

Abstract
In the 16 years since mutations to copper, zinc superoxide dismutase (SOD1) were first linked to familial amyotrophic lateral sclerosis (ALS), a multitude of apparently contradictory results have prevented any general consensus to emerge about the mechanism of toxicity. A decade ago, we showed that the loss of zinc from SOD1 results in the remaining copper in SOD1 to become extremely toxic to motor neurons in culture by a mechanism requiring nitric oxide. The loss of zinc causes SOD1 to become more accessible, more redox reactive, and a better catalyst of tyrosine nitration. Although SOD1 mutant proteins have a modestly reduced affinity for zinc, wild-type SOD1 can be induced to lose zinc by dialysis at slightly acidic pH. Our zinc-deficient hypothesis offers a compelling explanation for how mutant SOD1s have an increased propensity to become selectively toxic to motor neurons and also explains how wild-type SOD1 can be toxic in nonfamilial ALS patients. One critical prediction is that a therapeutic agent directed at zinc-deficient mutant SOD1 could be even more effective in treating sporadic ALS patients. Although transgenic mice experiments have yielded contradictory evidence to the zinc-deficient hypothesis, we will review more recent studies that support a role for copper in ALS. A more careful examination of the role of copper and zinc binding to SOD1 may help counter the growing disillusion in the ALS field about understanding the pathological role of SOD1. Antioxid. Redox Signal. 11, 1627–1639.
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating disease that results in motor neuron degeneration, paralysis, and inevitable death within 2–5 years of diagnosis (104). Every year ∼5,000 people are diagnosed with ALS, which is also known as Lou Gehrig's disease, with the typical age of onset ∼50–60. About 10% of ALS cases exhibit familial inheritance, but more commonly ALS is sporadic without a family history. Mutations to the copper, zinc superoxide dismutase protein (SOD1) occur in ∼20–25% of patients with familial ALS (32, 102). This widely quoted 2–3% estimate of SOD1 mutations in ALS was based on early limited data. More recent studies have suggested that SOD1 mutations may be more common and might account for as many as 7% of total ALS cases (3). The toxic gain-of-function of ALS-associated SOD1 mutations was firmly established when transgenic mice expressing large amounts of ALS mutant SOD1 proteins were shown to develop motor neuron disease (55). Recently, an SOD1 mutation has been recently discovered from genomic screens in five breeds of dogs that suffer from late onset degenerative myelopathy that resembles ALS (5). While there is no doubt that SOD1 can be toxic to motor neurons, the mechanism as to how mutant SOD1 leads to ALS is highly controversial and many questions remain, such as can SOD1 have a role in sporadic ALS (18).
This review focuses on the potential role that the loss of zinc may play in making the remaining copper in SOD1 toxic to motor neurons. We first proposed the zinc-deficient SOD1 hypothesis in 1997 (29, 30), and the hypothesis has drawn significant criticism over the years (14, 27, 128) to the point where it is now frequently not discussed in many ALS reviews on SOD1 toxicity. The underlying hypothesis is remarkably simple (Fig. 1); the loss of structural zinc partially disorganizes portions of the SOD1 protein that restrict access to copper which then becomes toxic. As described below, the zinc-deficient SOD1 hypothesis is a simple explanation for how many different mutations can cause the same disease, why people can carry SOD1 mutations for as long as 80 years before developing the disease, and why wild-type SOD1 can become toxic (10). Most of the evidence in favor of this hypothesis comes from our work with purified SOD1s, primary cultures of motor neurons, and co-cultures of motor neurons and astrocytes (6, 24, 25, 29, 30, 37–41, 90, 119). Results from these experiments have been confirmed and extended by other groups (44, 97, 98). A biochemical basis for peroxynitrite and tyrosine nitration by zinc-deficient SOD1 has been well established and several potential targets have been identified in vivo (30, 131). Through collaboration with the Tainer lab, we have recently determined the structure of zinc-deficient SOD1 (100), which is surprisingly revealing about its biochemical behavior and provides new insights into why SOD1 is a dominant disease. In addition, we have recently linked the increased production of superoxide and tyrosine nitration to mitochondrial dysfunction in motor neurons, oligodendrocytes, and astrocytes (23, 91, 101).
FIG. 1.

Proposed model for how SOD1 becomes toxic in ALS. SOD1 is normally present as a very stable dimer. However, zinc is held less strongly than copper to SOD1 and its loss partially disorganizes the protein and allows the copper to become toxic. Heterodimers of zinc-deficient SOD1 will be more stable than homodimeric zinc-deficient SOD1, because the Cu,Zn SOD1 subunit will stabilize the dimer interface. If the copper is lost, SOD1 tends to fall apart into monomers that are particularly prone to aggregation. This helps remove copper-containing zinc-deficient SOD1 and is predicted to reduce toxicity.
However, a variety of results largely based upon SOD1 transgenic mice experiments appear to contradict a role for either copper, nitric oxide, or tyrosine nitration in ALS (27, 121, 122, 124). The interpretation of these transgenic mice experiments is complex and we will argue that what appears to be the simplest interpretation could be incorrect. We believe that alternative interpretations of these SOD1 mice experiments can provide a cohesive explanation that supports the zinc-deficient SOD1 hypothesis.
The leading default hypothesis for SOD1 toxicity in ALS has become protein aggregation (17, 34, 123, 124). Many investigators have shown that SOD1 mutations have an increased propensity to aggregate, but the link to toxicity has not been established (27). However, SOD1 without copper or zinc is prone to forming unstable monomers that tend to aggregate (71, 95, 96). We will raise the possibility that aggregation of metal-deficient SOD1 may be protective by removing zinc-deficient SOD1 in vivo. In this view, aggregation would be a tombstone marking sites of degeneration, but not directly responsible for the cell death.
SOD1 Structure and Activity
Understanding the role of zinc in SOD1's structure and function requires a more detailed examination of subtle structural aspects. SOD1 is a small protein with 153 amino acids that remains associated tightly as a dimer of two identical subunits with the active sites facing in opposite directions (Fig. 2). The majority of the protein is folded into an eight-strand beta-barrel, where the extensive hydrogen bonding along the peptide backbone produces one of the most stable proteins known.
FIG. 2.

The key structural features of wild-type SOD1. The SOD1 loop containing the dimer interface, zinc binding, and disulfide subloops is shown in dark gray.
Two loops fold back onto the beta-barrel to form the active site (88, 113). The longer loop, extending from residue 49 to 83, has several important roles indicated by its division into the following subloops: dimer interface subloop, zinc binding subloop, and disulfide subloop (57). In the zinc binding subloop, a zinc ion is coordinated to His63, the bridging ligand between the zinc and copper sites, along with His71, His80, and Asp83. The copper ion is coordinated additionally with His46, His48, and His120. This loop is covalently held to the beta-barrel by an intrasubunit disulfide bridge from Cys57 of the disulfide subloop to Cys146 of the beta strand at the carboxyl terminal of SOD1 (57). The disulfide bridge is highly conserved among all eukaryotic SOD1s and is extremely resistant to reduction (4). Disulfide bonds are uncommon in intracellular proteins because of the reducing environment created by millimolar concentrations of glutathione and by sulfhydryl-reducing enzymes such as thioredoxin. However, this disulfide plays a crucial role in preventing the aggregation of metal-deficient SOD1 (51).
The second loop is known as the electrostatic loop, which assists the attraction of the anionic superoxide substrate into the active site (52). In addition, this loop contains Arg143 that has a dual role to anchor the main chain of the longer loop through hydrogen bonds and to orient the bound substrate (12, 45). This loop also becomes disrupted when SOD1 loses zinc, because this loop makes contact with the zinc-binding subloop (100).
SOD1 was identified by McCord and Fridovich to exhibit antioxidant activity by scavenging superoxide (79). Superoxide is produced in vivo by the catalyzed one electron reduction of molecular oxygen through various biological systems such as the mitochondrial electron transport chain, NADPH oxidases, and xanthine oxidase (48). SOD1 catalyzes the dismutation (disproportionation) of two superoxide (O2•−) molecules to molecular oxygen (O2) and hydrogen peroxide (H2O2) {reactions (1) and (2)} at near diffusion limited rates (66, 79). This two-step process involves the redox active copper ion which cycles from Cu2+ to Cu1+ and back to Cu2+. The first superoxide molecule transfers an electron to Cu2+, thereby reducing it to Cu1+. The bridging histidine 63 detaches from the reduced copper and binds a hydrogen ion (84). This hydrogen ion is transferred to the superoxide anion entering the active site, thereby neutralizing the negative charge on the second superoxide molecule and accelerating the second half of the catalytic cycle 10-fold (35). The zinc ion is not involved in the catalytic dismutation of superoxide but provides structural support for the active site.
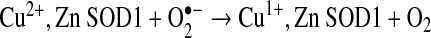 |
(1) |
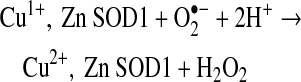 |
(2) |
SOD1 has long been recognized to be an unusually stable protein. With copper and zinc bound, SOD1 retains its activity in 6 M urea and survives heating to 80°C (46). However, it denatures much more quickly in the apo state in which copper and zinc are missing than when containing both metals, the holo state. Thus, mutations that diminish the binding of SOD1's metal cofactors will make the protein more susceptible to denaturation and aggregation.
