examples of normal skin (a), nodular melanoma (b), a melanoma metastasis (c) and superficial spreading melanoma (d) stained for PPARβ (rabbit polyclonal antibody and VIP as substrate, purple). Sections were counterstained with haematoxylin to visualise nuclei. Note the mostly nuclear expression of PPARβ in keratinocytes, melanocytes and hair follicles and the heterogenous, both nuclear and cytoplasmatic expression in tumoural melanocytic lesions. Within the same melanoma metastasis, regions of moderate (c (b)) and low (c (c)) PPARβ expression coexist. In superficial spreading melanoma, PPARβ expression dominated in the invasive front of the tumour (d). Arrows in (d (a)) indicate the position of the high-power magnifications of the invasive front (d, (b)) and adjacent tissue (epidermis) with melanocyte atypia (d (c)). No staining could be observed by replacing the first antibody with normal serum (e). Human colon sections served as additional positive (f (a)) and negative (f (b)) controls (DAB substrate, brown). Scale bars indicate 50 µm
PPARβ activation inhibits melanoma cell proliferation
To test the functional relevance of PPARβ expression in melanoma cells, we made use of an in vitro approach and treated human (A375) and mouse (B16) cells with low and increasing doses of the specific PPARβ agonists GW0742 or GW501516. Immunostainings for proliferating cell nuclear antigen and subsequent counting of positive cells revealed that already at a concentration of 100 nmol/l GW0742 as well as GW501516 proliferation of human and mouse melanoma cells was significantly reduced. This effect was even more pronounced at a concentration of 500 nmol/l GW0742 or GW501516 (Fig. 2a–d). Higher concentrations of 1 µmol/l or 2 µmol/l did not amplify the effect indicating receptor saturation at 500 nmol/l (data not shown). Inhibition of proliferation in response to pharmacological PPARβ activation was confirmed in enzyme-linked immunosorbent assay based 5-bromodeoxyuridine (BrdU) incorporation experiments (Fig. 2e–g). Retroviral transduction of B16 mouse melanoma cells with a dominant negative PPARβ isoform resulted in expression levels of approximately 175% of the dominant negative form compared to wild-type PPARβ levels (Fig. 3a). The transduction completely abolished the anti-proliferative effect of GW0742 (Fig. 3b). The experiment could not be performed on human melanoma cells because the transduction method for the dominant negative PPARβ isoform is rodent-specific [3]. Therefore, constructs containing siRNAs directed against human PPARβ were transfected in A375 cells. This approach knocked down PPARβ efficiently as confirmed by Western blot (Fig. 3c). The siRNA slightly increased proliferation under control conditions, which might result from blocking the effects of endogenous PPARβ ligands. The siRNA against PPARβ restored proliferation in the presence of different concentrations of GW0742 in A375 melanoma cells.
Fig. 2.
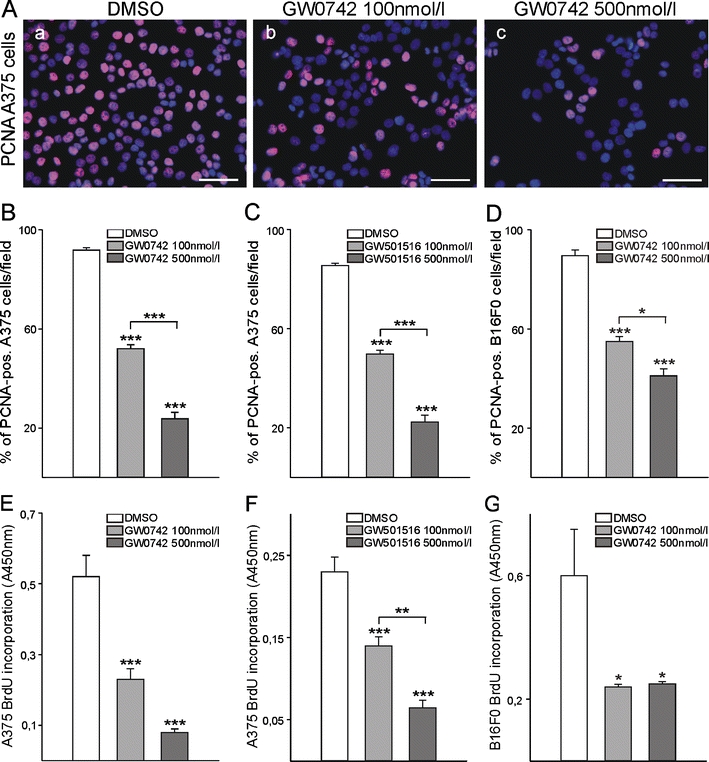
Pharmacological PPARβ activation in melanoma cell lines of human and murine origin. A375 (human) and B16F0 (mouse) melanoma cells were treated with different concentrations of two PPARβ agonists, GW0742, or GW501516, for 24 h. PPARβ activation inhibits melanoma cell proliferation. PPARβ-agonist-treated cells were immunostained with an anti-proliferating cell nuclear antigen (PCNA) antibody and counterstained with DAPI (a (a–c)). Cells in seven random optical fields were counted and the percentage of PCNA-positive cells determined (b–d; for each cell line and each agonist, n = 3, P < 0.001). Alternatively (e–g), cells were incubated with 5-bromodeoxyuridine (BrdU) followed by immunological detection of the incorporated BrdU (for each cell line and each agonist, n = 4, P < 0.001 for A375 cells and, P < 0.05 for B16F0 cells)
Fig. 3.
