

Abstract
The endothelium lining the inner surface of blood vessels of the cardiovascular system is constantly exposed to hemodynamic shear stress. The interaction between endothelial cells and hemodynamic shear stress has critical implications for atherosclerosis. Regions of arterial narrowing, curvatures, and bifurcations are especially susceptible to atherosclerotic lesion formation. In such areas, endothelial cells experience low, or oscillatory, shear stress. Corresponding changes in endothelial cell structure and function make them susceptible to the initiation and development of atherosclerosis. In contrast, blood flow with high laminar shear stress activates signal transductions as well as gene and protein expressions that play important roles in vascular homeostasis. In response to laminar shear stress, the release of vasoactive substances such as nitric oxide and prostacyclin decreases permeability to plasma lipoproteins as well as the adhesion of leukocytes, and inhibits smooth muscle cell proliferation and migration. In summary, different flow patterns directly determine endothelial cell morphology, metabolism, and inflammatory phenotype through signal transduction and gene and protein expression. Thus, high laminar shear stress plays a key role in the prevention of atherosclerosis through its regulation of vascular tone and long-term maintenance of the integrity and function of endothelial cells. Antioxid. Redox Signal. 11, 1669–1682.
Introduction
The endothelium consists of a monolayer of endothelial cells in the lumen of blood vessels. While it functions as a mechanical barrier, more importantly it maintains anti-inflammatory, anticoagulant properties, enables physiological control of vasoregulation, and modulates vascular permeability. As a result of their unique location, endothelial cells are exposed to a highly dynamic environment in which physiology, pathology, blood dynamics, and biomechanics converge with cellular and molecular biology. Within such an environment, shear stress is a key regulator of endothelial cells. Shear stress is the force per unit area created when a tangential force of blood flow acts on the endothelium. Shear-induced mechanotransduction transforms mechanical forces to biochemical responses, activating signal transduction and endothelium-dependent gene and protein expression that determine endothelial cell phenotype. Thus, shear stress is indispensable in the long-term maintenance of blood vessel tone and structure.
In the arterial circulation, localized hemodynamic shear stress determines site-specific susceptibility to atherosclerosis and its progression (17). In arteries, certain regions such as branches, bifurcations, arterial narrowing, and curvatures are more likely to develop atherosclerotic lesions, and endothelial cells are exposed to low and oscillatory shear stress (Fig. 1) (19, 20, 66, 123).
FIG. 1.

Flow pattern changes depending on the geometry of blood vessel. In “straight” regions of vasculature, endothelial cells experience ordered laminar shear stress, while at or near branch points and vascular bifurcations, endothelial cells experience low or oscillatory shear stress.
In contrast, in the straight part of the arterial tree where blood flow is unidirectional with high velocity of laminar shear stress (streamlined blood flow where viscous forces are predominant over inertial forces), there is less lesion formation (24, 25, 76, 123).
Mechanisms that link atherosclerotic lesion formation and regional flow patterna remain complicated and difficult to dissect. Recently, the effect of shear stress on plaque vulnerability has been studied with a perivascular shear stress modifier that generates regions of lowered, increased, or lowered/oscillatory shear stress in mouse carotid arteries (19). It has been found that atherosclerosis develops invariably in regions with low or oscillatory shear stress, while it is protected in regions with high shear stress. Regions of low shear stress form larger lesions containing few smooth muscle cells, less collagen, but more lipids and show more outward remodeling. These studies have demonstrated that lower shear stress and oscillatory shear stress are both essential for plaque formation. However, lower shear stress tends to induce larger lesions with vulnerable plaque, whereas oscillatory shear stress tends to induce stable lesions.
The interplay between shear stress and endothelial cells is critical for vascular homeostasis. Endothelial cells sense hemodynamic and hormonal stimuli, integrate them with signal transduction as well as gene and protein expression, and release vasoactive factors such as nitric oxide (NO), prostaglandins, hyperpolarizing factors, growth factors, and other related molecules to modulate the permeability to plasma lipoproteins, adhesion of leukocytes, and apoptosis (26, 27, 128). Abnormalities in endothelial cells' responsiveness to flow shear stress, or their failure to respond to particular types of blood flow patterns, contribute to vascular pathology, especially in the early stages of atherosclerosis (46, 70, 74, 132). This review provides a brief summary of the influence of hemodynamic shear stress on endothelial phenotype, in terms of their signal transduction, gene and protein expression, as well as structure and function.
Shear Stress-Induced Signal Transduction, Gene and Protein Expression, and Endothelial Cell Phenotype
Upon exposure to laminar shear stress, initiation of intracellular signal transduction, specifically the activation of protein kinases, is the second important molecular event in endothelial cells that modulates the activity of downstream target proteins and transcription factors, and hence the gene expression programs.
Laminar shear stress activates the signal transduction that leads to the gene and protein expression that acts against the development of atherosclerosis (15, 30, 33, 37, 55, 104, 129). In contrast, oscillatory shear stress triggers gene and protein expression in favor of the development of atherosclerosis (81, 101, 105, 107, 146).
As a result, laminar shear stress prevents endothelial cell activation through modulation of plasma lipoprotein permeability, protein expression in the leukocyte adhesion cascade, smooth muscle cell proliferation and migration, and the release of pro-thrombotic and anti-thrombotic factors, growth factors, and vasoactive substances (Fig. 2). Impairment of these shear stress-mediated processes through either physical damage or endothelial cell dysfunction under pathological conditions plays a central role in the pathogenesis of atherosclerosis.
FIG. 2.

Molecular mechanisms of atherosclerosis-protective effects of laminar shear stress. Laminar shear stress maintains the anti-inflammatory and anti-apoptosis phenotype of endothelial cells through its regulation of protein expression, release of vasoactive factors, permeability of lipoproteins, adhesion and migration of leukocytes, and coagulation.
Signal transduction, gene and protein expression, and atherosclerosis protective phenotype of endothelial cells in response to laminar shear stress
Laminar shear stress-induced NO production plays a key role in endothelial function and vascular homeostasis. In contrast, impaired endothelium-dependent vessel dilatation throughout the artery bed is the major characteristic of endothelial dysfunction that is the precursor to cardiovascular diseases including atherosclerosis.
The dominant mechanism responsible for endothelial dysfunction is the decrease in bioavailable NO, as well as the increase in reactive oxygen species (ROS) production and apoptosis. In addition to its central role in the regulation of vascular tone, NO inhibits several key steps in the initiation and development of atherosclerosis (15, 30, 37, 55, 69, 129).
The enzyme that catalyzes NO production in endothelial cells is endothelial nitric oxide synthase (eNOS). eNOS regulates diverse vascular functions, including the control of blood pressure, local vascular tone, and remodeling in response to shear stress (64, 65, 110, 117).
Although it is well known that exposure of endothelial cells to laminar shear stress stimulates the production of NO from eNOS in both cultured cells and intact vessels, the molecular mechanism by which shear stress regulates NO production is not yet clear (28, 29, 109). It appears that laminar shear stress regulates both the activity and protein expression of eNOS. eNOS activity is coordinately regulated by post-translational modifications, including phosphorylation, myristolation, palmitoylation, and subcellular localization (10). The best-characterized residues for eNOS phosphorylation are active site Ser 1177, Ser 635, and inhibitory sites Ser 116 and Thr 497 (9, 40, 94). Studies have shown that laminar shear stress stimulates serine/threonine kinase Akt (also known as protein kinase B or Rac kinase) phosphorylation on serine 473 in a vascular endothelial growth factor (VEGF) receptor-dependent manner. Activation of Akt in turn phosphorylates eNOS at Ser 1177, leading to eNOS activation and NO production (35, 44, 45, 69). However, there is also evidence that PKA contributes to shear stress-mediated eNOS phosphorylation and NO production independent of Akt (8, 11). Recent studies suggest that eNOS phosphorylation is dependent largely on its subcellular localization, and maybe the availability of the specific kinase in each location as well (10). Regardless of the upstream kinases, eNOS phosphorylation clearly plays an indispensable role on endothelial function in response to laminar shear stress.
Recently, eNOS transcription and expression has also been found to be decreased in atherosclerosis-prone regions of the mouse aorta with an in vitro disturbed flow model, suggesting that a sustained level of eNOS expression is necessary for the flow-mediated prevention of atherosclerosis, most likely because of its consistent supply of bioavailable NO (138).
Bioavailable NO production by laminar shear stress inhibits several key early events in the development of atherosclerosis. NO inhibits the expression of monocyte chemoattractant peptide-1 (MCP-1) and monocyte adhesion induced by cytokines and oxidized LDL, reduces vascular cell adhesion molecule-1 (VCAM-1) expression, prevents propagation of lipid oxidation, inhibits vascular smooth muscle cell proliferation, decreases platelet aggregation, and prevents cell death (15, 16, 37, 55, 129).
The role of NO in the prevention of monocytes adhesion in response to laminar shear stress is well studied. In the early stages of atherosclerosis, macrophages derived from intimal monocytes attach, spread, and migrate across the endothelium. Many studies support an inverse relationship between leukocyte adhesion and laminar shear stress. It seems probable that for conduit vessels, high shear stress inhibits leukocyte binding and chemoattractant protein expression, while low shear stress and flow reversal promote leukocyte binding and transmigration. MCP-1 signaling via its C–C motif chemokine receptor-2 has been shown to be important for monocyte adhesion to inflamed endothelium exposed to flow. There is a marked reduction in monocyte adhesion (∼45%) when human pulmonary artery endothelial cell-derived MCP-1 is either neutralized with the anti-MCP-1 antibody, or inhibited by translational arrest of MCP-1 mRNA transcripts with MCP-1 antisense oligomers (90).
MCP-1 gene expression in response to shear stress is the most immediate early gene activation. Studies have suggested that this gene is probably suppressed in endothelial cells exposed to a constant high shear stress (118). On exposure to laminar shear stress, MCP-1 expression is transiently increased 2- 3-fold in human umbilical vein endothelial cells, and then decreased to basal levels after an hour. Once the gene expression is fully suppressed, it remains so even after static incubation. Additionally, the temporal gradient in shear stress increases MCP-1 gene expression (5).
Further studies have suggested that NO inhibits MCP-1 through multiple mechanisms in response to laminar shear stress (15, 98). It appears that NO inhibits MCP-1 in a protein kinase C (PKC)-epsilon and extracellular signal-regulated kinase 1/2 (ERK1/2) dependent manner. Inhibition of NO production increases MCP-1 while eNOS overexpression decreases MCP-1 (98). Other studies have also demonstrated that the anti-inflammatory effect of NO is through exocytosis of Weibel–Palade bodies, which contain von Willebrand's factor and P-selectin, as well as platelet aggregation (77, 78, 89, 93). In addition, NO also prevents MCP-1 and cytokine-induced adhesion molecule expression through nuclear factor-κB (30, 129). All these findings suggest an anti-inflammatory effect of NO in response to laminar shear stress.