Mutations of SOD1
Originally 13 mutations were identified in SOD1 that linked this protein to familial ALS (102). Although SOD1 mutations only account for as few as 2–3% to as high as 7% of all ALS cases, this discovery has been the first significant clue for understanding the mechanism of ALS. Currently, over 130 mutations have been identified in this protein (http://alsod.iop.kcl.ac.uk/als/), the majority being missense point mutations where one residue is substituted for a different residue. Some sites can have as many as six different substitutions. The various mutations of SOD1 are found scattered throughout the protein including key areas of the dimer interface, active site, and beta barrel (Fig. 3). The consequence these mutations have on the protein is to destabilize the overall structure. Whereas the overall distribution of mutation sites appears almost random, most of the mutations affect side chains facing into the hydrophobic interior of the protein or in the dimer interface (70). In a few ALS affected families, ∼30 residues on the carboxyl terminal are deleted by several truncation mutations. This deletion will seriously disrupt the dimer interface and the stability of the zinc-binding loop but will leave the copper-binding residues intact. One of these mutants has been shown to be particularly virulent in a mouse transgenic model of ALS at very low levels of mutant SOD1 expression (64).
FIG. 3.

Mutations in SOD1. The location of all side chains in SOD1 currently identified as mutated in ALS (dark gray) versus no reported mutations (light gray) as viewed facing the active site (A) and from the top (B).
Mutations to the SOD1 gene exhibit an autosomal dominance with one exception, the D90A mutation, which has been found to be recessive in some families but dominant in others (63). The biophysical characteristics displayed by the mutations on SOD1 divide them into two groups based on their SOD1 activity and metal binding properties: metal-binding region and wild-type-like in that they can fold into fully functional SOD1s (56, 78). We showed that eight different wild-type-like mutant SOD1s containing copper and zinc were as capable as wild-type SOD1 to protect cultured motor neurons from death (37, 38). Yet, the wild-type-like SOD1 mutations can have the most severe phenotype. In North America, the A4V mutation, which belongs to the wild-type-like group, is the most common mutation and displays one of the fastest progressing forms of ALS, with under 1 year for survival after diagnosis (13). The age of onset and relative survival of SOD1-linked familial ALS widely varies and depends on the specific mutation. Notably, two different mutations, even at the same position on the SOD1 chain, can have strikingly different survival times. A correlation has been made between a greater decrease in SOD1's stability due to mutations with a rapid disease progression of SOD1-associated ALS (72).
Transgenic Animal Models of ALS
A toxic gain-of-function from mutations in SOD1 has been established as the initiation of familial ALS from the development and work of an animal model of ALS (55). Gurney et al. (55) showed that transgenic mice overexpressing human G93A SOD1 developed motor neuron degeneration, paralysis, and death within 3–4 months, while transgenic mice containing human wild-type SOD1 remained healthy. Interestingly, G93A SOD1 is a wild-type-like ALS mutant which forces a methyl group to protrude in a hairpin turn of one of the beta-sheets. In striking contrast, SOD1 with the A4V mutation in the transgenic mouse did not develop motor neuron degeneration, although this mutation causes the most rapid progression of disease in humans (55). The A4V mutation affects the dimer interface and substantial aggregation of SOD1 was observed in motor neurons in these mice, even though they did not develop motor neuron pathology.
Additional transgenic mouse models of ALS that have been produced and thoroughly studied are G85R SOD1 and G37R SOD1 (15). Both a G93A and an H46R-expressing SOD1 transgenic rats have been created to develop motor neuron disease (82). The most standard model used in mice and rats is the G93A SOD1 mutation. Expression levels of the human SOD1 gene in these animal models can greatly vary ranging from equivalent to 10-to-20 protein fold higher overexpression (18). The expression level coincides with the onset and length of progression of the disease. Although the origin of paralysis can vary in humans, the transgenic mice routinely develop weakness in the hind limb, and the disease pathology of ALS in mice closely coincides with that in humans. Interestingly, not all of the mutations show the same early stage pathology in the transgenic mice (117). The wild-type-like group more frequently displays mitochondrial vacuolation, whereas intracellular aggregates are observed in the metal binding group mutations. Whether this is a new lead into the disease progression of familial ALS or a limitation in the animal models remains to be seen.
SOD1 Aggregation
The processes leading to SOD1 aggregation in familial ALS have received considerable attention to a potential explanation for the toxic gain-of-function of mutant SOD1. Protein aggregates are found in various neurodegeneration diseases such as Alzheimer's, Parkinson's, and Huntington's diseases (103), which would anticipate an important role for protein aggregation in this disease. Further support for this hypothesis is provided by the common tendency of ALS mutant SOD1s to aggregate whether expressed in Escherichia coli (30), in transfected cells (53, 109), or in transgenic animals (16, 125).
Over the years, we have struggled with the propensity of mutant SOD1s to form inclusion bodies in E. coli. When ALS-mutant SOD1 is expressed in E. coli at 37°C, only the initial 2–5% of protein folds into soluble protein, with all subsequent protein expression trapped into inclusion bodies. Remarkably, reduction in the growth temperature to 18°C in order to slow the rate of SOD1 expression resulted in a dozen arbitrarily chosen ALS mutant SOD1s to fold into soluble and active protein (69). After considerable experimentation, we also learned that SOD1 could be refolded from the inclusion bodies by treating with a high concentration of sulfhydryl reducing agents and suspension in urea. Rapid removal of the urea in the presence of a sulfhydryl reducing agent allowed SOD1 to refold rapidly into a native conformation able to rebind metals (69).
Many reports have shown that SOD1 aggregates in transfected cells in culture, but the cells generally remain viable and cell death frequently does not correlate with aggregation (68). It is not clear how aggregates would cause motor neuron death in ALS, but some evidence suggests sequestration of chaperones, clogging of the proteasome, or binding to bcl-2 might be involved (85, 89). Generally, these cell-based assays have not considered whether copper or zinc has been incorporated into the protein.
Oxidation of mutant SOD1 has been shown to cause aggregation. The sole tryptophan on SOD1 is particularly susceptible to oxidation by carbonate radical and can cause covalent dimerization of SOD1 monomers (132, 133). However, it has been observed that mutant SOD1s do not need to be oxidatively damaged to aggregate. The metal-deficient apoSOD1 protein is easily denatured and prone to aggregation when it becomes monomeric (75). Therefore, SOD1 would aggregate after losing its metals, with zinc more likely to be lost first because it is bound 7000 times more weakly than copper (29).
Alternatively, we propose that the SOD1 protein aggregates may be a protective mechanism as a route to removing the toxic mutant SOD1 since SOD1 aggregation has not been found to correlate with motor neuron death (68, 73). Further support for this counter hypothesis to aggregation is illustrated by transgenic animal studies where wild-type SOD1 overexpressing mice were crossed with G93A mice (49). Instead of increasing the survival time as was postulated, the mice died more rapidly. In contradiction to a protective effect with the wild-type SOD1, it was instead thought to form heterodimers with the mutant G93A SOD1, thereby stabilizing it in solution. Additional support for a protective aggregation mechanism is the previously discussed A4V SOD1 mutant overexpressing mice which do not show the typical ALS progression phenotype despite the mutant SOD1 accumulated into protein aggregates (55).
Zinc-Deficient SOD1
Zinc serves a structural role in SOD1 and its importance to the proper functioning of SOD1 is underappreciated. Although zinc is not directly involved in electron transfer reactions with superoxide, it substantially affects the coordination of copper through the shared histidine that binds to both copper and zinc. Zinc is held ∼7,000-fold less tightly than copper within wild-type SOD1. Disruption in the SOD1 structure by mutations reduce the affinity for zinc to an even greater extent and causes zinc to be lost before copper (29).
The consequences of losing zinc in SOD1 are far greater on the structure of SOD1 than the effects so far reported for any ALS mutation with copper and zinc still bound to the protein. The redox properties of SOD1 are significantly altered with the loss of zinc. This can be visually observed by a change in color of SOD1, whether wild-type or mutant, with zinc present versus zinc deficient. Both wild-type and A4V SOD1 are green when they contain copper and zinc, and both are equally efficient at scavenging superoxide (37). The green color is due to the shared ligand histidine 63 bridging copper and zinc, which creates an additional shoulder in the 450 nm region. The loss of zinc from either SOD1 causes the protein to turn blue due to alterations in the environment of the bound copper. Cu,Zn SOD1 and zinc-deficient SOD1 both become colorless when the copper is reduced. Valentine's group observed that some ALS mutant SOD1s expressed in yeast were readily reduced by ascorbate and these became colorless, while others were not (126). Our lab has found that SOD1 reduction was dependent on whether the SOD1s were zinc deficient and not on whether it was a mutant or wild-type protein (37).
Zinc-deficient SOD1 can be reduced by ascorbate more than 3000 times faster than wild-type Cu,Zn SOD1 {reaction (3)} (37). The copper in zinc-deficient SOD1 has been postulated to be more accessible to intracellular reductants with the loss of zinc. The reduced copper would be reoxidized by molecular oxygen to produce superoxide {reaction (4)} and could continually be kept in a reduced state in the CNS, where ascorbate levels are millimolar in concentration and among the highest in the body.
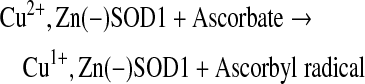 |
(3) |
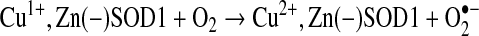 |
(4) |
The x-ray structure of a constitutively zinc-deficient SOD1 (Fig. 4) provides support for the effects of losing zinc on the aberrant redox activity as well as propensity for aggregation of SOD1 that have been reported (100). The loss of zinc induced far larger changes to the SOD1 structure than any other SOD1 mutant protein that has been structurally examined before. The most dramatic change is the disordering of the two largest loops that normally restricts access of copper to only superoxide but now can leave the copper more accessible to small molecules, rationalizing the rapid reduction of SOD1 by ascorbate. In addition, the dimer interaction was twisted by nine degrees in the zinc-deficient SOD1 compared to the wild-type SOD1, by far the largest alteration in the dimer interface ever observed [see Fig. 4 in Roberts et al. (100)]. The normally rigid C57–C146 disulfide bond that stabilizes the dimer interface was also found to have two different conformations, suggesting it may be more susceptible to reduction. Other changes in the position of Arg143 occurred that most closely resemble those found in a monomeric SOD1 where the dimer interface was disrupted by site directed mutagenesis. Thus, the loss of zinc induced changes in the dimer interface that are predicted to favor dissociation of dimers and to favor aggregation. These changes are more likely to occur in the presence of thiol reductants because the intramolecular disulfide bond is more accessible to reductants.