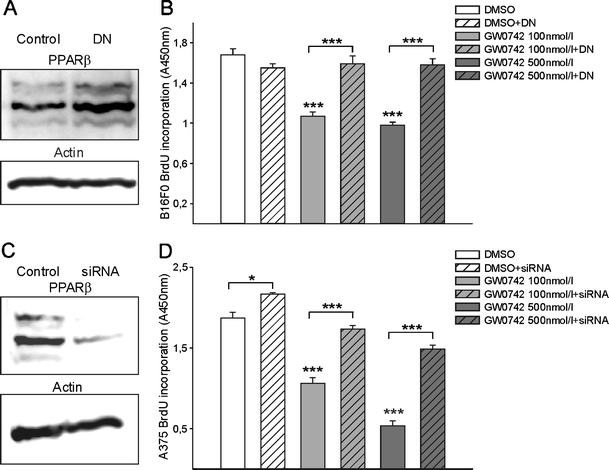
Retroviral transduction of B16F0 melanoma cells with a dominant negative isoform of PPARβ (PPARβDN) and siRNA transfection of A375 melanoma cells against PPARβ and subsequent treatment with a PPARβ agonist GW0742. Western blot for PPARβ from lysates of B16F0 cells without or with retroviral transduction of the PPARβDN (a). Note the increase in PPARβ in the transduced cells reflecting the level of the transduced dominant negative PPARβ expression. β-actin served as standard. Cells with or without retroviral transduction with the PPARβDN, were treated with the PPARβ agonist GW0742 for 24 h. Note that the strong growth inhibitory effect of GW0742 was completely abolished in the cells expressing the dominant negative isoform of PPARβ (n = 4, P < 0.001; b). Western blot from lysates of A375 cells transfected with siRNA constructs against human PPARβ (c). Silenced and control cells were treated with the PPARβ agonist GW0742 for 24 h. Silencing of PPARβ restores proliferation in the presence of different concentrations of the agonist (n = 8; d)
PPARβ activation is not inducing apoptosis in melanoma cells
To clarify whether apoptosis might contribute to the reduced cell number in response to PPARβ stimulation, we used TdT-dUTP terminal nick-end labelling with counterstaining of all cell nuclei with DAPI. This was followed by counting of TUNEL-positive cells and the total number of cells per optical field. Neither 100 nmol/l GW0742 or GW501516 nor 500 nmol/l of each of the specific PPARβ agonists had a significant influence on apoptosis of melanoma cells (Fig. 4).
Fig. 4.
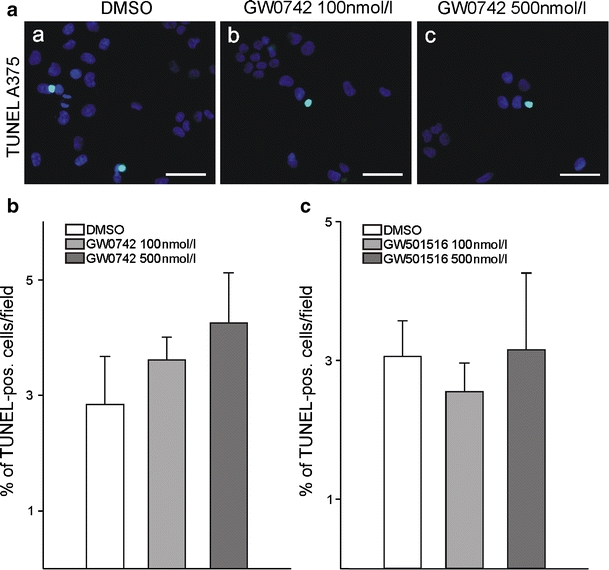
Apoptosis assay of melanoma cells after pharmacological PPARβ activation. TdT-dUTP terminal nick-end labelling (TUNEL) labelling of A375 melanoma cells was performed after 24 h of treatment with either GW0742 or GW501516. Nuclei were counterstained with DAPI (a). Cells in ten random fields were counted and the percentage of TUNEL-positive cells determined (b, c; n = 4)
PPARβ stimulation reduces expression of the Wilms’ tumour suppressor WT1 and its downstream targets nestin and zyxin in melanoma cells
As we reported recently that the Wilms’ tumour suppressor WT1 is associated with high melanoma cell proliferation [45], we investigated whether this pathway might be involved in the inhibition of proliferation in response to PPARβ activation. In quantitative RT-PCR analyses, we determined that WT1 RNA levels were reduced upon PPARβ stimulation in human (Fig. 5a) as well as in mouse (Fig. 5b) melanoma cells. Using Western blot analyses, we found that although PPARβ expression seems to be lower in the mouse cells compared to the human cell lines, in both cases stimulation of the receptor with different concentrations of GW0742 did not affect PPARβ protein expression. In contrast, WT1 expression was reduced in a concentration-dependent manner in A375 as well as B16 cells. This observation seems to be relevant for the reduced proliferation in response to PPARβ stimulation as also nestin and zyxin protein levels were reduced in response to GW0742 treatment of human and mouse melanoma cells (Fig. 5c, d). Both proteins are involved in melanoma cell proliferation [12, 23, 41] and we showed recently that they act downstream of WT1 in melanoma cells [45].
Fig. 5.