Lipoprotein transport and low-density lipoprotein (LDL) metabolism appears inversely related to the availability of bioavailable NO, although the specific molecular mechanism is less well-defined (16). This hypothesis is supported by observations that LDL accumulation within the vascular wall is preferentially localized to areas with oscillatory shear stress. In these areas eNOS expression is decreased (138). In contrast, LDL incorporation and permeability have been shown decreased in straight segments, in comparison to arterial branch points that experience high shear (7, 48, 49). These findings have also been confirmed by other investigators for different areas of the vasculature in both rabbits and pigs. Additional studies are required to define the mechanisms by which LDL accumulation is affected by NO in low and oscillatory shear stress.
NO production in response to laminar shear stress is at least partly responsible for flow-induced secretion of prostacyclin (135). Shear stress is the most powerful stimulus for the release of factors that inactivate the clotting cascade. Laminar shear stress-induced secretion of prostacyclin is the first documented response of endothelial cells to shear stress (58). Prostacyclin, also known as prostaglandin I2, is the most potent natural inhibitor of platelet aggregation. Secretion of prostacyclin from endothelial cells is enhanced in a cyclooxygenase-2 (COX-2) dependent manner. COX-2 is the enzyme that catalyzes the formation of prostacyclin. In parallel cultures, after exposure to pulsatile laminar shear stress, the prostacyclin level is significantly enhanced, concomitant with an increase in the transactivation of a COX-2 promoter (58, 127, 134). Further studies have demonstrated a positive role of NO because eNOS inhibitors N(G)-nitro-L-arginine methyl ester (100 μM, L-NAME) and N(G)-nitro-L-arginine (10 μM, LNA) cause ∼50% decreases in prostaglandin (135). This suggests that half of the laminar shear stress-induced production of prostacyclin is due to NO-dependent signaling.
The atherosclerosis-protective role of NO has also been demonstrated by studies on atherosclerotic lesion formation in apolipoprotein E (ApoE)/eNOS double knockout mice. eNOS deficiency increases atherosclerosis in Western-type diet-fed ApoE knockout mice (73, 80).
Laminar shear stress also maintains endothelium homeostasis through the activation of kinase signaling that promotes the viability of endothelial cells (23, 36, 71, 108). One critical molecule involved in cell survival is Akt. Activation of Akt by laminar shear stress not only contributes to the anti-apoptotic effects mediated by NO but also plays a key role in the suppression of apoptotic cell death due to either growth factor deprivation or inhibition of matrix adhesion and integrin-mediated signal transduction. Laminar shear stress activates Akt through phosphorylation of Akt in a phosphoinositide 3-kinase (PI3K)-dependent manner, which can be blocked by the PI3K inhibitors wortamannin and LY294002 (36). Upon activation, Akt phosphorylates pro-apoptotic molecule Bad on Ser 136 and Ser 112, which is required for Bad's association with 14-3-3 to sequester Bad in the cytoplasm, thereby preventing its translocation to the mitochondria to induce apoptosis. In contrast, overexpression of a dominant-negative Akt mutant significantly attenuates the apoptotic-suppressive effect of shear stress against serum depletion-induced apoptosis. These findings indicate a critical role for Akt in shear stress-mediated inhibition of endothelial cell apoptosis (36).
Recently the critical role of Akt in the prevention of atherosclerosis lesion formation has been well studied (38). On an ApoE knockout (ApoE−/−) background, deletion of Akt1 through genetic engineering increased aortic lesion expansion compared to that of ApoE−/− mice. Increased endothelial and macrophage apoptosis have been found in the vessel walls of double knockout mice (ApoE−/− Akt1−/−) in addition to increased inflammation and reduced eNOS phosphorylation, suggesting an indispensable role of Akt in cell survival and eNOS activation in vivo, particularly during the development of atherosclerosis.
Additionally, big mitogen-activated protein kinase 1 (BMK-1), which is potently stimulated by laminar shear stress, also induces phosphorylation of Bad and inhibits growth factor depletion-induced endothelial cell apoptosis. In contrast, inhibiting BMK-1 activity by overexpressing dominant-negative BMK-1 stimulates apoptosis. The role of BMK-1 in cell survival is further demonstrated by using the Bad mutant S112-136A, which abolishes the anti-apoptotic effect of constitutive active MEK-5, the upstream molecule of BMK-1 (108). These data suggest that kinase-mediated inhibition of Bad activation is likely an important mechanism involved in the anti-apoptotic effect of laminar shear stress (Fig. 3).
FIG. 3.

Laminar shear stress is anti-apoptotic through its activation of multiple signal transduction pathways. It inhibits TNFα-induced ASK1 activation by increasing the binding of 14-3-3 while it activates Akt and BMK-1, which inhibiting apoptotic molecule Bad. Furthermore, nitric oxide inactivation of caspase-3 may represent additional protective mechanism.
The gene expression profile of endothelial cells in response to different flow patterns has recently been studied intensively (81, 101, 105, 107, 146). Among transcription factors that integrate flow signaling and gene expression, the lung Kruppel-like factor (LKLF/KLF2), an endothelial transcription factor, has drawn great attention because of its critical role in mediating the anti-inflammatory effects of laminar shear stress (104). KLF2 expression is specifically induced by laminar shear stress, and it is indispensable for maintaining the atheroprotective, quiescent phenotype of endothelial cells (32).
KLF2 plays an important role at least partly because of its induction of eNOS and thrombomodulin, while reducing the expression of pro-atherogenic molecules MCP-1 and endothelin (33). Shear stress has been shown to regulate the generation of thrombomodulin, a potent activator of the protein C anticoagulant pathway and a surface receptor that binds to thrombin. Subjecting bovine aortic endothelial cells to moderate and elevated shear stress results in a mild and transient increase, followed by a significant decrease in thrombomodulin mRNA. In contrast, shear stress of low magnitude does not affect thrombomodulin mRNA levels, indicating the specific role of laminar shear stress (88). The effect of laminar shear stress in the regulation of thrombomodulin has further been confirmed in human umbilical vein endothelial cells. Laminar shear stress stimulates a sustained increase in thrombomodulin with its expression increasing ∼200% compared to cells maintained under static conditions (125). In addition, shear stress has also been shown to stimulate the expression of tissue plasminogen activators, as well as reduce the secretion of plasminogen activator inhibitor type-1 (34). In contrast, endothelial cells exposed to oscillatory shear stress fail to show increases in the levels of thrombomodulin and tissue plasminogen activator, further supporting the anticoagulation effect of laminar shear stress.
The protective effect of KLF2 has been further demonstrated by the fact that KLF2 overexpression dramatically induces eNOS gene expression and total enzymatic activity. In contrast, KLF2 inhibits the induction of VCAM-1 and E-selectin in response to various pro-inflammatory cytokines and subsequently, attenuates the rolling and attachment of inflammatory cells (115). The anti-inflammatory effect of KLF2 is likely due to its ability to inhibit ATF activity in response to laminar shear stress. This idea is supported by the finding that nuclear binding of ATF is dramatically reduced in a KLF2-dependent manner in human umbilical vein endothelial cells exposed to laminar shear stress (39).
Low and oscillatory shear stress regulate the endothelial secretion of several smooth muscle growth factors including platelet-derived growth factor A (PDGF-A), and endothelin-1, a growth factor acting synergistically with PDGF during smooth muscle cell proliferation. In an arteriovenous fistula model, increased mRNA levels of PDGF-A are associated with smooth muscle cell proliferation in areas that experience low blood flow compared to areas experiencing high flow (75). Similarly, a temporal gradient in shear (impulse flow and the onset of step flow) stimulates the expression of endothelial PDGF-A (5).
Endothelin-1 is a potent and long-lasting vasoconstrictor that is synthesized and released by endothelial cells. Shear stress suppresses mRNA transcript levels and the rate of endothelin-1 peptide release (86, 87, 116). More interestingly, siRNA knockdown of KLF2 specifically suppresses laminar shear stress-mediated downregulation of endothelin-1 expression, demonstrating KLF2's role in regulating the effect of shear stress on endothelin-1 (33). In addition to PDGF and endothelin-1, endothelial cells also release NO and transforming growth factor-beta (TGF-β) in response to laminar shear stress, both of which are inhibitors of vascular smooth muscle cell growth (12, 21, 28, 29).
Inhibition of atherosclerosis-prone signals by laminar shear stress
Inflammation events contribute to each step in the initiation and development of atherosclerosis. Tumor necrosis factor alpha (TNFα) stimulates signal transduction and gene expression that may promote endothelial dysfunction and atherosclerosis. For example, TNFα-induced activation of c-Jun N-terminal kinase (JNK) and p38 has been implicated in increasing endothelial cell production of matrix metalloproteinase, pro-coagulant factors, and components of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. These events can lead to oxidation, vascular remodeling, and thrombosis processes characteristic of the atherosclerotic lesion.
There has been controversy regarding the role of shear stress on JNK activation, which is likely due to the differences in flow apparatus and cell types (50, 83, 124). In human umbilical vein endothelial cells, laminar shear stress inhibits TNFα-induced JNK activation (124). Consistent with this finding, laminar shear stress has been shown to inhibit TNFα-mediated apoptosis. Several mechanisms seem to be involved in laminar shear stress inhibition of TNFα-induced JNK activation (Fig. 4). First, activation of ERK1/2 has been linked to cell growth and survival, while JNK/p38 is associated with apoptosis. Thus, it is likely that the anti-apoptotic effect of shear stress is mediated through selective activation of ERK1/2 and inhibition of JNK induced by TNFα. This idea is further supported by observations that PD98059, a known inhibitor of ERK1/2, abolishes shear stress-induced ERK1/2 activation, as well as TNFα-mediated JNK activation. Second, laminar shear stress inactivates upstream kinase of JNK, such as apoptosis signaling kinase 1 (ASK1). Laminar shear stress has been shown to enhance the interaction between 14-3-3 and ASK1, which inhibits ASK1 activation (85). It would be interesting to find out if Akt activation is required for the inhibition of ASK1 in response to laminar shear stress. Third, laminar shear stress may alter the activity of phosphatases that contribute to JNK inactivation. In fact, laminar shear stress affects the phosphatase activity of SHP-2, which may be important for TNFα-mediated inflammation (82). Most recently, mitogen-activated protein kinase phosphatase 1 (MKP-1), a negative regulator of JNK and p38, has been found to be activated by laminar shear stress (146). It appears that MKP-1 is important in laminar shear stress-mediated inhibition of VCAM-1 expression because gene silencing of MKP-1 abolishes its inhibitory effect. These data collectively suggest that changes in phosphatase activity might be another general mechanism of the atheroprotective effect of laminar shear stress.