FIG. 4.

X-ray structure of zinc-deficient SOD1 monomer. A ribbon diagram of one SOD1 subunit for zinc-deficient SOD1 is shown with the copper represented as a gray sphere. The two loops that normally limit access to the copper are too disordered to be visualized, but are illustrated with arbitrary dotted lines. The strand of the electrostatic loop hanging in space on the far left was resolved only because it packed against another dimer in the crystal. Adapted from the protein data bank entry 2R27 (100).
Oxidative and Nitrative Stress Catalyzed by SOD1
The one molecule that can compete with SOD1 for superoxide at physiologically relevant concentrations is nitric oxide, which reacts at a diffusion-limited rate with superoxide to form peroxynitrite (8, 59). Nitric oxide can diffuse at least ten times faster than a protein the size of SOD1; as a consequence, nitric oxide reacts at least ten times faster with superoxide than SOD1 can scavenge it. Because nitric oxide, like oxygen, diffuses through membranes faster than through aqueous solutions, the site of nitric oxide formation does not need to be in the same subcellular compartment as SOD1 or even in the same cell for the formation of peroxynitrite to occur (33).
Endogenous production of peroxynitrite by cultured motor neurons was found to activate apoptotic cascades (40). Motor neurons isolated from spinal cords of embryonic rats develop the phenotype of a mature motor neuron over a period of ∼1 week when grown in the presence of any of several different neurotrophic factors (40). If the trophic factors are withdrawn at any point, ∼50% of motor neurons undergo apoptosis (38). Motor neuron death requires the simultaneous production of both nitric oxide and superoxide (Fig. 5), implicating peroxynitrite as an early intermediate for activating apoptosis after trophic factor-deprivation (40). Motor neurons that induce neuronal nitric oxide synthase become immunoreactive for nitrotyrosine, a marker for peroxynitrite (60, 86, 118), before they undergo apoptosis. Inhibiting nitric oxide synthesis prevented apoptosis and the protection was lost when physiological concentration of nitric oxide, <100 nM, was generated extracellularly (76, 77, 106).
FIG. 5.

Proposed mechanism for the role of zinc-deficient SOD1 in the formation of peroxynitrite, leading to protein nitration and motor neuron apoptosis. Reduced copper in zinc-deficient SOD1 catalyzes the one-electron reduction of oxygen to superoxide. Nitric oxide reacts with superoxide at a diffusion-limited rate to form peroxynitrite. Zinc-deficient SOD1 will also catalyze the nitration of proteins such as neurofilaments. Nitration of neurofilaments interferes with their assembly, and disassembled neurofilaments have a particularly high affinity for zinc that can potentially sequester zinc from SOD1 (29, 30). Tyrosine nitration by peroxynitrite has now been strongly implicated in promoting motor neuron apoptosis and making astrocytes reactive (131). Reactive astrocytes also contribute to motor neuron death and might participate in the progressive death of motor neurons (6, 25).
Zinc-deficient SOD1 can be a slow but significant source of superoxide in motor neurons, because the copper will tend to stay close to 100% reduced by oxidizing small antioxidant molecules (37). Superoxide can be released by the reoxidation of reduced copper in SOD1 (120). Thus, the diffusion of nitric oxide into a compartment containing zinc-deficient SOD1 can be effectively converted to peroxynitrite. Experimentally, 10 μM of zinc-deficient SOD1 was shown to generate 1–2 μM of peroxynitrite per hour under physiologically relevant concentrations of nitric oxide (100 nM) (37).
Intracellular delivery of zinc-deficient SOD1s, either wild-type or four different ALS associated mutants, were shown to be toxic to motor neurons (37). The toxicity required endogenous nitric oxide production but was blocked by copper chelators. When replete with copper and zinc, both the wild-type and mutant SOD1s were not toxic and all protected motor neurons from trophic factor deprivation. These results strongly support a role for endogenous peroxynitrite interacting with zinc-deficient SOD1 in the toxicity that leads to motor neuron death. More recently, we have been able to show that inhibitors of tyrosine nitration also block motor neuron death induced by endogenous peroxynitrite formation (131).
Both Cu,Zn SOD1 and zinc-deficient SOD1 are able to use peroxynitrite to catalyze tyrosine nitration. The observation that bovine SOD1 turned yellow when exposed to peroxynitrite led to the discovery of tyrosine nitration as a biological process (11, 61, 62, 107). The addition of SOD1 and peroxynitrite to a brain homogenate surprisingly resulted in a few number of proteins that were nitrated (29). Actin, glial fibrillary acidic protein (GFAP), heat shock protein 90 (HSP90), and neurofilament light chain (NFL) were identified as the major nitrated bands (110, 131). Tyrosine nitration has been found in both sporadic and familial ALS patients and ALS animal models using antibodies, by HPLC, and more recently confirmed by mass spectrometry (1, 2, 7, 22, 26, 42, 43, 105, 114, 116).
Using mass spectrometry, the sites of nitration on NFL were mapped, which revealed that the rod domain of disassembled NFL was the most susceptible area to nitration (30). Nitration of NFL subunits can cause profound disruption of neurofilament structures (30). Because neurofilaments are the most abundant proteins in motor neurons, they may contribute to the specificity for death in vitro. When peroxynitrite is added to oxidized SOD1, zinc-deficient SOD1 is only slightly more effective than Cu,Zn SOD1 in catalyzing tyrosine nitration. With zinc-deficient SOD1 predominately reduced in vivo by the direct reaction of copper with intracellular reductants, it can become far more effective at catalyzing nitration than Cu,Zn SOD1 in vivo.
Neurofilaments may have another connection to zinc-deficient SOD1. They have a remarkably high affinity for zinc and could potentially sequester zinc from SOD1 to make it zinc deficient (30). Neurofilament proteins are particularly abundant in motor neurons in vivo and the genetic deletion of the NFL gene in a low expressing mutant SOD1 transgenic mouse was reported to increase survival by ∼45 days (127). Paradoxically, even better protection in another low expressing mutant SOD1 mouse was found by overexpressing the heaviest subunit of neurofilament (NFH) (28).
Low Activity SOD1 Mutations
Disease progression in transgenic mice expressing the human G85R SOD1 mutation is strikingly different from the G93A SOD1 mice (15). Motor neuron disease developed at a much lower SOD1 expression level and no increase in superoxide scavenging activity was observed in tissues from the G85R SOD1 transgenic mice (15). Since copper is required for SOD1 activity, it was interpreted from the G85R SOD1 ALS mouse model that copper was not necessary for mutant SOD1 to be toxic in vivo. Mutation of the equivalent residue in the endogenous mouse SOD1 also resulted in transgenic mice developing motor neuron degeneration (80, 81). However, when the G86R mouse SOD1 model of ALS was crossed with mice carrying the X-linked mottled/brindled (Mobr) mutation, which causes severe copper depletion in the spinal cord, these genetically copper-deficient mice were found to have an extended lifespan by ∼10 days (65). Our lab has currently been working with the G85R mutant SOD1 and found that the recombinant G85R SOD1 can bind both copper and zinc and exhibits superoxide scavenging activity (unpublished data). Recently, the crystal structure of G85R SOD1 was solved by Hart's group. The G85R SOD1 crystallized in several different conformations and several of these structures contained copper but were zinc deficient (20). The location of the mutation at the base of the zinc-binding site is consistent with the difficulty of zinc binding to the G85R mutant protein, potentially explaining why the G85R SOD1 mutant is toxic at much lower expression levels than G93A mutant SOD1.
Role of CCS in ALS
The zinc-deficient hypothesis depends upon copper still being bound to SOD1, although not necessarily to the copper site. Whereas the mechanism responsible for zinc insertion into SOD1 is unknown, the copper chaperone protein for SOD1 (CCS), first identified in yeast (31), is known to deliver copper to SOD1 (21). However, CCS is not absolutely essential for delivery of copper to human SOD1. The superoxide scavenging activity of endogenous wild-type SOD1 was decreased by ∼70–80% in knockout mice lacking CCS (130). These results show that CCS is the major source supplying copper to SOD1 in vivo, but also mandates that SOD1 must have an alternative means for acquiring copper in cells. Copper is strictly required to be in the active site to account for the 20–30% of the superoxide scavenging activity that remains (9).
In studies using ALS mutant SOD1 mice with the knockout of endogenous mouse CCS, CCS deletion did not significantly affect the survival in three different transgenic SOD1 mouse lines (111). CCS has been shown to preferentially insert copper into zinc-containing SOD1 (94), which would produce only Cu,Zn SOD1. However, human SOD1 is able to directly acquire copper in the absence of CCS from other potential sources such as copper-glutathione (21). Alternative mechanisms for copper acquisition would not necessarily require that zinc be bound, and thus could potentially contribute to the formation of zinc-deficient SOD1. The mitochondria are the richest stores of copper in the cell and may be another major copper source for mutant SOD1s to acquire copper without the assistance of CCS. This route of copper acquisition may explain why mitochondrial pathology is an early marker of injury in the SOD1 ALS mice (67, 129).