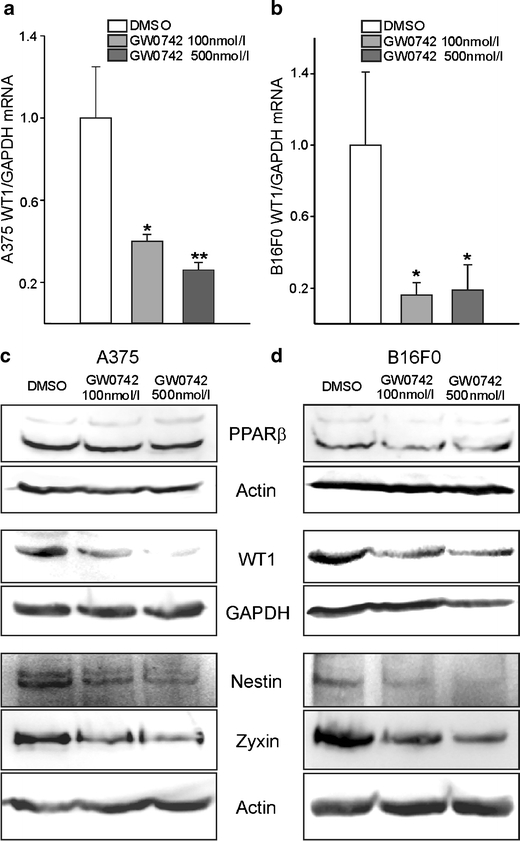
Pharmacological PPARβ activation decreases WT1 expression levels in melanoma cells. Quantitative reverse transcription (RT)-PCR for WT1 in A375 (a) and B16F0 (b) melanoma cells treated with GW0742 for 24 h. WT1 expression was normalised to GAPDH (n = 8, P < 0.01, P < 0.05). Western blot for PPARβ, WT1, nestin and zyxin from lysates of A375 (c) and B16F0 (d) melanoma cells treated with GW0742. Note that PPARβ expression remained stable upon pharmacological PPARβ activation, whereas WT1, nestin and zyxin expression was decreased. GAPDH and β-actin served as standards (n = 8 each)
PPARβ directly suppresses WT1 in melanoma cells
The down-regulation of WT1 in response to PPARβ activation raised the possibility that PPARβ might directly repress WT1. To test this hypothesis, we first analysed whether both proteins share an overlapping expression in the same cell types. Immunofluorescent double labelling showed that as reported, WT1 is not expressed in normal skin (Fig. 6 a, b) whereas WT1 and PPARβ share a partial overlapping expression in melanoma samples (Fig. 6 c–f). The only partial overlap is in agreement with a suppressor function of one protein on the other factor. Also in A375 melanoma cells, a partial overlap of PPARβ and WT1 could be detected (Fig. 7a). Addition of GW0742 significantly repressed the activity of the published WT1 promoter [42] in A375 melanoma cells (Fig. 7b). Transient co-transfection with a PPARβ expression construct repressed the activity of the WT1 promoter reporter construct to a comparable extend (Fig. 7c). The combination of transient co-transfection with a PPARβ expression construct and treatment with GW0742 had no additional effects on WT1 promoter activity, indicating a saturating effect of the individual approaches (data not shown). Using in silico analysis, a sequence region containing a predicted PPAR responsive element was identified in the WT1 promoter. This region shows a 62% homology from mouse to human and a 67% identity between mouse and zebrafish. In chromatin immunoprecipitation, we show that PPARβ protein associates with this region, whereas no interaction could be detected in the 3′UTR of WT1. An antibody against acetylated histone H3 was used to check for nucleosome integrity (Fig. 7d). Electrophoretic mobility shift assays confirmed binding of PPARβ to a 26 bp oligonucleotide from the WT1 promoter (Fig. 7e). The published PPARβ binding oligonucleotide from the acyl-CoA oxidase gene (Aco) [1] served as positive control. Incubation of the Aco/PPARβ/RxR complex with a polyclonal PPARβ antibody produced in rabbit supershifted the retardation band (lane 5 vs. 2). Under our reaction conditions, the binding affinity of the PPARbeta/RxR complex to the Aco oligonucleotide seems to be very high as a large excess of unlabelled competitor was needed to reduce the signal (lanes 3 and 4). Binding of the PPARbeta/RxR complex to the oligonucleotide from the WT1 promoter seems to be of lower affinity, as it was easier to compete. In this case, incubation with the same antibody decreased the retardation band intensity (lane 10 vs. 7). Deletion of the identified binding site from the WT1 promoter construct completely abolished repression by co-transfection of the PPARβ expression construct and by addition of GW0742 (Fig. 7f). In order to determine whether normal melanoma cell proliferation is indeed dependent of WT1 expression, transient transfection experiments with different WT1 splice variants were performed. Subsequently, the effects of GW0742 administration were investigated. In A375 melanoma cells over-expressing the WT1(−KTS), (+KTS) or a combination of both isoforms, no decrease of WT1 expression on the protein level could be observed after pharmacological activation of PPARβ (Fig. 7g). Over-expression of WT1(−KTS) stimulated nestin expression whereas WT1(+KTS) increased zyxin protein levels. Combination of both isoforms resulted in higher nestin and zyxin protein levels. Consistently, WT1 over-expression restored the proliferative potential of the melanoma cells also in the presence of the PPARβ activator GW0742 (Fig. 7h).
Fig. 6.