FIG. 4.
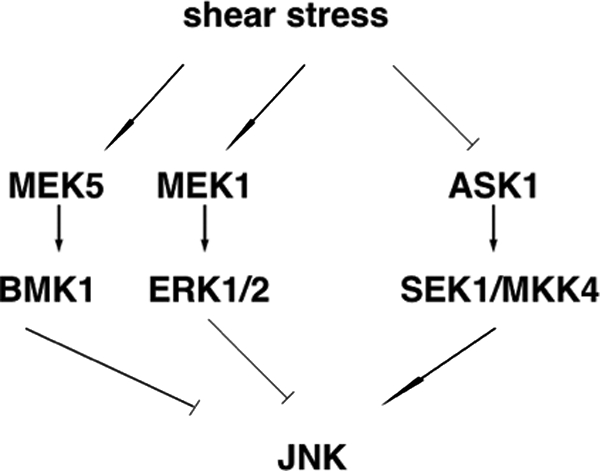
Molecular mechanisms of JNK inhibition by laminar shear stress. Laminar shear stress inhibits ASK1, an upstream kinase of JNK, and concomitantly activates ERK1/2 and BMK-1.
Laminar shear stress inhibits TNFα-mediated inflammatory events by modulating the expression level of pro-inflammatory proteins. Binding sites for ATF are enriched in KLF2, eNOS, and thrombomodulin, whereas TNFα-induced gene expression is mostly NF-κB-dependent (39). NF-κB and its inhibitor IκB are present in elevated levels in the cytoplasm of endothelial cells at sites exposed to disturbed blood flow (53).
Endothelial cells regulate leukocyte adhesion and migration into the blood vessel wall by modulating the secretion of chemotactic factors and the expression of cell surface molecules. Intercellular adhesion molecule-1 (ICAM-1) binds to β2-integrin on various white blood cell derivatives, while VCAM-1 mediates the adhesion of monocytes to the endothelium. VCAM-1 is also one of the earliest markers for fatty streaks and is upregulated in areas of the endothelium surrounding atherosclerotic plaques.
Leukocyte binding after exposure to shear is increased with upregulation in adhesion molecule expression. There is evidence showing that the VCAM-1 mRNA level is markedly decreased in endothelial cells exposed to laminar shear stress (2, 3, 96). In a disturbed shear system that models the in vivo hemodynamic characteristics of lesion-prone vascular regions, there is a distinctive, clustered-cell pattern of monocyte adhesion that strongly resembles in vivo leukocyte adhesion in the early and late stages of atherosclerosis. This clustered monocyte cell adhesion correlates with endothelial cell expression of ICAM-1 and E-selectin (13). In addition to studies that use cultured cells, there are also ex vivo studies on the effect of chronic shear stress on TNFα-induced inflammatory responses. In an ex vivo rabbit aorta organ culture system, TNFα-induced expression of VCAM-1 is downregulated by high shear stress (12 dynes/cm2) while low shear stress (0.4 dynes/cm2) has no such effect (141).
There are several proteins that bind and inhibit TNFα activation of ASK1, which then inhibits inflammatory molecule expression. Thioredoxin (Trx) is one of those proteins (111). Trx is a small thiol oxidoreductase of 12 KD whose enzymatic activity is dependent on two redox-sensitive cysteines (C32 and C35) located in its catalytic center. In its reduced form, Trx binds to the C-terminus of ASK1 and inhibits ASK1 kinase activity. TNFα activates ASK1 partly by dissociating Trx from ASK1. Trx itself is negatively regulated by its binding protein, thioredoxin interacting protein (TXNIP, also known as vitamin D upregulating protein 1, VDUP-1). TXNIP binds to the catalytic center cysteine of Trx to inhibit its activity (106). Studies using ex vivo aortic segments and cultured endothelial cells have shown that laminar shear stress decreases TXNIP protein expression in endothelial cells (∼50% downregulation compared to low shear) (142). The downregulation of TXNIP releases the catalytic cysteine of Trx and increases functional Trx, as evidenced by the increased Trx activity, while Trx protein levels remained unchanged. This is thought to enable increased binding of Trx to ASK1 and subsequently decrease phosphorylation of JNK and p38. It also blocks VCAM-1 expression induced by TNFα in response to laminar shear stress. Likewise, downregulation of TXNIP using RNA interference has been shown to increase the binding of Trx to ASK1, decrease JNK and p38 activation, and block TNFα -induced VCAM-1 expression. In contrast, overexpression of TXNIP increases the activation of JNK and p38. Increased expression of VCAM-1, together with other selectins, promotes leukocyte adhesion to the endothelium. These leukocytes will ultimately infiltrate below the endothelial cell surface, taking up lipid and becoming foam cells. Therefore, downregulation of TXNIP represents another atheroprotective mechanism mediated by laminar shear stress (Fig. 5). The finding that laminar shear stress inhibits JNK and TXNIP is thought to be significant, as it likely influences other aspects of endothelial function that contribute to atherosclerosis (54).
FIG. 5.
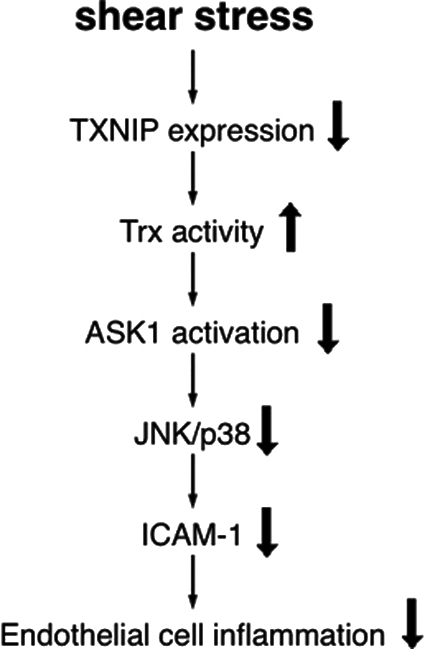
Laminar shear stress is anti-inflammatory through its downregulation of TXNIP and VCAM-1 expression. Downregulation of TXNIP increases Trx activity, which inhibits ASK1 and downstream molecules, such as JNK and p38. Downregulation of TXNIP also decreases TNFα-induced VCAM-1 expression.
Shear Stress-Mediated Redox Regulation in Endothelial Cells
Shear stress-mediated redox regulation is becoming an increasingly interesting field to study because of ROS's role in contributing to atherosclerosis. ROS increase leukocyte adhesion molecule expression, stimulate smooth muscle cell proliferation and cell migration, promote lipid oxidation, upregulate matrix metalloproteinase, and alter vasomotor activity (68).
In healthy vessels exposed to laminar shear stress, the endothelium exerts anticoagulant effects via its secretion of NO, thromodulin, heparin sulfate, tissue factor (TF) inhibitor, and annexin V. In contrast, oscillatory shear stress induces a procoagulant phenotype of endothelial cells that may favor the formation and progression of atherothrombotic lesions. Studies have shown significantly increased release of TF mRNA and protein expression in human endothelial cells exposed to oscillatory shear stress for 24 h, while the expression of TF inhibitor (TFPI) mRNA and protein remain unchanged relative to static conditions (91). Because TF initiates blood coagulation and contributes to vascular remodeling, it is a potential contributor to the development and progression of atherosclerosis. Conversely, cells exposed to unidirectional laminar shear show a decrease in TF activity along with a significant increase in TFPI mRNA and protein expression. It is interesting that increased expression of TF is mediated in part by ROS production in smooth muscle cells. Likewise, endothelial cells exposed to oscillatory shear stress fail to show increased expression of thrombomodulin and tissue plasminogen activator. Furthermore, when endothelial cells are exposed to oscillatory or low shear stress, the simultaneous production of superoxide O2– and nitric oxide anion NO• can facilitate peroxynitrite (ONOO–) formation and tyrosine nitration (63). Tyrosine nitration has been found in human atherosclerotic lesions and may predispose vascular areas exposed to oscillatory flow to atherosclerosis.
Monocyte adhesion is another key event in the early stages of atherosclerosis. Binding of circulating monocytes to the endothelium is dependent on the expression of adhesion molecules on the cell surface, which is regulated in a ROS-dependent manner through unknown mechanisms. The antioxidant ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one) inhibits TNFα-induced VCAM-1 and ICAM-1 expression in this regard (145). Chronic exposure of endothelial cells to oscillatory shear stress stimulates ROS generation through p47phox-dependent NAD(P)H oxidase, which leads to monocyte adhesion (119). Although there is also a transient increase in ROS generation upon exposure to laminar shear stress due to the activation of membrane-bound NADPH oxidase activation, sustained laminar shear stress maintains endothelial cells in a reduced environment, which inhibits leukocyte binding and inflammation by decreasing TNFα-induced expression of VCAM-1 and E-selectin. In contrast, extensive monocyte adhesion that co-localizes to areas of VCAM-1 expression is observed under low and oscillatory shear stress (3, 22, 100, 133).
ROS promote atherogenesis through several enzymes, including xanthine oxidase (OX), NADPH oxidase, and uncoupled eNOS (31, 67, 92). It has been proposed that NADPH oxidase maintains endothelial XO levels by regulating XO protein expression. Moreover, XO is responsible for the generation of ROS in response to oscillatory flow (92). Recent studies have also demonstrated that the expression of the vascular NADPH oxidase subunit NOX4 increases in response to oscillatory flow, leading to an increase in LDL oxidation relative to laminar shear stress (67).