A recent study by Elliott's group reported remarkable effects of overexpressing human CCS in the mutant G93A human SOD1 mouse model of ALS (108). Strikingly, motor neuron disease was greatly accelerated in the dual CCS/G93A SOD1 transgenic mice, who had a mean survival of 36 days, compared to 242 days for the transgenic G93A SOD1 mice. The survival of dual CCS/wildtype SOD1 transgenic mice and transgenic mice expressing just CCS were normal. The high levels of CCS present could generate greater copper incorporation into mutant SOD1, leading to greater copper toxicity. Although the transgenic mice expressing CCS die within 40 days, there was no evidence of SOD1 protein aggregation in these mice (92). These results indicate an important role for CCS through its interaction with mutant SOD1 in disease progression that needs to be further assessed.
Mutations to the Copper-Binding Ligands in the SOD1 Active Site
Two naturally occurring ALS mutations in humans directly affect the copper binding ligands of SOD1 (H46R and H48Q), which might suggest that copper is not necessary for SOD1 toxicity. However, Liochev and Fridovich showed that the H48Q mutant SOD1 was still able to bind copper in a redox active fashion (74). To further test the role of copper in mutant SOD1 toxicity, transgenic mice expressing mutant SOD1s with the combined H46R/H48Q double mutation and with all four copper-binding ligands (quad mutant) were developed, and both types of mice developed motor neuron disease (122, 124). The double mutant SOD1 transgenic mice with the highest expression levels of mutant SOD1 developed motor neuron degeneration within 4–6 months while the highest expressing quad mutant SOD1 mouse developed the disease between 8–11 months. Since histidine 63 is also a ligand for zinc, the zinc-binding site would also be affected in the quad mice. The double and quad mutant SOD1s were both reported to have no scavenging activity in superoxide activity gels. More recently, Borchelt et al. used radioactive labeled copper incorporation into transfected cells containing the double and quad mutant SOD1 (9, 121). They found no evidence for radioactive copper binding. However, their assay involved conditions using EDTA and 1% SDS, which we have found in vitro disrupts copper binding to recombinant mutant quad SOD1 (unpublished data).
Although studies with the dual and quad mice show that copper in the SOD1 copper binding site is not essential for SOD1 to be toxic to motor neurons, it does not prove that copper could not be bound in other locations on SOD1 (19). For over 30 years, copper has been known to bind in the zinc site of SOD1 (46, 87). The quad mutant SOD1 protein still contains two histidines and a negatively charged aspartate in the zinc-binding site that form a strong binding site for metals. Our lab has found that the quad mutant SOD1 can form a blue protein with copper tightly bound into the zinc-binding pocket and is much more redox reactive than when copper is in its normal site (unpublished data). The question of whether copper is involved in SOD1 toxicity in the quad mice is still unsettled, and does not rule out a role for copper in this disease.
Dietary Supplementation of Zinc
If zinc-deficient SOD1 is responsible for the toxic gain-of-function in SOD1 resulting in motor neuron degeneration, then supplementation with zinc may be protective while a zinc-deficient diet should accelerate the disease. An important role for zinc in the progression of this disease stems from research with metallothioneins that are major intracellular zinc-binding proteins that protect cells when zinc levels are low (112). The progression of ALS in G93A SOD1 transgenic mice was found to be accelerated when three major isoforms of metallothionein in the brain were genetically deleted (83, 93). In addition, two astrocytic isozymes for metallothionein were found to be as important as the neuronal isozyme for delaying ALS.
A study reported that high dosages of zinc administered in the drinking water of SOD1 transgenic mice accelerated ALS progression (54). This report appeared to contradict our theory that zinc supplementation should be protective if dietary zinc is deficient in the SOD1 transgenic mice. Examination of the zinc dosages shows that the study used extremely high concentrations of zinc, estimated to be 75 and 375 mg/kg/day. These levels of zinc are ∼15 and 75 times greater than the current recommendations established for rodents (99). Excessive levels of zinc are known to inhibit copper absorption, which inhibits copper-dependent ceruloplasmin and results in a potentially lethal anemia (47). We found that depletion of zinc in the G93A SOD1 mice accelerated the progression of ALS, and moderate supplementation of zinc provided protection (36). A dosage of 12 mg/kg/day extended life by 11 days in the SOD1 ALS mice compared to the zinc-deficient group (Fig. 6). Even at a slightly higher dosage of 18 mg/kg/day, disease progression was accelerated in more than half of the mice, but with the addition of 0.3 mg/kg/day copper this early progression was prevented.
FIG. 6.

Effects of zinc deficiency versus zinc supplementation on survival. Male G93A mice were placed on a zinc-deficient diet at 30 days. At day 50, mice were supplemented with 0 mg/L (0 mg/kg/day), 30 mg/L (6 mg/kg/day), 60 mg/L (12 mg/kg/day), 90 mg/L (18 mg/kg/day) zinc, or 90 mg/L (18 mg/kg/day) zinc plus 2 mg/L (0.3 mg/kg/day) copper in the drinking water (n = 9 for each group). Adapted from Ermilova et al. (36).
Copper Chelation
If the aberrant redox activity of copper in zinc-deficient SOD1 is responsible for the toxic gain-of-function of mutant SOD1, then focus on copper's role in this disease should be another route towards therapeutic treatment. This route would be through the chelation therapy of copper. The loss of zinc in either wild-type or mutant SOD1 greatly exposes the copper ion. Copper chelators should be able to access the copper in order to pull it out of the active site, therefore rendering the toxic zinc-deficient SOD1 to be harmless. Copper is essential for many processes, including insertion of iron in heme and for oxygen binding in cytochrome c oxidase, so it would be impossible to substantially deplete copper levels (47). Nevertheless, chelation therapy is already being investigated as a potential therapeutic strategy towards the treatment of various other neurodegeneration diseases such as Alzheimer's, Parkinson's, and prion disease (50).
Copper chelators have already been shown to slightly delay death in animal models of ALS. D-penicillamine, a copper chelator used in Wilson's disease (a genetic disorder which involves the accumulation of copper in tissue), was found to delay the onset and extend the survival of the G93A SOD1 mouse model of ALS (58). The delay of onset was by ∼7–10 days and the extension of survival by ∼10–11 days, depending on administration technique.
Recently, ammonium tetrathiomolybdate (TTM), a selective copper(II) chelator also used for the treatment of Wilson's disease, was found to delay onset by 10 days, slow progression by 11 days, and prolong survival in the G93A SOD1 mice by a more significant timeframe of 30 days (115). Our lab has found that TTM preferentially removes copper from zinc-deficient SOD1 compared to Cu,Zn SOD1 in vitro (unpublished data), providing a potential protective mode of action in the SOD1 ALS mice.
Overall, these animal studies support an important role for the redox activity of copper in the disease pathology of ALS. Even though these copper chelators did not show a significant protective effect, chelation therapy may still be a promising therapeutic strategy for the treatment of ALS. The efficacy of a copper chelator as a potential treatment for ALS could be greatly improved if optimized to specifically inactivate zinc-deficient SOD1, and it is relatively simple to adapt zinc-deficient SOD1 as a target for high throughput screening for therapeutic chelators.
Summary
Ultimately unraveling the disease mechanism of familial ALS with the aid of animal models could also illuminate the mechanism behind the progression of sporadic ALS. The zinc-deficient SOD1 theory provides a link between familial and sporadic ALS, explains how over a hundred different mutations could have the same common action, and why people can express the mutant protein for up to 80 years before developing the disease. The theory proposes that the mutations to SOD1 do not directly confer the toxic gain-of-function but rather increases the susceptibility to lose zinc and the zinc-deficient SOD1 is responsible for the death of motor neurons. Wild-type SOD1 could participate in sporadic ALS if it becomes zinc-deficient. The difference in affinities for zinc between the wild type and many ALS mutant SOD1s varies by only 5–50 fold (29). An important prediction of the zinc-deficient SOD1 hypothesis is that therapeutic approaches developed against zinc-deficient mutant SOD1 could be at least as beneficial to sporadic ALS patients without SOD1 mutations.
Acknowledgments
This work was financially supported in part by funding from the National Institute of Health grants NCCAM T32 AT002688 (K.A.T.), NS058628, AT002034 and ES00240.
Abbreviations
ALS, amyotrophic lateral sclerosis; CCS, copper chaperone protein for SOD1; GFAP, glial fibrillary acidic protein; O2•−, superoxide; H2O2, hydrogen peroxide; HSP90, heat shock protein 90; NFH, neurofilament heavy chain; NFL, neurofilament light chain; O2), molecular oxygen; SOD1, copper, zinc superoxide dismutase; TTM, ammonium tetrathiomolybdate.