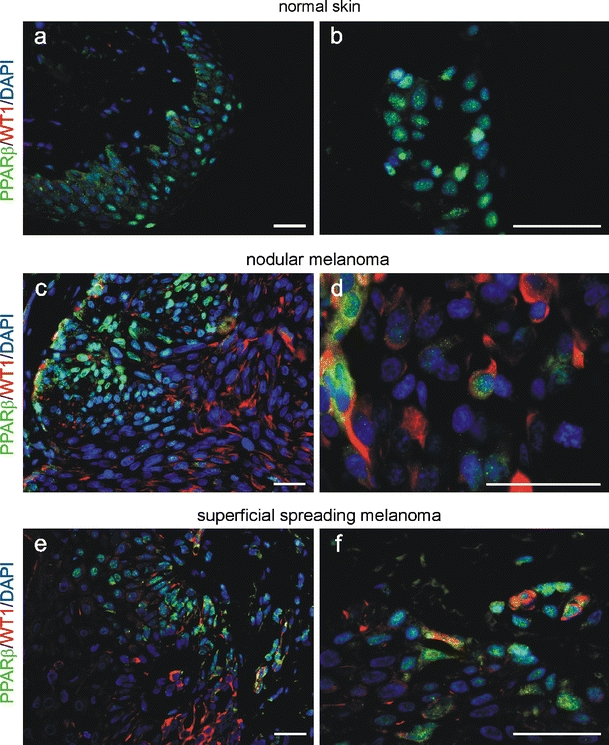
PPARβ and WT1 co-localise partially in melanoma. In normal skin (a, b), no signal for WT1 could be observed in immunofluorescence double-labelling. Partial co-localisation (yellow) of PPARβ (Cy2, green) and WT1 (Cy3, red) could be detected in melanoma (c–f). Nuclei were counterstained with DAPI (blue). Scale bars indicate 50 µm
Fig. 7.
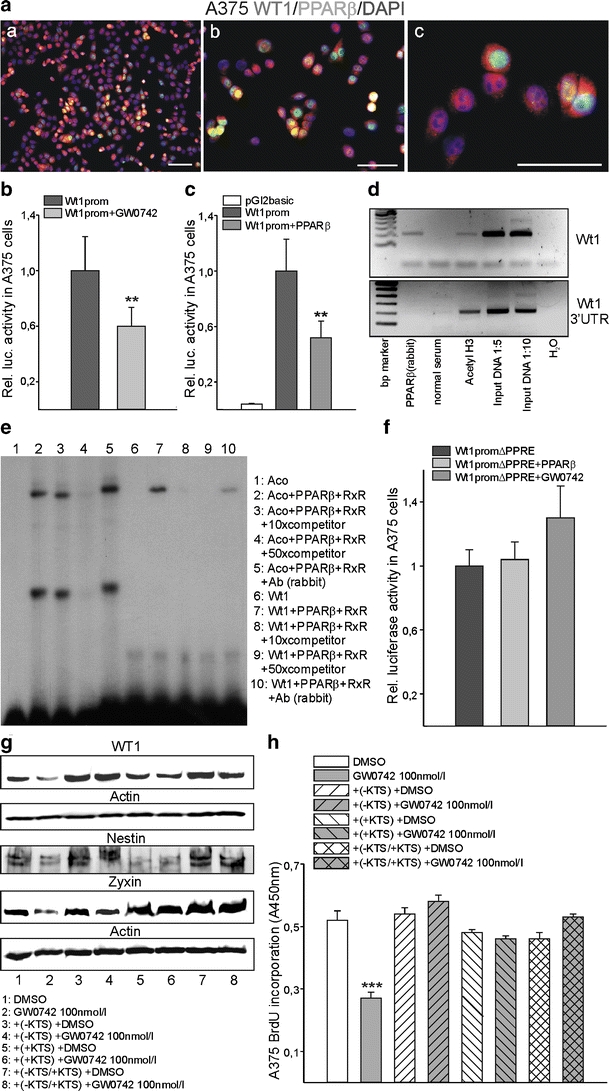
PPARβ represses WT1. Co-localisation of PPARβ (Cy2, green) and WT1 (Cy3, red) in A375 melanoma cells (a). Nuclei were counterstained with DAPI (blue). Scale bars indicate 50 µm. b Transient transfection of the WT1 promoter in a luciferase vector in the presence of vehicle or 200 nM GW0742 in A375 cells. Luciferase activities were normalised for the activity of co-transfected β-galactosidase (n = 8, P < 0.01). c Transient co-transfections of a WT1 promoter construct together with a PPARβ expression construct in A375 melanoma cells (n = 12, P < 0.01). d Chromatin immunoprecipitation to analyse PPARβ protein interaction with WT1 regulatory elements. PPARβ protein binds to the WT1 promoter (upper panel), but not to the 3’-untranslated region (UTR) of WT1 (lower panel). Input DNA and immunoprecipitates obtained with acetylated histone 3 antibody served as positive controls. Negative controls were performed with normal serum instead of specific antibodies and with DNase-free water for the PCR. e Electrophoretic mobility shift assay demonstrating binding of the PPARβ/RXRα complex to the predicted consensus element of the WT1 promoter (lane 7). Unlabelled oligonucleotide from the acyl-CoA oxidase gene in the indicated molar excess was used as competitor (lanes 3, 4, 8 and 9). Supershift assays were performed by incubating the binding reactions with a polyclonal anti-PPARβ antibody (Ab rabbit, lanes 5 and 10). An oligonucleotide from the acyl-CoA oxidase gene served as positive control [1]. f Transient co-transfections of a WT1 construct with deletion of the identified 26 bp consensus motif together with PPARβ expression constructs or in the presence of 200 nM GW0742 in A375 melanoma cells (n = 12). Note that the 26 bp deletion abolished transactivation of the WT1 promoter by PPARβ or GW0742. g Western blot for WT1, nestin and zyxin from lysates of A375 cells transiently transfected with the WT1 (−KTS) or (+KTS) variant or a combination of both isoforms in a 50:50% ratio. Cells were subsequently treated with vehicle or 100 nM GW0742. Note that the decrease of WT1 expression by pharmacological PPARβ activation is abolished in the WT1 over-expressing cells. β-actin served as standard. h BrdU incorporation assay of the WT1 over-expressing cells, without or with pharmacological PPARβ activation. The growth inhibitory effect of GW0742 is abolished in the cells over-expressing any of the WT1 splice variants (n = 4, P < 0.001)
Discussion
Recently, PPARβ activation became in focus as an interesting novel approach for the treatment of diabetes, metabolic syndrome, associated cardiovascular diseases and a potential tumour therapy [5, 25, 26, 39, 43]. Interestingly, an association between increased risk of melanoma and high body mass index has been described [34] whereas exercise appeared to have a protective effect against melanoma development [37]. Whether PPARβ is involved in these associations is presently unclear, but several studies have shown that PPARβ activation mimics exercise effects in heart, skeletal muscle and adipose tissue [3, 30, 43]. Our present data using immunohistochemistry on human melanoma samples clearly show that PPARβ is expressed in these tumours. In normal skin, PPARβ expression in vivo had been described already earlier in keratinocytes, hair follicles, sebaceous and eccrine glands (reviewed in [28]), which is in agreement with our findings. PPARβ expression in melanocytes had been shown by PCR from cultured melanocytes in vitro [21]; however, the expression and function of PPARβ in melanocytes and melanoma remained elusive. We show here that PPARβ is expressed in melanocytes and melanoma cells in vivo. In melanomas, the expression pattern was heterogeneous. Especially in superficial spreading melanomas, PPARβ was confined to the tumorigenic vertical growth phase component of the tumour. Moreover, in all melanomas analysed, we