Laminar shear stress modulates the expression of antioxidant enzymes such as superoxide dismutase (SOD) and glutathione peroxidase (GPX-1), as well as the redox status of proteins. Glutathione (GSH, γ-glutamyl-cysteinyl-glycine) is the major low-molecular-weight thiol antioxidant in endothelial cells. It serves as a substrate for GPX to eliminate lipid hydroperoxides and H2O2, whereby it is converted to glutathione disulfide (GSSG). Laminar shear stress induces upregulation of GSH/GSSG, which keeps endothelial cells in a reduced condition. Normally, endothelial cells are in a reduced state because GSH exists in very high concentrations (mM) while GSSG is maintained at levels <1% of total GSH. Steady laminar shear stress inhibits H2O2-induced JNK activation, partly by increasing GSH/GSSG in a glutathione reductase (GR)-dependent manner. This is supported by the fact that the GR inhibitor, but not the thioredoxin reductase inhibitor, blocks the inhibitory effect of laminar shear stress on H2O2-mediated JNK activation (61). It appears that laminar shear stress mediates changes in the glutathione redox cycle by reducing GSSG through GR. L-buthionine-(S,R)-sulfoximine (BSO), which inhibits GSH synthesis, can partly reverse the protective effects of shear stress (57). In contrast, GSH levels decrease when cells are exposed to oscillatory shear stress (95). Although it is not clear how shear stress regulates the glutathione system, the impact is profound, because it is at least partly responsible for the inhibition of both H2O2 and TNFα-induced cell death.
As previously mentioned, laminar shear stress increases Trx activity by downregulating TXNIP. Trx serves both as a redox sensor and a redox-regulating molecule in keeping the thiol homeostasis of the cell; specifically, it acts as a general protein disulfide oxidoreductase and interacts with a broad range of proteins. This reaction is based on reversible oxidation of two cysteine thiol groups of Trx to a disulfide, accompanied by the transfer of two electrons and two protons (140).
In addition to Trx, GPX-1 is also regulated by laminar shear stress. GPX-1 belongs to a family of selenoproteins and plays an important role in the defense mechanisms against oxidative damage by catalyzing the reduction of a variety of hydroperoxides, with glutathione as the reducing substrate. Both GPX-1 mRNA and its activity are upregulated by laminar shear stress in a time- and force-dependent manner. Interestingly, this response is shear stress specific, because tensile stress changes neither the expression nor the activity of GPX-1 (126).
Laminar shear stress not only detoxifies hydroperoxides through increased expression and activity of GPX-1 but also provides protection to superoxide. SOD is the enzyme that catalyzes the decomposition of a superoxide into hydrogen peroxide and oxygen. Of the three forms of atheroprotective SOD found in the vascular wall (MnSOD, extracellular SOD, and Cu/Zn SOD), the expression of all of them are upregulated when endothelial cells are exposed to laminar shear stress (1, 122, 139).
Therefore, laminar shear stress keeps endothelial cells in a reduced condition by activating antioxidant enzymes (Fig. 6). GR provides reducing power by converting GSSG to GSH without changing the total glutathione level. Increased expression of SOD can detoxify superoxide by converting superoxide to H2O2. The H2O2 is further detoxified by GPX-1, which is also activated by laminar shear stress.
FIG. 6.

Laminar shear stress maintains the reduced environment of endothelium by increasing antioxidant enzyme activity of the glutaredoxin (Grx) system. Laminar shear stress increases the ratio of GSH/GSSG by activating GR. It also increases the activity of GPX-1 and Grx that are important in maintaining the redox homeostasis.
NF-E2-related factor 2 (Nrf-2) plays an essential role in the protection of endothelial cells through antioxidant response element (ARE)-mediated gene expression of phase II detoxification antioxidant proteins. Although both oscillatory and laminar shear stress increase Nrf-2 nuclear translocation, only laminar shear stress stabilizes Nrf-2 and induces Nrf-2-mediated gene expression (62, 136, 137). In human umbilical vein endothelial cells, Nrf-2 regulated genes such as heme oxygenase 1, NAD(P)H quinone oxidoreductase1, glutamate-cysteine ligase modifier subunit, and ferritin heavy chain are induced by laminar shear stress but inhibited by Nrf-2 siRNA (136, 137). Further studies have shown that laminar shear stress stabilizes Nrf-2 protein level through lipid peroxidation (136). However, the mechanism by which Nrf-2 is activated in response to laminar shear stress requires further investigation.
S-nitrosylation is a redox-dependent post-translational modification that is highly regulated in endothelial cells. In the presence of endogenous NO, free -SH groups in proteins can be modified to -SNO (120, 121). When exposed to laminar shear stress, the level of S-nitrosylated proteins increases significantly, in part because shear stress is an important stimulus for NO production in endothelial cells (28, 29, 59). Because S-nitrosylation is a redox-sensitive modification that regulates protein function, it may partly explain certain changes in protein activity in response to laminar shear stress. To date, proteins that are known to be regulated by S-nitrosylation include the catalytic p17 subunit of caspase-3, Trx, and the GTPase p21ras (59). Increased protein S-nitrosylation results in the inhibition of caspase-3 activity, as well as the enhancement of Trx and GTPase p21 activity. Increased S-nitrosylation of various proteins may become a novel signaling mechanism regulated by laminar shear stress.
Mechanosensing of Shear Stress
Endothelial cells are very sensitive to shear stress. Upon exposure to laminar shear stress, endothelial cells deform, transmit the stress, transform mechanical forces to biochemical activities, and activate signal transduction. They adjust themselves not only biologically but also morphologically. Prolonged exposure to laminar shear stress leads to the reorientation of endothelial cells so that their longitudinal axes are parallel to the direction of blood flow (Fig. 7). During these processes, the involvement of cytoskeleton is required for stress transmission in order to link the luminal surface to junctions and focal adhesions. In addition, increased membrane fluidity may have some implications in the modulation of conformation and interaction among membrane proteins (14). In contrast, endothelial cells do not experience reorientation in response to low or oscillatory shear stress (101, 105, 107).
FIG. 7.

Endothelial cells sense the mechanic forces and adjust themselves morphologically. Cultured human umbilical vein endothelial cells were exposed to laminar shear stress (12 dynes/cm2) for 24 h in a parallel flow system. Cells undergo reorientation, with their longitudinal axis parallel to the direction of flow.
The mechanisms by which the physical forces generated by blood flow are sensed and transduced into biological signals by endothelial cells are still unclear. However, several proteins are known to be important mediators in mechanotransduction. Some of these proteins are located at the surface of endothelial cells, where they can directly sense and transmit mechanical forces across the membrane to the interior of the cells. Meanwhile, nuclear membrane is also subjected to the transmission of stress mediated by the cytoskeleton.
Membrane proteins that are likely to sense the mechanical forces include integrins, platelet endothelial cell adhesion molecule-1 (PECAM-1), tyrosine kinase receptors, particularly VEGF receptor 2 (VEGFR2), G-proteins, and ion channels. Other possible flow sensors are local membrane structure such as caveolae, gap junctions, membrane lipids, and glycocalyx.
In many cases, these mechanosignaling proteins are associated with well-defined signal transduction pathways that regulate various aspects of cell function. For example, integrins integrate mechanical forces with cytoskeleton proteins, which transmit and modulate the tension between focal adhesion sites, membrane receptors, and the extracellular matrix (56, 84, 130). Therefore, when endothelial cells are aligned with laminar shear stress, there is decreased deformability and increased resistance to detachment.
In the case of PECAM-1, it undergoes rapid changes in response to flow shear stress to serve as mechanoreceptors (43, 72). PECAM-1's role in mechanosensing has been demonstrated by its tyrosine phosphorylation by directly applying mechanical force to PECAM-1 (103). In a similar way, shear stress induces a rapid tyrosine phosphorylation (<30 s) of PECAM-1 accompanied by the binding of its cytoplasmic tail to the phosphatase SHP-2. Knockdown of either PECAM-1 or SHP-2 abrogates the activation of ERK1/2 by shear stress. Additionally, VEGFR2 may mediate mechanotransduction. It is activated very quickly (1–2 min) after the onset of laminar flow, becoming tyrosine phosphorylated. It then forms membrane clusters and binds to Shc and other adaptor proteins, which trigger the mechanotransduction (18, 69).
An important issue raised by these studies is the exact nature of the coordination and differential roles of these molecules in sensing the mechanical forces. The pathway upstream of integrin activation has been studied. It appears that PECAM-1, vascular endothelial cell cadherin, and VEGFR2 comprise a mechanosensory complex. More interestingly, the presence of these proteins is sufficient to confer responsiveness to flow in heterologous cells. In support of these observations in vivo, there has been no activation of NF-kappaB (NF-κB) or downstream inflammatory genes in regions of oscillatory shear stress in PECAM-1-knockout mice. Therefore, this mechanosensing pathway is believed to be necessary for the earliest-known events in atherogenesis (131).
The second relevant question is what determines the endothelial cells' response to the magnitudes and types of flow, and how endothelial cells distinguish laminar from oscillatory shear stress. The G proteins are one of the earliest known membrane-bound proteins that respond to shear stress (4, 52, 79, 99). When endothelial cells are exposed to shear stress, G proteins are activated within one second (52). NO production upon the onset of shear stress is both calcium-and G protein-dependent (79). More importantly, G proteins may also play a role in distinguishing flow profiles (51). Gαq and Gi respond specifically to temporal gradients of shear stress in artificial phospholipid bilayers in the absence of cytoskeleton elements (51). Studies have shown that Gαq links to PECAM-1 at cell–cell junctions while the temporal gradient has been found to be a potent mediator of their dissociation (144). These studies implicate G proteins as a primary sensor of shear stress.
Many studies have also suggested that ion channels play important roles in this process. Ion channel activation is the most rapid response of endothelial cells to shear stress, occurring almost immediately upon the onset of flow. The potassium channel in endothelial cells is the first ion channel identified using whole-cell patch-clamp recordings of single arterial endothelial cells exposed to a controlled level of laminar shear stress in capillary flow tubes (102). Recently, this channel has been cloned and expressed in Xenopus oocytes. Its sensitivity to flow appears to be preserved (42, 60). Blocking mechanosensitive K+ channels with barium chloride or tetraethylammonium inhibits the shear-mediated increase in NO production and TGF-β release, suggesting that transmembrane ion flux and intracellular ion homeostasis are important mediators of the endothelial cell response to shear stress (42). A second type of known flow-sensitive ion channel is a chloride (Cl−) channel, whose activation leads to cell membrane depolarization (6, 97). More interestingly, recent studies have shown that blocking the flow-activated Cl− current abolishes flow-induced Akt phosphorylation in bovine aorta endothelial cells, whereas blocking flow-sensitive K+ currents has no effect. This suggests that flow-activated Cl− channels play an important role in regulating flow-mediated signaling in endothelial cells (47). In addition to these two channels, an ion channel that is more permeable to calcium than to sodium, can also be activated by laminar shear stress (113, 114).