References
- 1.Abe K. Pan L-H. Watanabe M. Kato T. Itoyama Y. Induction of nitrotyrosine-like immunoreactivity in the lower motor neuron of amyotrophic lateral sclerosis. Neurosci Lett. 1995;199:152–154. doi: 10.1016/0304-3940(95)12039-7. [DOI] [PubMed] [Google Scholar]
- 2.Abe K. Pan LH. Watanabe M. Konno H. Kato T. Itoyama Y. Upregulation of protein-tyrosine nitration in the anterior horn cells of amyotrophic lateral sclerosis. Neurol Res. 1997;19:124–128. doi: 10.1080/01616412.1997.11740784. [DOI] [PubMed] [Google Scholar]
- 3.Andersen PM. Sims KB. Xin WW. Kiely R. O'Neill G. Ravits J. Pioro E. Harati Y. Brower RD. Levine JS. Heinicke HU. Seltzer W. Boss M. Brown RH., Jr Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: A decade of discoveries, defects and disputes. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:62–73. doi: 10.1080/14660820310011700. [DOI] [PubMed] [Google Scholar]
- 4.Arnesano F. Banci L. Bertini I. Martinelli M. Furukawa Y. O'Halloran T V. The unusually stable quaternary structure of human Cu,Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J Biol Chem. 2004;279:47998–48003. doi: 10.1074/jbc.M406021200. [DOI] [PubMed] [Google Scholar]
- 5.Awano T. Johnson GS. Wade CM. Katz ML. Johnson GC. Taylor JF. Perloski M. Biagi T. Baranowska I. Long S. March PA. Olby NJ. Shelton GD. Khan S. O'Brien DP. Lindblad–Toh K. Coates JR. Genome-wide association analysis reveals a SOD1 mutation in canine degenerative myelopathy that resembles amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2009;106:2794–2799. doi: 10.1073/pnas.0812297106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barbeito LH. Pehar M. Cassina P. Vargas MR. Peluffo H. Viera L. Estevez AG. Beckman JS. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res Brain Res Rev. 2004;47:263–274. doi: 10.1016/j.brainresrev.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Beal MF. Ferrante RJ. Browne SE. Matthews RT. Kowall NW. Brown RH., Jr Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann Neurol. 1997;42:646–654. doi: 10.1002/ana.410420416. [DOI] [PubMed] [Google Scholar]
- 8.Beckman JS. Beckman TW. Chen J. Marshall PM. Freeman BA. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury by nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beckman JS. Estevez AG. Barbeito L. Crow JP. CCS knockout mice establish an alternative source of copper for SOD in ALS. Free Radic Biol Med. 2002;33:1433–1435. doi: 10.1016/s0891-5849(02)01092-4. [DOI] [PubMed] [Google Scholar]
- 10.Beckman JS. Estevez AG. Crow JP. Barbeito L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001;24:S15–20. doi: 10.1016/s0166-2236(00)01981-0. [DOI] [PubMed] [Google Scholar]
- 11.Beckman JS. Ischiropoulos H. Zhu L. van der Woerd M. Smith C. Chen J. Harrison J. Martin JC. Tsai M. Kinetics of superoxide dismutase- and iron-catalyzed nitration of phenolics by peroxynitrite. Arch Biochem Biophys. 1992;298:438–445. doi: 10.1016/0003-9861(92)90432-v. [DOI] [PubMed] [Google Scholar]
- 12.Beyer WF., Jr. Fridovich I. Mullenbach GT. Hallewell R. Examination of the role of arginine-143 in the human copper and zinc superoxide dismutase by site-specific mutagenesis. J Biol Chem. 1987;262:11182–11187. [PubMed] [Google Scholar]
- 13.Broom WJ. Johnson DV. Auwarter KE. Iafrate AJ. Russ C. Al–Chalabi A. Sapp PC. McKenna–Yasek D. Andersen PM. Brown RH., Jr SOD1A4V-mediated ALS: absence of a closely linked modifier gene and origination in Asia. Neurosci Lett. 2008;430:241–245. doi: 10.1016/j.neulet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Bruijn LI. Beal MF. Becher MW. Schulz JB. Wong PC. Price DL. Cleveland DW. Elevated free nitrotyrosine levels, but not protein-bound nitrotyrosine or hydroxyl radicals, throughout amyotrophic lateral sclerosis (ALS)-like disease implicate tyrosine nitration as an aberrant in vivo property of one familial ALS-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci USA. 1997;94:7606–7611. doi: 10.1073/pnas.94.14.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruijn LI. Becher MW. Lee MK. Anderson KL. Jenkins NA. Copeland NG. Sisodia SS. Rothstein JD. Borchelt DR. Price DL. Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 16.Bruijn LI. Cleveland DW. Mechanisms of selective motor neuron death in ALS: Insights from transgenic mouse models of motor neuron disease. Neuropathol Appl Neurobiol. 1996;22:373–387. doi: 10.1111/j.1365-2990.1996.tb00907.x. [DOI] [PubMed] [Google Scholar]
- 17.Bruijn LI. Houseweart MK. Kato S. Anderson KL. Anderson SD. Ohama E. Reaume AG. Scott RW. Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 18.Bruijn LI. Miller TM. Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- 19.Bush AI. Is ALS caused by an altered oxidative activity of mutant superoxide dismutase? Nat Neurosci 5: 919; author reply. 2002:919–920. doi: 10.1038/nn1002-919a. [DOI] [PubMed] [Google Scholar]
- 20.Cao X. Antonyuk SV. Seetharaman SV. Whitson LJ. Taylor AB. Holloway SP. Strange RW. Doucette PA. Valentine JS. Tiwari A. Hayward LJ. Padua S. Cohlberg JA. Hasnain SS. Hart PJ. Structures of the G85R variant of SOD1 in familial amyotrophic lateral sclerosis. J Biol Chem. 2008;283:16169–16177. doi: 10.1074/jbc.M801522200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carroll MC. Girouard JB. Ulloa JL. Subramaniam JR. Wong PC. Valentine JS. Culotta VC. Mechanisms for activating Cu- and Zn-containing superoxide dismutase in the absence of the CCS Cu chaperone. Proc Natl Acad Sci USA. 2004;101:5964–5969. doi: 10.1073/pnas.0308298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Casoni F. Basso M. Massignan T. Gianazza E. Cheroni C. Salmona M. Bendotti C. Bonetto V. Protein nitration in a mouse model of familial amyotrophic lateral sclerosis: Possible multifunctional role in the pathogenesis. J Biol Chem. 2005;280:16295–16304. doi: 10.1074/jbc.M413111200. [DOI] [PubMed] [Google Scholar]
- 23.Cassina P. Cassina A. Pehar M. Castellanos R. Gandelman M. de Leon A. Robinson KM. Mason RP. Beckman JS. Barbeito L. Radi R. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci. 2008;28:4115–4122. doi: 10.1523/JNEUROSCI.5308-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cassina P. Pehar M. Vargas MR. Castellanos R. Barbeito AG. Estevez AG. Thompson JA. Beckman JS. Barbeito L. Astrocyte activation by fibroblast growth factor-1 and motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. 2005;93:38–46. doi: 10.1111/j.1471-4159.2004.02984.x. [DOI] [PubMed] [Google Scholar]
- 25.Cassina P. Peluffo H. Pehar M. Martinez–Palma L. Ressia A. Beckman JS. Estevez AG. Barbeito L. Peroxynitrite triggers a phenotypic transformation in spinal cord astrocytes that induces motor neuron apoptosis. J Neurosci Res. 2002;67:21–29. doi: 10.1002/jnr.10107. [DOI] [PubMed] [Google Scholar]
- 26.Cha CI. Chung YH. Shin CM. Shin DH. Kim YS. Gurney ME. Lee KW. Immunocytochemical study on the distribution of nitrotyrosine in the brain of the transgenic mice expressing a human Cu/Zn SOD mutation. Brain Res. 2000;853:156–161. doi: 10.1016/s0006-8993(99)02302-1. [DOI] [PubMed] [Google Scholar]
- 27.Cleveland DW. Liu J. Oxidation versus aggregation. How do SOD1 mutants cause ALS? Nat Med. 2000;6:1320–1321. doi: 10.1038/82122. [DOI] [PubMed] [Google Scholar]
- 28.Couillard–Després S. Zhu Q. Wong PC. Price DL. Cleveland DW. Julien J-P. Protective effect of neurofilament heavy gene overexpression in motor neuron disease induced by mutant superoxide dismutase. Proc Natl Acad Sci USA. 1998;95:9626–9630. doi: 10.1073/pnas.95.16.9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crow JP. Sampson JB. Zhuang Y. Thompson JA. Beckman JS. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J Neurochem. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- 30.Crow JP. Ye YZ. Strong M. Kirk M. Barnes S. Beckman JS. Superoxide dismutase catalyzes nitration of tyrosines by peroxynitrite in the rod and head domains of neurofilament-L. J Neurochem. 1997;69:1945–1953. doi: 10.1046/j.1471-4159.1997.69051945.x. [DOI] [PubMed] [Google Scholar]
- 31.Culotta VC. Klomp LWJ. Strain J. Casareno RLB. Krems B. Gitlin JD. The copper chaperone for superoxide dismutase. J Biol Chem. 1997;272:23469–23472. doi: 10.1074/jbc.272.38.23469. [DOI] [PubMed] [Google Scholar]
- 32.Deng H-X. Hentati A. Tainer J. Igbal Z. Cayabyab A. Hung W–Y. Getzoff E. Hu P. Herzfeldt B. Roos R. Warner C. Deng G. Soriano E. Smyth C. Parge H. Ahmed A. Roses A. Hallewell R. Pericak–Vance M. Siddique T. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 33.Denicola A. Souza JM. Radi R. Lissi E. Nitric oxide diffusion in membranes determined by fluorescence quenching. Arch Biochem Biophys. 1996;328:208–212. doi: 10.1006/abbi.1996.0162. [DOI] [PubMed] [Google Scholar]