observed a nuclear and cytoplasmic granular staining, in contrast to the normal skin where the expression was nuclear. At present, it is unclear what functional significance this cytoplasmic localisation might have. Interestingly, a similar phenomenon has been described for the WT1 transcription factor. Shuttling of WT1 between the nucleus and the cytoplasm has been reported recently [32]. WT1 protein in the cytoplasm interacts with splicing factors thereby promoting post-transcriptional modifications [6]. Especially in tumours, WT1 localises predominantly to the cytoplasm [19]. Although PPARβ in melanoma is localised also to some extent in the cytoplasm, a potential role as a splicing factor/co-activator remains to be determined.
To investigate the functional role of PPARβ in melanoma, we used different melanoma cell lines of human and mouse origin and treated the cells independently with two different highly specific PPARβ agonists. In agreement with reported results for a breast cancer and another melanoma cell line [15], we observed an inhibition of proliferation. This was confirmed independently by labelling for PCNA and measurements of BrdU incorporation by ELISA. Surprisingly, in contrast to the study mentioned above, we observed this effect already significantly earlier and at a 100-fold lower dose of the specific PPARβ agonist. Whether theses discrepancies are due to different cell culture conditions or variations in the expression levels of PPARβ in the cell lines used remains uncertain. Although we observed certain variability in the growth inhibitory effect of PPARβ activation, the inhibition of proliferation was reproducible using two different PPARβ agonists in both cell lines tested. To test whether the observed inhibition of proliferation was specifically mediated via PPARβ receptors, we repeated the pharmacological PPARβ stimulation with GW0742 in B16F0 cells retrovirally transduced with a dominant negative isoform of PPARβ and in A375 cells transfected with a siRNA against PPARβ. Of note, the retrovirally transduced or silenced cells did not respond to the growth inhibitory effect of GW0742. Thus, proliferation of melanoma cells seems to be inhibited specifically by PPARβ activation.
Several other molecules have been implicated in melanoma cell proliferation, e.g. nestin [12, 23] and zyxin [41] correlate positively with melanoma proliferation. Recently, we showed that the Wilms’ tumour suppressor WT1 is important for melanoma cell proliferation via activation of nestin and zyxin [45]. WT1 is a transcription factor originally identified as a tumour suppressor based on mutational inactivation in nephroblastoma [18]. However, expression of WT1 in different adult tumours and tumour-derived cell lines has been described, suggesting that WT1 might also be able to act as an oncogene (for review, see [19]).
Interestingly, here we show that pharmacological PPARβ activation decreases WT1 expression on the RNA and protein levels. As reported recently using a siRNA approach, inhibition of WT1 results in reduced melanoma cell proliferation without affecting apoptosis [45], which was confirmed in the present study. To test for a possible direct regulation of WT1 by PPARβ, we performed double-labelling experiments. The only partial overlapping expression pattern of PPARβ and WT1 in melanoma in vivo and in melanoma cells in vitro and our observed down-regulation of WT1 in response to PPARβ activation suggested that PPARβ might act as a repressor of WT1. Several lines of evidence confirmed that WT1 represents a relevant target of PPARβ in melanoma cells. First, transient transfection experiments showed that the activity of the WT1 promoter was significantly inhibited by pharmacological PPARβ activation. Second, PPARβ co-transfection also inhibited the activity of the WT1 promoter indicating a specific effect of PPARβ on the WT1 promoter. Third, in chromatin immunoprecipitation experiments we confirmed direct binding of PPARβ to the WT1 promoter sequence, but not to the 3′-UTR of WT1. Fourth, in electrophoretic mobility shift assays, we identified a 26 bp element in the WT1 promoter, which directly binds to the PPARβ/RxRα protein complex. Finally, mutation of the identified binding site abolished repression by PPARβ or the agonist GW0742.