Recently, some specialized structures have been identified to extend considerably further into the lumen of blood flow. For example, glycocalyx is a highly charged, glycoprotein-rich extension of the cell surface. The interaction between the glycocalyx and the membrane lipids may also play a role in mechanosensing (112). Interestingly, its thickness and distribution is particularly important because selective cleavage of glycocalyx components, glycosaminoglycan heparan sulfate, has been found to abolish both flow-mediated NO production and monolayer realignment (41, 143). Further studies are required to obtain details regarding their mechanical properties and distribution in arteries.
Concluding Remarks
Laminar shear stress is of utmost importance in maintaining vascular homeostasis. In blood vessel regions with low or oscillatory shear stress, events such as increased leukocyte adhesion, lipoprotein uptake, smooth muscle cell migration, and increased ROS generation contribute to atherosclerosis. However, these activities are inhibited by laminar shear stress. Laminar shear stress also activates signal transduction pathways and gene expression to suppress inflammation and atherosclerosis. Finally, it is responsible for maintaining the redox homeostasis of the blood vessel wall. Taken together, all of the above show that laminar shear stress is atherosclerosis-protective, while low and oscillatory shear stresses are closely associated with the pathogenesis of atherosclerosis.
Acknowledgments
This work was supported by NIH Grant HL077789 to Bradford C. Berk and AHA grant 0530195N to Shi Pan.
Abbreviations
ApoE, apolipoprotein E; ASK1, apoptosis signaling kinase 1; BMK-1, big mitogen-activated protein kinase 1; BSO, L-buthionine-(S,R)-sulfoximine; ebselen, 2-phenyl-1,2-benzisoselenazol-3(2H)-one); ERK, extracellular signal-regulated kinase; eNOS, endothelial nitric oxide synthesis; GPX, glutathione peroxidase; GR, glutathione reductase; GSH, glutathione; GSSG, oxidized glutathione; Grx, glutaredoxin; ICAM-1, intercellular adhesion molecule-1; JNK, c-Jun N-terminal kinase; KLF2, lung Kruppel-like factor 2; MCP-1, monocyte chemotactic protein 1; NADPH, nicotinamide adenine dinucleotide phosphate; NO, nitric oxide; OX, xanthine oxidase; Nrf-2, NF-E2-related factor 2; PDGF, platelet derived growth factor; PECAM-1, PI3K, phosphoinositide 3-kinase; PKC, protein kinase C; platelet endothelial cell adhesion molecule-1; SOD, superoxide dismutase; TF, tissue factor; TGF-β transforming growth factor-beta; TNF-α, tumor necrosis factor; Trx, thioredoxin; TXNIP, thioredoxin interacting protein; VCAM-1, vascular cell adhesion molecule-1; VDUP-1, vitamin D upregulating protein 1.
References
- 1.Ai L. Rouhanizadeh M. Wu JC. Takabe W. Yu H. Alavi M. Li R. Chu Y. Miller J. Heistad DD. Hsiai TK. Shear stress influences spatial variations in vascular Mn-SOD expression: Implication for LDL nitration. Am J Physiol Cell Physiol. 2008;294:C1576–1585. doi: 10.1152/ajpcell.00518.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ando J. Tsuboi H. Korenaga R. Takada Y. Toyama S–N. Miyasaka M. Kamiya A. Down-regulation of vascular adhesion molecule-1 by fluid shear stress in cultured mouse endothelial cells. Ann NY Acad Sci. 1995;748:148–156. doi: 10.1111/j.1749-6632.1994.tb17314.x. [DOI] [PubMed] [Google Scholar]
- 3.Ando J. Tsuboi H. Korenaga R. Takada Y. Toyama S-N. Miyasaka M. Kamiya A. Shear stress inhibits adhesion of cultured mouse endothelial cells to lymphocytes by downregulating VCAM-1 expression. Am J Physiol. 1994;267:C679–687. doi: 10.1152/ajpcell.1994.267.3.C679. [DOI] [PubMed] [Google Scholar]
- 4.Bao X. Lu C. Frangos JA. Mechanism of temporal gradients in shear-induced ERK1/2 activation and proliferation in endothelial cells. Am J Physiol Heart Circ Physiol. 2001;281:H22–29. doi: 10.1152/ajpheart.2001.281.1.H22. [DOI] [PubMed] [Google Scholar]
- 5.Bao X. Lu C. Frangos JA. Temporal gradient in shear but not steady shear stress induces PDGF-A and MCP-1 expression in endothelial cells: role of NO, NF kappa B, and egr-1. Arterioscler Thromb Vasc Biol. 1999;19:996–1003. doi: 10.1161/01.atv.19.4.996. [DOI] [PubMed] [Google Scholar]
- 6.Barakat AI. Leaver EV. Pappone PA. Davies PF. A flow-activated chloride-selective membrane current in vascular endothelial cells. Circ Res. 1999;85:820–828. doi: 10.1161/01.res.85.9.820. [DOI] [PubMed] [Google Scholar]
- 7.Berceli SA. Warty VS. Sheppeck RA. Mandarino WA. Tanksale SK. Borovetz HS. Hemodynamics and low density lipoprotein metabolism. Rates of low density lipoprotein incorporation and degradation along medial and lateral walls of the rabbit aorto-iliac bifurcation. Arteriosclerosis. 1990;10:686–694. doi: 10.1161/01.atv.10.5.686. [DOI] [PubMed] [Google Scholar]
- 8.Boo YC. Hwang J. Sykes M. Michell BJ. Kemp BE. Lum H. Jo H. Shear stress stimulates phosphorylation of eNOS at Ser(635) by a protein kinase A-dependent mechanism. Am J Physiol Heart Circ Physiol. 2002;283:H1819–1828. doi: 10.1152/ajpheart.00214.2002. [DOI] [PubMed] [Google Scholar]
- 9.Boo YC. Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: Role of protein kinases. Am J Physiol Cell Physiol. 2003;285:C499–508. doi: 10.1152/ajpcell.00122.2003. [DOI] [PubMed] [Google Scholar]
- 10.Boo YC. Kim HJ. Song H. Fulton D. Sessa W. Jo H. Coordinated regulation of endothelial nitric oxide synthase activity by phosphorylation and subcellular localization. Free Radic Biol Med. 2006;41:144–153. doi: 10.1016/j.freeradbiomed.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 11.Boo YC. Sorescu G. Boyd N. Shiojima I. Walsh K. Du J. Jo H. Shear stress stimulates phosphorylation of endothelial nitric-oxide synthase at Ser1179 by Akt-independent mechanisms. Role of protein kinase A. J Biol Chem. 2002;277:3388–3396. doi: 10.1074/jbc.M108789200. [DOI] [PubMed] [Google Scholar]
- 12.Buga GM. Gold ME. Fukuto JM. Ignarro LJ. Shear stress-induced release of nitric oxide from endothelial cells grown on beads. Hypertension. 1991;17:187–193. doi: 10.1161/01.hyp.17.2.187. [DOI] [PubMed] [Google Scholar]
- 13.Burns MP. DePaola N. Flow-conditioned HUVECs support clustered leukocyte adhesion by coexpressing ICAM-1 and E-selectin. Am J Physiol Heart Circ Physiol. 2005;288:H194–204. doi: 10.1152/ajpheart.01078.2003. [DOI] [PubMed] [Google Scholar]
- 14.Butler PJ. Norwich G. Weinbaum S. Chien S. Shear stress induces a time- and position-dependent increase in endothelial cell membrane fluidity. Am J Physiol Cell Physiol. 2001;280:C962–969. doi: 10.1152/ajpcell.2001.280.4.C962. [DOI] [PubMed] [Google Scholar]
- 15.Cai H. Harrison DG. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 16.Cardona–Sanclemente LE. Born GV. Effect of inhibition of nitric oxide synthesis on the uptake of LDL and fibrinogen by arterial walls and other organs of the rat. Br J Pharmacol. 1995;114:1490–1494. doi: 10.1111/j.1476-5381.1995.tb13375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caro CG. Fitz-Gerald JM. Schroter RC. Arterial wall shear and distribution of early atheroma in man. Nature. 1969;223:1159–1160. doi: 10.1038/2231159a0. [DOI] [PubMed] [Google Scholar]
- 18.Chen KD. Li YS. Kim M. Li S. Yuan S. Chien S. Shyy JY. Mechanotransduction in response to shear stress. Roles of receptor tyrosine kinases, integrins, and Shc. J Biol Chem. 1999;274:18393–18400. doi: 10.1074/jbc.274.26.18393. [DOI] [PubMed] [Google Scholar]
- 19.Cheng C. Tempel D. van Haperen R. van der Baan A. Grosveld F. Daemen MJ. Krams R. de Crom R. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation. 2006;113:2744–2753. doi: 10.1161/CIRCULATIONAHA.105.590018. [DOI] [PubMed] [Google Scholar]
- 20.Cheng C. van Haperen R. de Waard M. van Damme LC. Tempel D. Hanemaaijer L. van Cappellen GW. Bos J. Slager CJ. Duncker DJ. van der Steen AF. de Crom R. Krams R. Shear stress affects the intracellular distribution of eNOS: Direct demonstration by a novel in vivo technique. Blood. 2005;106:3691–3698. doi: 10.1182/blood-2005-06-2326. [DOI] [PubMed] [Google Scholar]
- 21.Cucina A. Sterpetti AV. Borrelli V. Pagliei S. Cavallaro A. D'Angelo LS. Shear stress induces transforming growth factor-beta 1 release by arterial endothelial cells. Surgery. 1998;123:212–217. [PubMed] [Google Scholar]
- 22.Cybulsky MI. Gimbrone MA., Jr Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991;251:788–791. doi: 10.1126/science.1990440. [DOI] [PubMed] [Google Scholar]
- 23.Dai G. Vaughn S. Zhang Y. Wang ET. Garcia-Cardena G. Gimbrone MA., Jr Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ Res. 2007;101:723–733. doi: 10.1161/CIRCRESAHA.107.152942. [DOI] [PubMed] [Google Scholar]
- 24.Davies PF. Endothelial cells, hemodynamic forces, and the localization of atherosclerosis. In: Ryan US, editor. Endothelial Cells. Boca Raton, Florida: CRC; 1988. pp. 123–139. [Google Scholar]
- 25.Davies PF. Spatial hemodynamics, the endothelium, and focal atherogenesis: A cell cycle link? [editorial; comment] Circ Res. 2000;86:114–116. doi: 10.1161/01.res.86.2.114. [DOI] [PubMed] [Google Scholar]