- 34.Durham HD. Roy J. Dong L. Figlewicz DA. Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropath Exp Neuro. 1997;56:523–530. doi: 10.1097/00005072-199705000-00008. [DOI] [PubMed] [Google Scholar]
- 35.Ellerby LM. Cabelli DE. Graden JA. Valentine JS. Copper-zinc superoxide dismutase: Why not pH-dependent? J Am Chem Soc. 1996;118:6556–6561. [Google Scholar]
- 36.Ermilova IP. Ermilov VB. Levy M. Ho E. Pereira C. Beckman JS. Protection by dietary zinc in ALS mutant G93A SOD transgenic mice. Neurosci Lett. 2005;379:42–46. doi: 10.1016/j.neulet.2004.12.045. [DOI] [PubMed] [Google Scholar]
- 37.Estévez AG. Crow JP. Sampson JB. Reiter C. Zhuang Y–X. Richardson GJ. Tarpey MM. Barbeito L. Beckman JS. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286:2498–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- 38.Estévez AG. Sampson JB. Zhuang Y–X. Spear N. Richardson GJ. Crow JP. Tarpey MM. Barbeito L. Beckman JS. Liposome-delivered superoxide dismutase prevents nitric oxide-dependent motor neuron death induced by trophic factor withdrawal. Free Radic Biol Med. 2000;28:437–446. doi: 10.1016/s0891-5849(99)00261-0. [DOI] [PubMed] [Google Scholar]
- 39.Estévez AG. Spear N. Manuel SM. Barbeito L. Radi R. Beckman JS. Role of endogenous nitric oxide and peroxynitrite formation in the survival and death of motor neurons in culture. In: Mize R, editor; Friedlander M, editor. Progress in Brain Research. Amsterdam: Elsevier; 1998. pp. 269–280. [DOI] [PubMed] [Google Scholar]
- 40.Estévez AG. Spear N. Manuel SM. Radi R. Henderson CE. Barbeito L. Beckman JS. Nitric oxide and superoxide contribute to motor neuron apoptosis induced by trophic factor deprivation. J Neurosci. 1998;18:923–931. doi: 10.1523/JNEUROSCI.18-03-00923.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Estévez AG. Spear N. Thompson JA. Cornwell TL. Radi R. Barbeito L. Beckman JS. Nitric oxide dependent production of cGMP supports the survival of rat embryonic motor neurons cultured with brain derived neurotrophic factor. J Neurosci. 1998;18:3708–3714. doi: 10.1523/JNEUROSCI.18-10-03708.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferrante RJ. Browne SE. Shinobu LA. Bowling AC. Baik MJ. MacGarvey U. Kowall NW. Brown RH., Jr Beal MF. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem. 1997;69:2064–2074. doi: 10.1046/j.1471-4159.1997.69052064.x. [DOI] [PubMed] [Google Scholar]
- 43.Ferrante RJ. Shinobu LA. Schulz JB. Matthews RT. Thomas CE. Kowall NW. Gurney ME. Beal MF. Increased 3-nitrotyrosine and oxidative damage in mice with a human copper/zinc superoxide dismutase mutation. Ann Neurol. 1997;42:326–334. doi: 10.1002/ana.410420309. [DOI] [PubMed] [Google Scholar]
- 44.Ferri A. Nencini M. Casciati A. Cozzolino M. Angelini DF. Longone P. Spalloni A. Rotilio G. Carri MT. Cell death in amyotrophic lateral sclerosis: Interplay between neuronal and glial cells. FASEB J. 2004;18:1261–1263. doi: 10.1096/fj.03-1199fje. [DOI] [PubMed] [Google Scholar]
- 45.Fisher CL. Cabelli DE. Tainer JA. Hallewell RA. Getzoff ED. The role of arginine 143 in the electrostatics and mechanism of Cu,Zn superoxide dismutase: Computational and experimental evaluation by mutational analysis. Proteins. 1994;19:24–34. doi: 10.1002/prot.340190105. [DOI] [PubMed] [Google Scholar]
- 46.Forman JH. Fridovich I. On the stability of bovine superoxide dismutase. J Biol Chem. 1973;248:2645–2649. [PubMed] [Google Scholar]
- 47.Fox PL. The copper-iron chronicles: The story of an intimate relationship. Biometals. 2003;16:9–40. doi: 10.1023/a:1020799512190. [DOI] [PubMed] [Google Scholar]
- 48.Fridovich I. Biological effects of the superoxide radical. Arch Biochem Biophys. 1986;247:1–11. doi: 10.1016/0003-9861(86)90526-6. [DOI] [PubMed] [Google Scholar]
- 49.Fukada K. Nagano S. Satoh M. Tohyama C. Nakanishi T. Shimizu A. Yanagihara T. Sakoda S. Stabilization of mutant Cu/Zn superoxide dismutase (SOD1) protein by coexpressed wild SOD1 protein accelerates the disease progression in familial amyotrophic lateral sclerosis mice. Eur J Neurosci. 2001;14:2032–2036. doi: 10.1046/j.0953-816x.2001.01828.x. [DOI] [PubMed] [Google Scholar]
- 50.Gaeta A. Hider RC. The crucial role of metal ions in neurodegeneration: The basis for a promising therapeutic strategy. Br J Pharmacol. 2005;146:1041–1059. doi: 10.1038/sj.bjp.0706416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Getzoff ED. Tainer JA. Stempien MM. Bell GI. Hallewell RA. Evolution of CuZn superoxide dismutase and the Greek key ß-barrel structural motif. Proteins. 1989;5:322–336. doi: 10.1002/prot.340050408. [DOI] [PubMed] [Google Scholar]
- 52.Getzoff ED. Tainer JA. Weiner PK. Kollman PA. Richardson JS. Richardson DC. Electrostatic recognition between superoxide and copper, zinc superoxide dismutase. Nature (Lond) 1983;306:287–290. doi: 10.1038/306287a0. [DOI] [PubMed] [Google Scholar]
- 53.Ghadge GD. Lee JP. Bindokas VP. Jordan J. Ma L. Miller RJ. Roos RP. Mutant superoxide dismutase-1-linked familial amyotrophic lateral sclerosis: Molecular mechanisms of neuronal death and protection. J Neurosci. 1997;17:8756–8766. doi: 10.1523/JNEUROSCI.17-22-08756.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Groeneveld GJ. de Leeuw van Weenen J. van Muiswinkel FL. Veldman H. Veldink JH. Wokke JH. Bar PR. van den Berg LH. Zinc amplifies mSOD1-mediated toxicity in a transgenic mouse model of amyotrophic lateral sclerosis. Neurosci Lett. 2003;352:175–178. doi: 10.1016/j.neulet.2003.08.062. [DOI] [PubMed] [Google Scholar]
- 55.Gurney ME. Pu H. Chiu AY. Dal Corto MC. Polchow CY. Alexander DD. Caliendo J. Hentati A. Kwon YW. Deng H-X. Chen W. Zhai P. Sufit RL. Siddique T. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 56.Hayward LJ. Rodriguez JA. Kim JW. Tiwari A. Goto JJ. Cabelli DE. Valentine JS. Brown RH., Jr Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J Biol Chem. 2002;277:15923–15931. doi: 10.1074/jbc.M112087200. [DOI] [PubMed] [Google Scholar]
- 57.Hornberg A. Logan DT. Marklund SL. Oliveberg M. The coupling between disulphide status, metallation and dimer interface strength in Cu/Zn superoxide dismutase. J Mol Biol. 2007;365:333–342. doi: 10.1016/j.jmb.2006.09.048. [DOI] [PubMed] [Google Scholar]
- 58.Hottinger AF. Fine EG. Gurney ME. Zurn AD. Aebischer P. The copper chelator d-penicillamine delays onset of disease and extends survival in a transgenic mouse model of familial amyotrophic lateral sclerosis. Eur J Neurosci. 1997;9:1548–1551. doi: 10.1111/j.1460-9568.1997.tb01511.x. [DOI] [PubMed] [Google Scholar]
- 59.Huie RE. Padmaja S. The reaction rate of nitric oxide with superoxide. Free Rad Res Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 60.Ischiropoulos H. Biological tyrosine nitration: a pathophysiological function of nitric oxide and reactive oxygen species. Arch Biochem Biophys. 1998;356:1–11. doi: 10.1006/abbi.1998.0755. [DOI] [PubMed] [Google Scholar]
- 61.Ischiropoulos H. Zhu L. Beckman JS. Peroxynitrite formation from macrophage-derived nitric oxide. Arch Biochem Biophys. 1992;298:446–451. doi: 10.1016/0003-9861(92)90433-w. [DOI] [PubMed] [Google Scholar]
- 62.Ischiropoulos H. Zhu L. Chen J. Tsai M. Martin JC. Smith CD. Beckman JS. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- 63.Jonsson PA. Backstrand A. Andersen PM. Jacobsson J. Parton M. Shaw C. Swingler R. Shaw PJ. Robberecht W. Ludolph AC. Siddique T. Skvortsova VI. Marklund SL. CuZn-superoxide dismutase in D90A heterozygotes from recessive and dominant ALS pedigrees. Neurobiol Dis. 2002;10:327–333. doi: 10.1006/nbdi.2002.0508. [DOI] [PubMed] [Google Scholar]
- 64.Jonsson PA. Ernhill K. Andersen PM. Bergemalm D. Brannstrom T. Gredal O. Nilsson P. Marklund SL. Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis. Brain. 2004;127:73–88. doi: 10.1093/brain/awh005. [DOI] [PubMed] [Google Scholar]
- 65.Kiaei M. Bush AI. Morrison BM. Morrison JH. Cherny RA. Volitakis I. Beal MF. Gordon JW. Genetically decreased spinal cord copper concentration prolongs life in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2004;24:7945–7950. doi: 10.1523/JNEUROSCI.2000-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klug D. Rabani J. Fridovich I. A direct demonstration of the catalytic action of superoxide dismutase through the use of pulse radiolysis. J Biol Chem. 1972;247:4839–4842. [PubMed] [Google Scholar]