Interestingly, over-expression of WT1 in melanoma cells restored the proliferative potential of these tumour cells overcoming the growth inhibitory effects of PPARβ activation. This confirms our recent finding that WT1 is required for melanoma cell proliferation [45].
To test whether PPARβ activation affected not only WT1, but also WT1 downstream target genes, we analysed protein levels of zyxin and nestin [45], which are both involved in melanoma cell proliferation. The dose-dependent reduction of nestin and zyxin upon pharmacological PPARβ activation suggests that not only WT1, but also its downstream signalling in melanoma cells is consistently affected by PPARβ stimulation. Transient over-expression of WT1(−KTS) resulted in upregulation of nestin, which is consistent with a previous report [46], whereas over-expression of WT1(+KTS) stimulated zyxin expression. Whether zyxin represents a direct target of WT1(+KTS) remains to be determined in future studies. As nestin [12, 23] and zyxin [41] are both involved in melanoma cell proliferation, it is not surprising that over-expression of either WT1(−KTS) or WT1(+KTS) restored melanoma cell proliferation. In these WT1 over-expression experiments, pharmacological PPARβ activation did not have an effect on melanoma cell proliferation, since the different WT1 expression constructs are under control of a CMV promoter, but did not include the WT1 promoter sequence.
Although some molecules regulating WT1 have been reported, e.g. Pax2/8 [7, 13], Pea3 [10], NF-κB [8] or Hif-1α [42], the repression of WT1 by PPARβ and subsequent inhibition of melanoma cell proliferation is of special interest, as specific pharmacological activators of PPARβ already exist. Whether pharmacological PPARβ activation in vivo is sufficient to inhibit melanoma growth will be clarified in further studies. Nevertheless, it is likely that PPARβ activation might inhibit melanoma growth as it has been shown that colon carcinoma cells express PPARβ; and colon carcinogenesis could be inhibited by the PPARβ agonist GW0742 [26].
In summary, we have shown here that PPARβ is expressed in melanocytes and melanoma in vivo. PPARβ activation inhibits melanoma cell proliferation, which involves transcriptional repression of WT1.
Acknowledgements
The human A375 cell line was provided by F. Tarantini (University of Florence, Italy), the mouse B16F0 cell line by R. Ballotti (INSERM U895, Faculty of Medicine, Nice). GW0742 was a gift from T.M. Willson (GlaxoSmithKline). We thank G. Manfroni, G. Visciano and B. Szczepaniak for technical assistance. The authors thank K. D. Wagner for critical reading of the manuscript and helpful comments. N. Wagner was the recipient of a fellowship from the Fondation de France. The study was financially supported by the Fondation Cœur et Artères and the Association pour la Recherche sur le Cancer.
Conflict of interest
The authors declare no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Jean-François Michiels and Christophe Perrin contributed equally.
References
- 1.Amri EZ, Bonino F, Ailhaud G, Abumrad NA, Grimaldi PA. Cloning of a protein that mediates transcriptional effects of fatty acids in preadipocytes. Homology to peroxisome proliferator-activated receptors. J Biol Chem. 1995;270:2367–2371. doi: 10.1074/jbc.270.5.2367. [DOI] [PubMed] [Google Scholar]
- 2.Balch CM, Buzaid AC, Soong SJ, et al. Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. J Clin Oncol. 2001;19:3635–3648. doi: 10.1200/JCO.2001.19.16.3635. [DOI] [PubMed] [Google Scholar]
- 3.Bastie C, Luquet S, Holst D, Jehl-Pietri C, Grimaldi PA. Alterations of peroxisome proliferator-activated receptor delta activity affect fatty acid-controlled adipose differentiation. J Biol Chem. 2000;275:38768–38773. doi: 10.1074/jbc.M006450200. [DOI] [PubMed] [Google Scholar]
- 4.Berwick M, Wiggins C. The current epidemiology of cutaneous malignant melanoma. Front Biosci. 2006;11:1244–1254. doi: 10.2741/1877. [DOI] [PubMed] [Google Scholar]
- 5.Burkart EM, Sambandam N, Han X, et al. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. 2007;117:3930–3939. doi: 10.1172/JCI32578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies RC, Calvio C, Bratt E, Larsson SH, Lamond AI, Hastie ND. WT1 interacts with the splicing factor U2AF65 in an isoform-dependent manner and can be incorporated into spliceosomes. Genes Dev. 1998;12:3217–3225. doi: 10.1101/gad.12.20.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dehbi M, Gharemani M, Lechner M, Dressler G, Pelletier J. The paired-box transcription factor, PAX2, positively modulates expression of the Wilms’ tumor suppressor gene (WT1) Oncogene. 1996;13:447–453. [PubMed] [Google Scholar]