- 26.Davies PF. Remuzzi A. Gordon EJ. Dewey CF., Jr. Gimbrone MA., Jr Turbulent fluid shear stress induces vascular endothelial cell turnover in vitro. Proc Natl Acad Sci USA. 1986;83:2114–2117. doi: 10.1073/pnas.83.7.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davies PF. Robotewskyj A. Griem ML. Endothelial cell adhesion in real time. Measurements in vitro by tandem scanning confocal image analysis. J Clin Invest. 1993;91:2640–2652. doi: 10.1172/JCI116503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis ME. Cai H. Drummond GR. Harrison DG. Shear stress regulates endothelial nitric oxide synthase expression through c-Src by divergent signaling pathways. Circ Res. 2001;89:1073–1080. doi: 10.1161/hh2301.100806. [DOI] [PubMed] [Google Scholar]
- 29.Davis ME. Grumbach IM. Fukai T. Cutchins A. Harrison DG. Shear stress regulates endothelial nitric-oxide synthase promoter activity through nuclear factor kappaB binding. J Biol Chem. 2004;279:163–168. doi: 10.1074/jbc.M307528200. [DOI] [PubMed] [Google Scholar]
- 30.De Caterina R. Libby P. Peng HB. Thannickal VJ. Rajavashisth TB. Gimbrone MA., Jr Shin WS. Liao JK. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96:60–68. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Keulenaer GW. Alexander RW. Ushio–Fukai M. Ishizaka N. Griendling KK. Tumour necrosis factor alpha activates a p22phox-based NADH oxidase in vascular smooth muscle. Biochem J. 1998;329:653–657. doi: 10.1042/bj3290653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dekker RJ. van Soest S. Fontijn RD. Salamanca S. de Groot PG. VanBavel E. Pannekoek H. Horrevoets AJ. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kruppel-like factor (KLF2) Blood. 2002;100:1689–1698. doi: 10.1182/blood-2002-01-0046. [DOI] [PubMed] [Google Scholar]
- 33.Dekker RJ. Van Thienen JR. de Jager SC. Elderkamp YW. Seppen J. de Vries CJ. Biessen EA. van Berkel JC. Pannekoek H. Horrevoets AJ. Endothelial KLF2 links local arterial shear stress levels to the expression of vascular tone-regulating genes. Am J Pathol. 2005;167:609–618. doi: 10.1016/S0002-9440(10)63002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diamond SL. Eskin SG. McIntire LV. Fluid flow stimulates tissue plasminogen activator secretion by cultured human endothelial cells. Science. 1989;243:1483–1485. doi: 10.1126/science.2467379. [DOI] [PubMed] [Google Scholar]
- 35.Dimmeler S. Fleming I. Fisslthaler B. Hermann C. Busse R. Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt- dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 36.Dimmeler S. Haendeler J. Rippmann V. Nehls M. Zeiher AM. Shear stress inhibits apoptosis of human endothelial cells. FEBS Lett. 1996;399:71–74. doi: 10.1016/s0014-5793(96)01289-6. [DOI] [PubMed] [Google Scholar]
- 37.Dimmeler S. Hermann C. Galle J. Zeiher AM. Upregulation of superoxide dismutase and nitric oxide synthase mediates the apoptosis-suppressive effects of shear stress on endothelial cells. Arterioscler Thromb Vasc Biol. 1999;19:656–664. doi: 10.1161/01.atv.19.3.656. [DOI] [PubMed] [Google Scholar]
- 38.Fernandez–Hernando C. Ackah E. Yu J. Suarez Y. Murata T. Iwakiri Y. Prendergast J. Miao RQ. Birnbaum MJ. Sessa WC. Loss of Akt1 leads to severe atherosclerosis and occlusive coronary artery disease. Cell Metab. 2007;6:446–457. doi: 10.1016/j.cmet.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fledderus JO. van Thienen JV. Boon RA. Dekker RJ. Rohlena J. Volger OL. Bijnens AP. Daemen MJ. Kuiper J. van Berkel TJ. Pannekoek H. Horrevoets AJ. Prolonged shear stress and KLF2 suppress constitutive pro-inflammatory transcription through inhibition of ATF2. Blood. 2007;109:4249–4257. doi: 10.1182/blood-2006-07-036020. [DOI] [PubMed] [Google Scholar]
- 40.Fleming I. Fisslthaler B. Dimmeler S. Kemp BE. Busse R. Phosphorylation of Thr(495) regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res. 2001;88:E68–75. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]
- 41.Florian JA. Kosky JR. Ainslie K. Pang Z. Dull RO. Tarbell JM. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ Res. 2003;93:e136–142. doi: 10.1161/01.RES.0000101744.47866.D5. [DOI] [PubMed] [Google Scholar]
- 42.Forsyth SE. Hoger A. Hoger JH. Molecular cloning and expression of a bovine endothelial inward rectifier potassium channel. FEBS Lett. 1997;409:277–282. doi: 10.1016/s0014-5793(97)00514-0. [DOI] [PubMed] [Google Scholar]
- 43.Fujiwara K. Masuda M. Osawa M. Kano Y. Katoh K. Is PECAM-1 a mechanoresponsive molecule? Cell Struct Funct. 2001;26:11–17. doi: 10.1247/csf.26.11. [DOI] [PubMed] [Google Scholar]
- 44.Fulton D. Gratton JP. McCabe TJ. Fontana J. Fujio Y. Walsh K. Franke TF. Papapetropoulos A. Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gallis B. Corthals GL. Goodlett DR. Ueba H. Kim F. Presnell SR. Figeys D. Harrison DG. Berk BC. Aebersold R. Corson MA. Identification of flow-dependent endothelial nitric-oxide synthase phosphorylation sites by mass spectrometry and regulation of phosphorylation and nitric oxide production by the phosphatidylinositol 3-kinase inhibitor LY294002. J Biol Chem. 1999;274:30101–30108. doi: 10.1074/jbc.274.42.30101. [DOI] [PubMed] [Google Scholar]
- 46.Garcia–Cardena G. Comander JI. Blackman BR. Anderson KR. Gimbrone MA. Mechanosensitive endothelial gene expression profiles: Scripts for the role of hemodynamics in atherogenesis? Ann NY Acad Sci. 2001;947:1–6. [PubMed] [Google Scholar]
- 47.Gautam M. Shen Y. Thirkill TL. Douglas GC. Barakat AI. Flow-activated chloride channels in vascular endothelium. Shear stress sensitivity, desensitization dynamics, and physiological implications. J Biol Chem. 2006;281:36492–36500. doi: 10.1074/jbc.M605866200. [DOI] [PubMed] [Google Scholar]
- 48.Gerrity RG. Richardson M. Somer JB. Bell FP. Schwartz CJ. Endothelial cell morphology in areas of in vivo Evans blue uptake in the aorta of young pigs. II. Ultrastructure of the intima in areas of differing permeability to proteins. Am J Pathol. 1977;89:313–334. [PMC free article] [PubMed] [Google Scholar]
- 49.Gerrity RG. Schwartz CJ. Structural correlates of arterial endothelial permeability in the Evans blue model. Prog Biochem Pharmacol. 1977;13:134–137. [PubMed] [Google Scholar]
- 50.Go YM. Patel RP. Maland MC. Park H. Beckman JS. Darley–Usmar VM. Jo H. Evidence for peroxynitrite as a signaling molecule in flow-dependent activation of c-Jun NH(2)-terminal kinase. Am J Physiol. 1999;277:H1647–H1653. doi: 10.1152/ajpheart.1999.277.4.H1647. [DOI] [PubMed] [Google Scholar]
- 51.Gudi S. Nolan JP. Frangos JA. Modulation of GTPase activity of G proteins by fluid shear stress and phospholipid composition. Proc Natl Acad Sci USA. 1998;95:2515–2519. doi: 10.1073/pnas.95.5.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gudi SR. Clark CB. Frangos JA. Fluid flow rapidly activates G proteins in human endothelial cells. Involvement of G proteins in mechanochemical signal transduction. Circ Res. 1996;79:834–839. doi: 10.1161/01.res.79.4.834. [DOI] [PubMed] [Google Scholar]
- 53.Hajra L. Evans AI. Chen M. Hyduk SJ. Collins T. Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA. 2000;97:9052–9057. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harrison DG. The shear stress of keeping arteries clear. Nat Med. 2005;11:375–376. doi: 10.1038/nm0405-375. [DOI] [PubMed] [Google Scholar]
- 55.Harrison DG. Widder J. Grumbach I. Chen W. Weber M. Searles C. Endothelial mechanotransduction, nitric oxide and vascular inflammation. J Intern Med. 2006;259:351–363. doi: 10.1111/j.1365-2796.2006.01621.x. [DOI] [PubMed] [Google Scholar]
- 56.Helmke BP. Davies PF. The cytoskeleton under external fluid mechanical forces: hemodynamic forces acting on the endothelium. Ann Biomed Eng. 2002;30:284–296. doi: 10.1114/1.1467926. [DOI] [PubMed] [Google Scholar]
- 57.Hermann C. Zeiher AM. Dimmeler S. Shear stress inhibits H2O2-induced apoptosis of human endothelial cells by modulation of the glutathione redox cycle and nitric oxide synthase. Arterioscler Thromb Vasc Biol. 1997;17:3588–3592. doi: 10.1161/01.atv.17.12.3588. [DOI] [PubMed] [Google Scholar]
- 58.Herschman HR. Function and regulation of prostaglandin synthase 2. Adv Exp Med Biol. 1999;469:3–8. doi: 10.1007/978-1-4615-4793-8_1. [DOI] [PubMed] [Google Scholar]
- 59.Hoffmann J. Dimmeler S. Haendeler J. Shear stress increases the amount of S-nitrosylated molecules in endothelial cells: Important role for signal transduction. FEBS Lett. 2003;551:153–158. doi: 10.1016/s0014-5793(03)00917-7. [DOI] [PubMed] [Google Scholar]
- 60.Hoger JH. Ilyin VI. Forsyth S. Hoger A. Shear stress regulates the endothelial Kir2.1 ion channel. Proc Natl Acad Sci USA. 2002;99:7780–7785. doi: 10.1073/pnas.102184999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hojo Y. Saito Y. Tanimoto T. Hoefen RJ. Baines CP. Yamamoto K. Haendeler J. Asmis R. Berk BC. Fluid shear stress attenuates hydrogen peroxide-induced c-Jun NH2- terminal kinase activation via a glutathione reductase-mediated mechanism. Circ Res. 2002;91:712–718. doi: 10.1161/01.res.0000037981.97541.25. [DOI] [PubMed] [Google Scholar]
- 62.Hosoya T. Maruyama A. Kang MI. Kawatani Y. Shibata T. Uchida K. Warabi E. Noguchi N. Itoh K. Yamamoto M. Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J Biol Chem. 2005;280:27244–27250. doi: 10.1074/jbc.M502551200. [DOI] [PubMed] [Google Scholar]