- 67.Kong J. Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee JP. Gerin C. Bindokas VP. Miller R. Ghadge G. Roos RP. No correlation between aggregates of Cu/Zn superoxide dismutase and cell death in familial amyotrophic lateral sclerosis. J Neurochem. 2002;82:1229–1238. doi: 10.1046/j.1471-4159.2002.01056.x. [DOI] [PubMed] [Google Scholar]
- 69.Leinweber B. Barofsky E. Barofsky DF. Ermilov V. Nylin K. Beckman JS. Aggregation of ALS mutant superoxide dismutase expressed in Escherichia coli. Free Radic Biol Med. 2004;36:911–918. doi: 10.1016/j.freeradbiomed.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 70.Lindberg MJ. Bystrom R. Boknas N. Andersen PM. Oliveberg M. Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)-associated SOD1 mutants. Proc Natl Acad Sci USA. 2005;102:9754–9759. doi: 10.1073/pnas.0501957102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lindberg MJ. Normark J. Holmgren A. Oliveberg M. Folding of human superoxide dismutase: Disulfide reduction prevents dimerization and produces marginally stable monomers. Proc Natl Acad Sci USA. 2004;101:15893–15898. doi: 10.1073/pnas.0403979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lindberg MJ. Tibell L. Oliveberg M. Common denominator of Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis: Decreased stability of the apo state. Proc Natl Acad Sci USA. 2002;99:16607–16612. doi: 10.1073/pnas.262527099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lino MM. Schneider C. Caroni P. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J Neurosci. 2002;22:4825–4832. doi: 10.1523/JNEUROSCI.22-12-04825.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liochev SI. Chen LL. Hallewell RA. Fridovich I. Superoxide-dependent peroxidase activity of H48Q: A superoxide dismutase variant associated with familial amyotrophic lateral sclerosis. Arch Biochem Biophys. 1997;346:263–268. doi: 10.1006/abbi.1997.0298. [DOI] [PubMed] [Google Scholar]
- 75.Lynch SM. Boswell SA. Colon W. Kinetic stability of Cu/Zn superoxide dismutase is dependent on its metal ligands: Implications for ALS. Biochemistry. 2004;43:16525–16531. doi: 10.1021/bi048831v. [DOI] [PubMed] [Google Scholar]
- 76.Malinski T. Bailey F. Zhang ZG. Chopp M. Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1993;13:355–358. doi: 10.1038/jcbfm.1993.48. [DOI] [PubMed] [Google Scholar]
- 77.Malinski T. Taha Z. Nitric oxide release from a single cell measured in situ by a porphyrinic-based microsensor. Nature. 1992;358:676–678. doi: 10.1038/358676a0. [DOI] [PubMed] [Google Scholar]
- 78.Marklund SL. Andersen PM. Forsgren L. Nilsson P. Ohlsson PI. Wikander G. Oberg A. Normal binding and reactivity of copper in mutant superoxide dismutase isolated from amyotrophic lateral sclerosis patients. J Neurochem. 1997;69:675–681. doi: 10.1046/j.1471-4159.1997.69020675.x. [DOI] [PubMed] [Google Scholar]
- 79.McCord JM. Fridovich I. Superoxide dismutase: An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 80.Morrison BM. Gordon JW. Ripps ME. Morrison JH. Quantitative immunocytochemical analysis of the spinal cord in G86R superoxide dismutase transgenic mice: neurochemical correlates of selective vulnerability. J Comp Neurol. 1996;373:619–631. doi: 10.1002/(SICI)1096-9861(19960930)373:4<619::AID-CNE9>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 81.Morrison BM. Janssen WG. Gordon JW. Morrison JH. Time course of neuropathology in the spinal cord of G86R superoxide dismutase transgenic mice. J Comp Neurol. 1998;391:64–77. doi: 10.1002/(sici)1096-9861(19980202)391:1<64::aid-cne6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 82.Nagai M. Aoki M. Miyoshi I. Kato M. Pasinelli P. Kasai N. Brown RH., Jr. Itoyama Y. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: Associated mutations develop motor neuron disease. J Neurosci. 2001;21:9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nagano S. Satoh M. Sumi H. Fujimura H. Tohyama C. Yanagihara T. Sakoda S. Reduction of metallothioneins promotes the disease expression of familial amyotrophic lateral sclerosis mice in a dose-dependent manner. Eur J Neurosci. 2001;13:1363–1370. doi: 10.1046/j.0953-816x.2001.01512.x. [DOI] [PubMed] [Google Scholar]
- 84.Ogihara NL. Parge HE. Hart PJ. Weiss MS. Goto JJ. Crane BR. Tsang J. Slater K. Roe JA. Valentine JS. Eisenberg D. Tainer JA. Unusual trigonal-planar copper configuration revealed in the atomic structure of yeast copper-zinc superoxide dismutase. Biochemistry. 1996;35:2316–2321. doi: 10.1021/bi951930b. [DOI] [PubMed] [Google Scholar]
- 85.Okado–Matsumoto A. Fridovich I. Amyotrophic lateral sclerosis: A proposed mechanism. Proc Natl Acad Sci USA. 2002;99:9010–9014.. doi: 10.1073/pnas.132260399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pacher P. Beckman JS. Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pantoliano MW. McDonnell PJ. Valentine JS. Reversible loss of metal ions from the zinc binding site of copper-zinc superoxide dismutase. The low pH transition. J Am Chem Soc. 1979;101:6454–6456. [Google Scholar]
- 88.Parge HE. Hallewell RA. Tainer JA. Atomic structures of wild-type and thermostable mutant recombinant human Cu,Zn superoxide dismutase. Proc Natl Acad Sci USA. 1992;89:6109–6113. doi: 10.1073/pnas.89.13.6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pasinelli P. Belford ME. Lennon N. Bacskai BJ. Hyman BT. Trotti D. Brown RH., Jr Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 90.Pehar M. Cassina P. Vargas MR. Castellanos R. Viera L. Beckman JS. Estevez AG. Barbeito L. Astrocytic production of nerve growth factor in motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. 2004;89:464–473. doi: 10.1111/j.1471-4159.2004.02357.x. [DOI] [PubMed] [Google Scholar]
- 91.Pehar M. Vargas MR. Robinson KM. Cassina P. Diaz-Amarilla PJ. Hagen TM. Radi R. Barbeito L. Beckman JS. Mitochondrial superoxide production and nuclear factor erythroid 2-related factor 2 activation in p75 neurotrophin receptor-induced motor neuron apoptosis. J Neurosci. 2007;27:7777–7785. doi: 10.1523/JNEUROSCI.0823-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Proescher JB. Son M. Elliott JL. Culotta VC. Biological effects of CCS in the absence of SOD1 enzyme activation: Implications for disease in a mouse model for ALS. Hum Mol Genet. 2008;17:1728–1737. doi: 10.1093/hmg/ddn063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Puttaparthi K. Gitomer WL. Krishnan U. Son M. Rajendran B. Elliott JL. Disease progression in a transgenic model of familial amyotrophic lateral sclerosis is dependent on both neuronal and non-neuronal zinc binding proteins. J Neurosci. 2002;22:8790–8796. doi: 10.1523/JNEUROSCI.22-20-08790.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rae TD. Schmidt PJ. Pufahl RA. Culotta VC. O'Halloran TV. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 95.Rakhit R. Crow JP. Lepock JR. Kondejewski LH. Cashman NR. Chakrabartty A. Monomeric Cu,Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J Biol Chem. 2004;279:15499–15504. doi: 10.1074/jbc.M313295200. [DOI] [PubMed] [Google Scholar]
- 96.Rakhit R. Robertson J. Velde CV. Horne P. Ruth DM. Griffin J. Cleveland DW. Cashman NR. Chakrabartty A. An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat Med. 2007;13:754–759. doi: 10.1038/nm1559. [DOI] [PubMed] [Google Scholar]
- 97.Raoul C. Buhler E. Sadeghi C. Jacquier A. Aebischer P. Pettmann B. Henderson CE. Haase G. Chronic activation in presymptomatic amyotrophic lateral sclerosis (ALS) mice of a feedback loop involving Fas, Daxx, and FasL. Proc Natl Acad Sci USA. 2006;103:6007–6012. doi: 10.1073/pnas.0508774103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Raoul C. Estevez AG. Nishimune H. Cleveland DW. deLapeyriere O. Henderson CE. Haase G. Pettmann B. Motoneuron death triggered by a specific pathway downstream of Fas. potentiation by ALS-linked SOD1 mutations. Neuron. 2002;35:1067–1083. doi: 10.1016/s0896-6273(02)00905-4. [DOI] [PubMed] [Google Scholar]
- 99.Reeves PG. Nielsen FH. Fahey GC., Jr AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J Nutr. 1993;123:1939–1951. doi: 10.1093/jn/123.11.1939. [DOI] [PubMed] [Google Scholar]
- 100.Roberts BR. Tainer JA. Getzoff ED. Malencik DA. Anderson SR. Bomben VC. Meyers KR. Karplus PA. Beckman JS. Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS. J Mol Biol. 2007;373:377–390. doi: 10.1016/j.jmb.2007.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Robinson KM. Janes MS. Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protoc. 2008;3:941–947. doi: 10.1038/nprot.2008.56. [DOI] [PubMed] [Google Scholar]