- 8.Dehbi M, Hiscott J, Pelletier J. Activation of the wt1 Wilms’ tumor suppressor gene by NF-kB. Oncogene. 1998;16:2033–2039. doi: 10.1038/sj.onc.1201747. [DOI] [PubMed] [Google Scholar]
- 9.de Vries E, Coebergh JW. Cutaneous malignant melanoma in Europe. Eur J Cancer. 2004;40:2355–2366. doi: 10.1016/j.ejca.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Discenza MT, Vaz D, Hassell JA, Pelletier J. Activation of the WT1 tumor suppressor gene promoter by Pea3. FEBS Lett. 2004;560:183–191. doi: 10.1016/S0014-5793(04)00104-8. [DOI] [PubMed] [Google Scholar]
- 11.Eastham LL, Mills CN, Niles RM. PPARalpha/gamma expression and activity in mouse and human melanocytes and melanoma cells. Pharm Res. 2008;25:1327–1333. doi: 10.1007/s11095-007-9524-9. [DOI] [PubMed] [Google Scholar]
- 12.Flørenes VA, Holm R, Myklebost O, Lendahl U, Fodstad O. Expression of the neuroectodermal intermediate filament nestin in human melanomas. Cancer Res. 1994;54:354–356. [PubMed] [Google Scholar]
- 13.Fraizer GC, Shimamura R, Zhang X, Saunders FG. PAX 8 Regulates Human WT1 Transcription through a Novel DNA Binding Site. J Biol Chem. 1997;272:30678–30687. doi: 10.1074/jbc.272.49.30678. [DOI] [PubMed] [Google Scholar]
- 14.Garbe C, Eigentler TK. Diagnosis and treatment of cutaneous melanoma: state of the art 2006. Melanoma Res. 2007;17:117–127. doi: 10.1097/CMR.0b013e328042bb36. [DOI] [PubMed] [Google Scholar]
- 15.Girroir EE, Hollingshead HE, Billin AN, et al. Peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) ligands inhibit growth of UACC903 and MCF7 human cancer cell lines. Toxicology. 2008;243:236–243. doi: 10.1016/j.tox.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grabacka M, Placha W, Plonka PM, et al. Inhibition of melanoma metastases by fenofibrate. Arch Dermatol Res. 2004;296:54–58. doi: 10.1007/s00403-004-0479-y. [DOI] [PubMed] [Google Scholar]
- 17.Grabacka M, Plonka PM, Urbanska K, Reiss K. Peroxisome proliferator-activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin Cancer Res. 2006;12:3028–3036. doi: 10.1158/1078-0432.CCR-05-2556. [DOI] [PubMed] [Google Scholar]
- 18.Haber DA, Buckler AJ, Glaser T, et al. An internal deletion within an 11p13 zinc finger gene contributes to the development of Wilms’ tumour. Cell. 1990;61:1257–1269. doi: 10.1016/0092-8674(90)90690-G. [DOI] [PubMed] [Google Scholar]
- 19.Hohenstein P, Hastie ND. The many facets of the Wilms’ tumour gene, WT1. Hum Mol Genet. 2006;15:R196–R201. doi: 10.1093/hmg/ddl196. [DOI] [PubMed] [Google Scholar]
- 20.Huin C, Corriveau L, Bianchi A, Keller JM, Collet P, Krémarik-Bouillaud P, et al. Differential expression of peroxisome proliferator-activated receptors (PPARs) in the developing human fetal digestive tract. J Histochem Cytochem. 2000;48:603–611. doi: 10.1177/002215540004800504. [DOI] [PubMed] [Google Scholar]
- 21.Kang HY, Chung E, Lee M, Cho Y, Kang WH. Expression and function of peroxisome proliferator-activated receptors in human melanocytes. Br J Dermatol. 2004;150:462–468. doi: 10.1111/j.1365-2133.2004.05844.x. [DOI] [PubMed] [Google Scholar]
- 22.Kim DJ, Murray IA, Burns AM, Gonzalez FJ, Perdew GH, Peters JM. Peroxisome proliferator-activated receptor-beta/delta inhibits epidermal cell proliferation by down-regulation of kinase activity. J Biol Chem. 2005;280:9519–9527. doi: 10.1074/jbc.M413808200. [DOI] [PubMed] [Google Scholar]
- 23.Klein WM, Wu BP, Zhao S, Wu H, Klein-Szanto AJ, Tahan SR. Increased expression of stem cell markers in malignant melanoma. Mod Pathol. 2007;20:102–107. doi: 10.1038/modpathol.3800720. [DOI] [PubMed] [Google Scholar]
- 24.Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature. 1992;358:771–774. doi: 10.1038/358771a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luquet S, Lopez-Soriano J, Holst D, Fredenrich A, Melki J, Rassoulzadegan M, et al. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J. 2003;17:2299–2301. doi: 10.1096/fj.03-0269fje. [DOI] [PubMed] [Google Scholar]
- 26.Marin HE, Peraza MA, Billin AN, et al. Ligand activation of peroxisome proliferator-activated receptor beta inhibits colon carcinogenesis. Cancer Res. 2006;66:4394–4401. doi: 10.1158/0008-5472.CAN-05-4277. [DOI] [PubMed] [Google Scholar]
- 27.Michalik L, Desvergne B, Tan NS, et al. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)alpha and PPARbeta mutant mice. J Cell Biol. 2001;154:799–814. doi: 10.1083/jcb.200011148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michalik L, Wahli W. Peroxisome proliferator-activated receptors (PPARs) in skin health, repair and disease. Biochim Biophys Acta. 2007;1771:991–998. doi: 10.1016/j.bbalip.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 29.Mössner R, Schulz U, Krüger U, et al. Agonists of peroxisome proliferator-activated receptor gamma inhibit cell growth in malignant melanoma. J Invest Dermatol. 2002;119:576–582. doi: 10.1046/j.1523-1747.2002.01861.x. [DOI] [PubMed] [Google Scholar]