- 63.Hsiai TK. Hwang J. Barr ML. Correa A. Hamilton R. Alavi M. Rouhanizadeh M. Cadenas E. Hazen SL. Hemodynamics influences vascular peroxynitrite formation: Implication for low-density lipoprotein apo-B-100 nitration. Free Radic Biol Med. 2007;42:519–529. doi: 10.1016/j.freeradbiomed.2006.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang PL. Huang Z. Mashimo H. Bloch KD. Moskowitz MA. Bevan JA. Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 65.Huang PL. Lo EH. Genetic analysis of NOS isoforms using nNOS and eNOS knockout animals. Prog Brain Res. 1998;118:13–25. doi: 10.1016/s0079-6123(08)63197-0. [DOI] [PubMed] [Google Scholar]
- 66.Huo Y. Wischgoll T. Kassab GS. Flow patterns in three-dimensional porcine epicardial coronary arterial tree. Am J Physiol Heart Circ Physiol. 2007;293:H2959–2970. doi: 10.1152/ajpheart.00586.2007. [DOI] [PubMed] [Google Scholar]
- 67.Hwang J. Ing MH. Salazar A. Lassegue B. Griendling K. Navab M. Sevanian A. Hsiai TK. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: Implication for native LDL oxidation. Circ Res. 2003;93:1225–1232. doi: 10.1161/01.RES.0000104087.29395.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hwang J. Saha A. Boo YC. Sorescu GP. McNally JS. Holland SM. Dikalov S. Giddens DP. Griendling KK. Harrison DG. Jo H. Oscillatory shear stress stimulates endothelial production of O2- from p47phox-dependent NAD(P)H oxidases, leading to monocyte adhesion. J Biol Chem. 2003;278:47291–47298. doi: 10.1074/jbc.M305150200. [DOI] [PubMed] [Google Scholar]
- 69.Jin ZG. Ueba H. Tanimoto T. Lungu AO. Frame MD. Berk BC. Ligand independent activation of VEGF receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ Res. 2003;93:354–363. doi: 10.1161/01.RES.0000089257.94002.96. [DOI] [PubMed] [Google Scholar]
- 70.Jurewicz M. McDermott DH. Sechler JM. Tinckam K. Takakura A. Carpenter CB. Milford E. Abdi R. Human T and natural killer cells possess a functional renin-angiotensin system: Further mechanisms of angiotensin II-induced inflammation. J Am Soc Nephrol. 2007;18:1093–1102. doi: 10.1681/ASN.2006070707. [DOI] [PubMed] [Google Scholar]
- 71.Kaiser D. Freyberg MA. Friedl P. Lack of hemodynamic forces triggers apoptosis in vascular endothelial cells. Biochem Biophys Res Commun. 1997;231:586–590. doi: 10.1006/bbrc.1997.6146. [DOI] [PubMed] [Google Scholar]
- 72.Kano Y. Katoh K. Fujiwara K. Lateral zone of cell-cell adhesion as the major fluid shear stress-related signal transduction site. Circ Res. 2000;86:425–433. doi: 10.1161/01.res.86.4.425. [DOI] [PubMed] [Google Scholar]
- 73.Knowles JW. Reddick RL. Jennette JC. Shesely EG. Smithies O. Maeda N. Enhanced atherosclerosis and kidney dysfunction in eNOS(-/-)Apoe(-/-) mice are ameliorated by enalapril treatment. J Clin Invest. 2000;105:451–458. doi: 10.1172/JCI8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kol A. Lichtman AH. Finberg RW. Libby P. Kurt–Jones EA. Cutting edge: heat shock protein (HSP) 60 activates the innate immune response: CD14 is an essential receptor for HSP60 activation of mononuclear cells. J Immunol. 2000;164:13–17. doi: 10.4049/jimmunol.164.1.13. [DOI] [PubMed] [Google Scholar]
- 75.Kraiss LW. Geary RL. Mattsson EJ. Vergel S. Au YP. Clowes AW. Acute reductions in blood flow and shear stress induce platelet-derived growth factor-A expression in baboon prosthetic grafts. Circ Res. 1996;79:45–53. doi: 10.1161/01.res.79.1.45. [DOI] [PubMed] [Google Scholar]
- 76.Ku DN. Giddens DP. Zarins CK. Glagov S. Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arteriosclerosis. 1985;5:293–302. doi: 10.1161/01.atv.5.3.293. [DOI] [PubMed] [Google Scholar]
- 77.Kubes P. Kurose I. Granger DN. NO donors prevent integrin-induced leukocyte adhesion but not P-selectin-dependent rolling in postischemic venules. Am J Physiol. 1994;267:H931–937. doi: 10.1152/ajpheart.1994.267.3.H931. [DOI] [PubMed] [Google Scholar]
- 78.Kubes P. Suzuki M. Granger DN. Nitric oxide: An endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kuchan MJ. Jo H. Frangos JA. Role of G proteins in shear stress-mediated nitric oxide production by endothelial cells. Am J Physiol. 1994;267:C753–758. doi: 10.1152/ajpcell.1994.267.3.C753. [DOI] [PubMed] [Google Scholar]
- 80.Kuhlencordt PJ. Gyurko R. Han F. Scherrer–Crosbie M. Aretz TH. Hajjar R. Picard MH. Huang PL. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–454. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- 81.Lehoux S. Tedgui A. Cellular mechanics and gene expression in blood vessels. J Biomech. 2003;36:631–643. doi: 10.1016/s0021-9290(02)00441-4. [DOI] [PubMed] [Google Scholar]
- 82.Lerner–Marmarosh N. Yoshizumi M. Che W. Surapisitchat J. Kawakatsu H. Akaike M. Ding B. Huang Q. Yan C. Berk BC. Abe JI. Inhibition of tumor necrosis factor-[alpha]-induced SHP-2 phosphatase activity by shear stress: a mechanism to reduce endothelial inflammation. Arterioscler Thromb Vasc Biol. 2003;23:1775–1781. doi: 10.1161/01.ATV.0000094432.98445.36. [DOI] [PubMed] [Google Scholar]
- 83.Li YS. Shyy JY. Li S. Lee J. Su B. Karin M. Chien S. The Ras-JNK pathway is involved in shear-induced gene expression. Mol Cell Biol. 1996;16:5947–5954. doi: 10.1128/mcb.16.11.5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu Y. Chen BP. Lu M. Zhu Y. Stemerman MB. Chien S. Shyy JY. Shear stress activation of SREBP1 in endothelial cells is mediated by integrins. Arterioscler Thromb Vasc Biol. 2002;22:76–81. doi: 10.1161/hq0102.101822. [DOI] [PubMed] [Google Scholar]
- 85.Liu Y. Yin G. Surapisitchat J. Berk BC. Min W. Laminar flow inhibits TNF-induced ASK1 activation by preventing dissociation of ASK1 from its inhibitor 14-3-3. J Clin Invest. 2001;107:917–923. doi: 10.1172/JCI11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Malek A. Izumo S. Physiological fluid shear stress causes downregulation of endothelin-1 mRNA in bovine aortic endothelium. Am J Physiol. 1992;263:C389–C396. doi: 10.1152/ajpcell.1992.263.2.C389. [DOI] [PubMed] [Google Scholar]
- 87.Malek AM. Greene AL. Izumo S. Regulation of endothelin-1 gene by fluid shear stress is transcriptionally mediated and independent of protein kinase-C and cAMP. Proc Natl Acad Sci USA. 1993;90:5999–6003. doi: 10.1073/pnas.90.13.5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Malek AM. Jackman R. Rosenberg RD. Izumo S. Endothelial expression of thrombomodulin is reversibly regulated by fluid shear stress. Circ Res. 1994;74:852–860. doi: 10.1161/01.res.74.5.852. [DOI] [PubMed] [Google Scholar]
- 89.Matsushita K. Morrell CN. Cambien B. Yang SX. Yamakuchi M. Bao C. Hara MR. Quick RA. Cao W. O'Rourke B. Lowenstein JM. Pevsner J. Wagner DD. Lowenstein CJ. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell. 2003;115:139–150. doi: 10.1016/s0092-8674(03)00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maus U. Henning S. Wenschuh H. Mayer K. Seeger W. andLohmeyer J. Role of endothelial MCP-1 in monocyte adhesion to inflamed human endothelium under physiological flow. Am J Physiol Heart Circ Physiol. 2002;283:H2584–2591. doi: 10.1152/ajpheart.00349.2002. [DOI] [PubMed] [Google Scholar]
- 91.Mazzolai L. Silacci P. Bouzourene K. Daniel F. Brunner H. Hayoz D. Tissue factor activity is upregulated in human endothelial cells exposed to oscillatory shear stress. Thromb Haemost. 2002;87:1062–1068. [PubMed] [Google Scholar]
- 92.McNally JS. Davis ME. Giddens DP. Saha A. Hwang J. Dikalov S. Jo H. Harrison DG. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am J Physiol Heart Circ Physiol. 2003;285:H2290–H2297. doi: 10.1152/ajpheart.00515.2003. [DOI] [PubMed] [Google Scholar]
- 93.Michel T. Feron O. Nitric oxide synthases: Which, where, how, and why? J Clin Invest. 1997;100:2146–2151. doi: 10.1172/JCI119750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Michell BJ. Chen Zp Z. Tiganis T. Stapleton D. Katsis F. Power DA. Sim AT. Kemp BE. Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J Biol Chem. 2001;276:17625–17628. doi: 10.1074/jbc.C100122200. [DOI] [PubMed] [Google Scholar]
- 95.Mueller CF. Widder JD. McNally JS. McCann L. Jones DP. Harrison DG. The role of the multidrug resistance protein-1 in modulation of endothelial cell oxidative stress. Circ Res. 2005;97:637–644. doi: 10.1161/01.RES.0000183734.21112.b7. [DOI] [PubMed] [Google Scholar]
- 96.Nagel T. Resnick N. Atkinson WJ. Dewey CF., Jr. Gimbrone MA., Jr Shear stress selectively upregulates intercellular adhesion molecule-1 expression in cultured human vascular endothelial cells. J Clin Invest. 1994;94:885–891. doi: 10.1172/JCI117410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nakao M. Ono K. Fujisawa S. Iijima T. Mechanical stress-induced Ca2+ entry and Cl- current in cultured human aortic endothelial cells. Am J Physiol. 1999;276:C238–249. doi: 10.1152/ajpcell.1999.276.1.C238. [DOI] [PubMed] [Google Scholar]
- 98.Ni CW. Wang DL. Lien SC. Cheng JJ. Chao YJ. Hsieh HJ. Activation of PKC-epsilon and ERK1/2 participates in shear-induced endothelial MCP-1 expression that is repressed by nitric oxide. J Cell Physiol. 2003;195:428–434. doi: 10.1002/jcp.10259. [DOI] [PubMed] [Google Scholar]