- 102.Rosen DR. Siddique T. Patterson D. Figlewicz DA. Sapp P. Hentati A. Donaldson D. Goto J. O'Regan JP. Deng H–X. Rahmani Z. Krizus A. McKenna–Yasek D. Cayabyab A. Gaston SM. Berger R. Tanszi RE. Halperin JJ. Herzfeldt B. Van den Bergh R. Hung W–Y. Bird T. Deng G. Mulder DW. Smyth C. Lang NG. Soriana E. Pericak–Vance MA. Haines J. Rouleau GA. Gusella JS. Horvitz HR. Brown RH., Jr Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 103.Ross CA. Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 104.Rowland LP. Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 105.Sasaki S. Warita H. Abe K. Iwata M. Inducible nitric oxide synthase (iNOS) and nitrotyrosine immunoreactivity in the spinal cords of transgenic mice with a G93A mutant SOD1 gene. J Neuropathol Exp Neurol. 2001;60:839–846. doi: 10.1093/jnen/60.9.839. [DOI] [PubMed] [Google Scholar]
- 106.Shibuki K. Okada D. Endogenous nitric oxide release required for long-term synaptic depression in the cerebellum. Nature (Lond) 1991;349:326–329. doi: 10.1038/349326a0. [DOI] [PubMed] [Google Scholar]
- 107.Smith CD. Carson M. van der Woerd M. Chen J. Ischiropoulos H. Beckman JS. Crystal structure of peroxynitrite-modified bovine Cu,Zn superoxide dismutase. Arch Biochem Biophys. 1992;299:350–355. doi: 10.1016/0003-9861(92)90286-6. [DOI] [PubMed] [Google Scholar]
- 108.Son M. Puttaparthi K. Kawamata H. Rajendran B. Boyer PJ. Manfredi G. Elliott JL. Overexpression of CCS in G93A-SOD1 mice leads to accelerated neurological deficits with severe mitochondrial pathology. Proc Natl Acad Sci USA. 2007;104:6072–6077. doi: 10.1073/pnas.0610923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stathopulos PB. Rumfeldt JA. Scholz GA. Irani RA. Frey HE. Hallewell RA. Lepock JR. Meiering EM. Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc Natl Acad Sci USA. 2003;100:7021–7026. doi: 10.1073/pnas.1237797100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Strong MJ. Sopper MM. Crow JP. Strong WL. Beckman JS. Nitration of the low molecular weight neurofilament is equivalent in sporadic amyotrophic lateral sclerosis and control cervical spinal cord. Biochem Biophys Res Commun. 1998;248:157–164. doi: 10.1006/bbrc.1998.8930. [DOI] [PubMed] [Google Scholar]
- 111.Subramaniam JR. Lyons WE. Liu J. Bartnikas TB. Rothstein J. Price DL. Cleveland DW. Gitlin JD. Wong PC. Mutant SOD1 causes motor neuron disease independent of copper chaperone-mediated copper loading. Nat Neurosci. 2002;5:301–307. doi: 10.1038/nn823. [DOI] [PubMed] [Google Scholar]
- 112.Suhy DA. Simon KD. Linzer DI. O'Halloran TV. Metallothionein is part of a zinc-scavenging mechanism for cell survival under conditions of extreme zinc deprivation. J Biol Chem. 1999;274:9183–9192. doi: 10.1074/jbc.274.14.9183. [DOI] [PubMed] [Google Scholar]
- 113.Tainer JA. Getzoff ED. Been KM. Richardson JS. Richardson DC. Determination and analysis of the 2 Å structure of copper, zinc superoxide dismutase. J Mol Biol. 1982;160:181–217. doi: 10.1016/0022-2836(82)90174-7. [DOI] [PubMed] [Google Scholar]
- 114.Tohgi H. Abe T. Yamazaki K. Murata T. Ishizaki E. Isobe C. Remarkable increase in cerebrospinal fluid 3-nitrotyrosine in patients with sporadic amotrophic lateral sclerosis. Ann Neurol. 1999;46:129–131. doi: 10.1002/1531-8249(199907)46:1<129::aid-ana21>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 115.Tokuda E. Ono S. Ishige K. Watanabe S. Okawa E. Ito Y. Suzuki T. Ammonium tetrathiomolybdate delays onset, prolongs survival, and slows progression of disease in a mouse model for amyotrophic lateral sclerosis. Exp Neurol. 2008;213:122–128. doi: 10.1016/j.expneurol.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 116.Urushitani M. Shimohama S. The role of nitric oxide in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2:71–81. doi: 10.1080/146608201316949415. [DOI] [PubMed] [Google Scholar]
- 117.Valentine JS. Doucette PA. Zittin Potter S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem. 2005;74:563–593. doi: 10.1146/annurev.biochem.72.121801.161647. [DOI] [PubMed] [Google Scholar]
- 118.Van der Vliet A. Eiserich JP. O'Neill CA. Halliwell B. Cross CE. Tyrosine modification by reactive nitrogen species: A closer look. Arch Biochem Biophys. 1995;319:341–349. doi: 10.1006/abbi.1995.1303. [DOI] [PubMed] [Google Scholar]
- 119.Vargas MR. Pehar M. Cassina P. Martinez–Palma L. Thompson JA. Beckman JS. Barbeito L. Fibroblast growth factor-1 induces heme oxygenase-1 via nuclear factor erythroid 2-related factor 2 (Nrf2) in spinal cord astrocytes: Consequences for motor neuron survival. J Biol Chem. 2005;280:25571–25579. doi: 10.1074/jbc.M501920200. [DOI] [PubMed] [Google Scholar]
- 120.Viglino P. Scarpa M. Coin F. Rotilio G. Rigo A. Oxidation of reduced Cu,Zn superoxide dismutase by molecular oxygen. Biochem J. 1986;237:305–308. doi: 10.1042/bj2370305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang J. Caruano–Yzermans A. Rodriguez A. Scheurmann JP. Slunt HH. Cao X. Gitlin J. Hart PJ. Borchelt DR. Disease-associated mutations at copper ligand histidine residues of superoxide dismutase 1 diminish the binding of copper and compromise dimer stability. J Biol Chem. 2007;282:345–352. doi: 10.1074/jbc.M604503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang J. Slunt H. Gonzales V. Fromholt D. Coonfield M. Copeland NG. Jenkins NA. Borchelt DR. Copper-binding-site-null SOD1 causes ALS in transgenic mice: Aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet. 2003;12:2753–2764. doi: 10.1093/hmg/ddg312. [DOI] [PubMed] [Google Scholar]
- 123.Wang J. Xu G. Borchelt DR. High molecular weight complexes of mutant superoxide dismutase 1: Age-dependent and tissue-specific accumulation. Neurobiol Dis. 2002;9:139–148. doi: 10.1006/nbdi.2001.0471. [DOI] [PubMed] [Google Scholar]
- 124.Wang J. Xu G. Gonzales V. Coonfield M. Fromholt D. Copeland NG. Jenkins NA. Borchelt DR. Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol Dis. 2002;10:128–138. doi: 10.1006/nbdi.2002.0498. [DOI] [PubMed] [Google Scholar]
- 125.Watanabe M. Dykes–Hoberg M. Culotta VC. Price DL. Wong PC. Rothstein JD. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis. 2001;8:933–941. doi: 10.1006/nbdi.2001.0443. [DOI] [PubMed] [Google Scholar]
- 126.Wiedau–Pazos M. Goto JJ. Rabizadeh S. Gralla EB. Roe JA. Lee MK. Valentine JS. Bredesen DE. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science. 1996;271:515–518. doi: 10.1126/science.271.5248.515. [DOI] [PubMed] [Google Scholar]
- 127.Williamson TL. Bruijn LI. Zhu Q. Anderson KL. Anderson SD. Julien JP. Cleveland DW. Absence of neurofilaments reduces the selective vulnerability of motor neurons and slows disease caused by a familial amyotrophic lateral sclerosis-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci USA. 1998;95:9631–9636. doi: 10.1073/pnas.95.16.9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Williamson TL. Corson LB. Huang L. Burlingame A. Liu J. Bruijn LI. Cleveland DW. Toxicity of ALS-linked SOD1 mutants. Science. 2000;288:399. doi: 10.1126/science.288.5465.399a. [DOI] [PubMed] [Google Scholar]
- 129.Wong PC. Pardo CA. Borchelt DR. Lee MK. Copeland NG. Jenkins NA. Sisodia SS. Cleveland DW. Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 130.Wong PC. Waggoner D. Subramaniam JR. Tessarollo L. Bartnikas TB. Culotta VC. Price DL. Rothstein J. Gitlin JD. Copper chaperone for superoxide dismutase is essential to activate mammalian Cu/Zn superoxide dismutase. Proc Natl Acad Sci USA. 2000;10:1073. doi: 10.1073/pnas.040461197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ye Y. Quijano C. Robinson KM. Ricart KC. Strayer AL. Sahawneh MA. Shacka JJ. Kirk M. Barnes S. Accavitti–Loper MA. Radi R. Beckman JS. Estevez AG. Prevention of peroxynitrite-induced apoptosis of motor neurons and PC12 cells by tyrosine-containing peptides. J Biol Chem. 2007;282:6324–6337. doi: 10.1074/jbc.M610800200. [DOI] [PubMed] [Google Scholar]
- 132.Zhang H. Andrekopoulos C. Joseph J. Crow J. Kalyanaraman B. The carbonate radical anion-induced covalent aggregation of human copper, zinc superoxide dismutase, and alpha-synuclein: Intermediacy of tryptophan- and tyrosine-derived oxidation products. Free Radic Biol Med. 2004;36:1355–1365. doi: 10.1016/j.freeradbiomed.2004.02.038. [DOI] [PubMed] [Google Scholar]
- 133.Zhang H. Joseph J. Gurney M. Becker D. Kalyanaraman B. Bicarbonate enhances peroxidase activity of Cu,Zn-superoxide dismutase. Role of carbonate anion radical and scavenging of carbonate anion radical by metalloporphyrin antioxidant enzyme mimetics. J Biol Chem. 2002;277:1013–1020. doi: 10.1074/jbc.M108585200. [DOI] [PubMed] [Google Scholar]