- 30.Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicol CJ, Yoon M, Ward JM, et al. PPARgamma influences susceptibility to DMBA-induced mammary, ovarian and skin carcinogenesis. Carcinogenesis. 2004;25:1747–1755. doi: 10.1093/carcin/bgh160. [DOI] [PubMed] [Google Scholar]
- 32.Niksic M, Slight J, Sanford JR, Caceres JF, Hastie ND. The Wilms' tumour protein (WT1) shuttles between nucleus and cytoplasm and is present in functional polysomes. Hum Mol Genet. 2004;13(4):463–471. doi: 10.1093/hmg/ddh040. [DOI] [PubMed] [Google Scholar]
- 33.Norton AJ, Jordan S, Yeomans P. Brief, high-temperature heat denaturation (pressure cooking): a simple and effective method of antigen retrieval for routinely processed tissues. J Pathol. 1994;4:371–379. doi: 10.1002/path.1711730413. [DOI] [PubMed] [Google Scholar]
- 34.Odenbro A, Gillgren P, Bellocco R, Boffetta P, Håkansson N, Adami J. The risk for cutaneous malignant melanoma, melanoma in situ and intraocular malignant melanoma in relation to tobacco use and body mass index. Br J Dermatol. 2007;156:99–105. doi: 10.1111/j.1365-2133.2006.07537.x. [DOI] [PubMed] [Google Scholar]
- 35.Rees JL. Melanoma: what are the gaps in our knowledge. PLoS Med. 2008;5:e122. doi: 10.1371/journal.pmed.0050122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sher T, Yi HF, McBride OW, Gonzalez FJ. cDNA cloning, chromosomal mapping, and functional characterization of the human peroxisome proliferator activated receptor. Biochemistry. 1993;32:5598–5604. doi: 10.1021/bi00072a015. [DOI] [PubMed] [Google Scholar]
- 37.Shors AR, Solomon C, McTiernan A, White E. Melanoma risk in relation to height, weight, and exercise (United States) Cancer Causes Control. 2001;12:599–606. doi: 10.1023/A:1011211615524. [DOI] [PubMed] [Google Scholar]
- 38.Tan NS, Michalik L, Noy N, et al. Critical roles of PPAR beta/delta in keratinocyte response to inflammation. Genes Dev. 2001;15:3263–3277. doi: 10.1101/gad.207501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanaka T, Yamamoto J, Iwasaki S, et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci U S A. 2003;100:15924–15929. doi: 10.1073/pnas.0306981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thuillier P, Anchiraico GJ, Nickel KP, et al. Activators of peroxisome proliferator-activated receptor-alpha partially inhibit mouse skin tumor promotion. Mol Carcinog. 2000;29:134–142. doi: 10.1002/1098-2744(200011)29:3<134::AID-MC2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 41.van der Gaag EJ, Leccia MT, Dekker SK, Jalbert NL, Amodeo DM, Byers HR. Role of zyxin in differential cell spreading and proliferation of melanoma cells and melanocytes. J Invest Dermatol. 2002;118:246–254. doi: 10.1046/j.0022-202x.2001.01657.x. [DOI] [PubMed] [Google Scholar]
- 42.Wagner KD, Wagner N, Wellmann S, et al. Oxygen-regulated expression of the Wilms' tumor suppressor Wt1 involves hypoxia-inducible factor-1 (HIF-1) FASEB J. 2003;17:1364–1366. doi: 10.1096/fj.02-1065fje. [DOI] [PubMed] [Google Scholar]
- 43.Wagner N, Jehl-Piétri C, Lopez P, et al. Peroxisome proliferator-activated receptor beta stimulation induces rapid cardiac growth and angiogenesis via direct activation of calcineurin. Cardiovasc Res. 2009;83:61–71. doi: 10.1093/cvr/cvp106. [DOI] [PubMed] [Google Scholar]
- 44.Wagner N, Michiels JF, Schedl A, Wagner KD. The Wilms’ tumour suppressor WT1 is involved in endothelial cell proliferation and migration: expression in tumour vessels in vivo. Oncogene. 2008;26:3662–3672. doi: 10.1038/sj.onc.1211044. [DOI] [PubMed] [Google Scholar]
- 45.Wagner N, Panelos J, Massi D, Wagner KD. The Wilms’ tumor suppressor WT1 is associated with melanoma proliferation. Pflugers Arch. 2008;455:839–847. doi: 10.1007/s00424-007-0340-1. [DOI] [PubMed] [Google Scholar]
- 46.Wagner N, Wagner KD, Scholz H, Kirschner KM, Schedl A. Intermediate filament protein nestin is expressed in developing kidney and heart and might be regulated by the Wilms’ tumor suppressor Wt1. Am J Physiol Regul Integr Comp Physiol. 2006;291:R779–R787. doi: 10.1152/ajpregu.00219.2006. [DOI] [PubMed] [Google Scholar]
- 47.Wagner N, Wagner KD, Theres H, Englert C, Schedl A, Scholz H. Coronary vessel development requires activation of the TrkB neurotrophin receptor by the Wilms’ tumor transcription factor Wt1. Genes Dev. 2005;21:2631–2642. doi: 10.1101/gad.346405. [DOI] [PMC free article] [PubMed] [Google Scholar]