- 99.Ohno M. Gibbons GH. Dzau VJ. Cooke JP. Shear stress elevates endothelial cGMP. Role of a potassium channel and G protein coupling. Circulation. 1993;88:193–197. doi: 10.1161/01.cir.88.1.193. [DOI] [PubMed] [Google Scholar]
- 100.Ohtsuka A. Ando J. Korenaga R. Kamiya A. Toyamasorimachi N. Miyasaka M. The effect of flow on the expression of vascular adhesion molecule-1 by cultured mouse endothelial cells. Biochem Biophys Res Commun. 1993;193:303–310. doi: 10.1006/bbrc.1993.1624. [DOI] [PubMed] [Google Scholar]
- 101.Ohura N. Yamamoto K. Ichioka S. Sokabe T. Nakatsuka H. Baba A. Shibata M. Nakatsuka T. Harii K. Wada Y. Kohro T. Kodama T. Ando J. Global analysis of shear stress-responsive genes in vascular endothelial cells. J Atheroscler Thromb. 2003;10:304–313. doi: 10.5551/jat.10.304. [DOI] [PubMed] [Google Scholar]
- 102.Olesen SP. Clapham DE. Davies PF. Haemodynamic shear stress activates a K+ current in vascular endothelial cells. Nature. 1988;331:168–170. doi: 10.1038/331168a0. [DOI] [PubMed] [Google Scholar]
- 103.Osawa M. Masuda M. Kusano K. Fujiwara K. Evidence for a role of platelet endothelial cell adhesion molecule-1 in endothelial cell mechanosignal transduction: Is it a mechanoresponsive molecule? J Cell Biol. 2002;158:773–785. doi: 10.1083/jcb.200205049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Parmar KM. Larman HB. Dai G. Zhang Y. Wang ET. Moorthy SN. Kratz JR. Lin Z. Jain MK. Gimbrone MA., Jr. Garcia–Cardena G. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116:49–58. doi: 10.1172/JCI24787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Passerini AG. Polacek DC. Shi C. Francesco NM. Manduchi E. Grant GR. Pritchard WF. Powell S. Chang GY. Stoeckert CJ., Jr. Davies PF. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc Natl Acad Sci USA. 2004;101:2482–2487. doi: 10.1073/pnas.0305938101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Patwari P. Higgins LJ. Chutkow WA. Yoshioka J. Lee RT. The interaction of thioredoxin with Txnip: Evidence for formation of a mixed disulfide by disulfide exchange. J Biol Chem. 2006;28:21884–21891. doi: 10.1074/jbc.M600427200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Peters DG. Zhang XC. Benos PV. Heidrich–O'Hare E. Ferrell RE. Genomic analysis of immediate/early response to shear stress in human coronary artery endothelial cells. Physiol Genomics. 2002;12:25–33. doi: 10.1152/physiolgenomics.00016.2002. [DOI] [PubMed] [Google Scholar]
- 108.Pi X. Yan C. Berk BC. Big mitogen-activated protein kinase (BMK1)/ERK5 protects endothelial cells from apoptosis. Circ Res. 2003;11:11. doi: 10.1161/01.RES.0000112406.27800.6F. [DOI] [PubMed] [Google Scholar]
- 109.Rubanyi GM. Romero JC. Vanhoutte PM. Flow-induced release of endothelium-derived relaxing factor. Am J Physiol. 1986;19:H1145–H1149. doi: 10.1152/ajpheart.1986.250.6.H1145. [DOI] [PubMed] [Google Scholar]
- 110.Rudic RD. Shesely EG. Maeda N. Smithies O. Segal SS. Sessa WC. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J Clin Invest. 1998;101:731–736. doi: 10.1172/JCI1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Saitoh M. Nishitoh H. Fujii M. Takeda K. Tobiume K. Sawada Y. Kawabata M. Miyazono K. Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal- regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Satoh A. Toida T. Yoshida K. Kojima K. Matsumoto I. New role of glycosaminoglycans on the plasma membrane proposed by their interaction with phosphatidylcholine. FEBS Lett. 2000;477:249–252. doi: 10.1016/s0014-5793(00)01746-4. [DOI] [PubMed] [Google Scholar]
- 113.Schwarz G. Callewaert G. Droogmans G. Nilius B. Shear stress-induced calcium transients in endothelial cells from human umbilical cord veins. J Physiol Lond. 1992;458:527–538. doi: 10.1113/jphysiol.1992.sp019432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schwarz G. Droogmans G. Nilius B. Shear stress induced membrane currents and calcium transients in human vascular endothelial cells. Pflugers Arch. 1992;421:394–396. doi: 10.1007/BF00374230. [DOI] [PubMed] [Google Scholar]
- 115.SenBanerjee S. Lin Z. Atkins GB. Greif DM. Rao RM. Kumar A. Feinberg MW. Chen Z. Simon DI. Luscinskas FW. Michel TM. Gimbrone MA., Jr. Garcia–Cardena G. Jain MK. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199:1305–1315. doi: 10.1084/jem.20031132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sharefkin JB. Diamond SL. Eskin SG. McIntire LV. Dieffenbach CW. Fluid flow decreases preproendothelin mRNA levels and suppresses endothelin-1 peptide release in cultured human endothelial cells. J Vasc Surg. 1991;14:1–9. [PubMed] [Google Scholar]
- 117.Shesely EG. Maeda N. Kim HS. Desai KM. Krege JH. Laubach VE. Sherman PA. Sessa WC. Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shyy YJ. Hsieh HJ. Usami S. Chien S. Fluid shear stress induces a biphasic response of human monocyte chemotactic protein 1 gene expression in vascular endothelium. Proc Natl Acad Sci USA. 1994;91:4678–4682. doi: 10.1073/pnas.91.11.4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sorescu GP. Song H. Tressel SL. Hwang J. Dikalov S. Smith DA. Boyd NL. Platt MO. Lassegue B. Griendling KK. Jo H. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a nox1-based NADPH oxidase. Circ Res. 2004;95:773–779. doi: 10.1161/01.RES.0000145728.22878.45. [DOI] [PubMed] [Google Scholar]
- 120.Stamler JS. Lamas S. Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 121.Stamler JS. Simon DI. Osborne JA. Mullins ME. Jaraki O. Michel T. Singel DJ. Loscalzo J. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci USA. 1992;89:444–448. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sun D. Huang A. Yan EH. Wu Z. Yan C. Kaminski PM. Oury TD. Wolin MS. Kaley G. Reduced release of nitric oxide to shear stress in mesenteric arteries of aged rats. Am J Physiol Heart Circ Physiol. 2004;286:H2249–2256. doi: 10.1152/ajpheart.00854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Suo J. Oshinski JN. Giddens DP. Blood flow patterns in the proximal human coronary arteries: relationship to atherosclerotic plaque occurrence. Mol Cell Biomech. 2008;5:9–18. [PubMed] [Google Scholar]
- 124.Surapisitchat J. Hoefen RJ. Pi X. Yoshizumi M. Yan C. Berk BC. Fluid shear stress inhibits TNF-alpha activation of JNK but not ERK1/2 or p38 in human umbilical vein endothelial cells: Inhibitory crosstalk among MAPK family members. Proc Natl Acad Sci USA. 2001;98:6476–6481. doi: 10.1073/pnas.101134098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Takada Y. Shinkai F. Kondo S. Yamamoto S. Tsuboi H. Korenaga R. Ando J. Fluid shear stress increases the expression of thrombomodulin by cultured human endothelial cells. Biochem Biophys Res Commun. 1994;205:1345–1352. doi: 10.1006/bbrc.1994.2813. [DOI] [PubMed] [Google Scholar]
- 126.Takeshita S. Inoue N. Ueyama T. Kawashima S. Yokoyama M. Shear stress enhances glutathione peroxidase expression in endothelial cells. Biochem Biophys Res Commun. 2000;273:66–71. doi: 10.1006/bbrc.2000.2898. [DOI] [PubMed] [Google Scholar]
- 127.Topper JN. Cai J. Falb D. Gimbrone MA., Jr Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase-2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up-regulated by steady laminar shear stress. Proc Natl Acad Sci USA. 1996;93:10417–10422. doi: 10.1073/pnas.93.19.10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Traub O. Berk BC. Laminar shear stress: mechanisms by which endothelial cells transduce an atheroprotective force. Arterioscler Thromb Vasc Biol. 1998;18:677–685. doi: 10.1161/01.atv.18.5.677. [DOI] [PubMed] [Google Scholar]
- 129.Tsao PS. Buitrago R. Chan JR. Cooke JP. Fluid flow inhibits endothelial adhesiveness. Nitric oxide and transcriptional regulation of VCAM-1. Circulation. 1996;94:1682–1689. doi: 10.1161/01.cir.94.7.1682. [DOI] [PubMed] [Google Scholar]
- 130.Tzima E. del Pozo MA. Shattil SJ. Chien S. Schwartz MA. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J. 2001;20:4639–4647. doi: 10.1093/emboj/20.17.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tzima E. Irani–Tehrani M. Kiosses WB. Dejana E. Schultz DA. Engelhardt B. Cao G. DeLisser H. Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 132.Van der Heiden K. Hierck BP. Krams R. de Crom R. Cheng C. Baiker M. Pourquie MJ. Alkemade FE. DeRuiter MC. Gittenberger–de Groot AC. Poelmann RE. Endothelial primary cilia in areas of disturbed flow are at the base of atherosclerosis. Atherosclerosis. 2008;196:542–550. doi: 10.1016/j.atherosclerosis.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 133.Walpola PL. Gotlieb AI. Cybulsky MI. Langille BL. Expression of ICAM-1 and VCAM-1 and monocyte adherence in arteries exposed to altered shear stress [published erratum appears in Arterioscler Thromb Vasc Biol 1995: 15: 429] Arterioscler Thromb Vasc Biol. 1995;15:2–10. doi: 10.1161/01.atv.15.1.2. [DOI] [PubMed] [Google Scholar]
- 134.Walshe TE. Ferguson G. Connell P. O'Brien C. Cahill PA. Pulsatile flow increases the expression of eNOS, ET-1, and prostacyclin in a novel in vitro coculture model of the retinal vasculature. Invest Ophthalmol Vis Sci. 2005;46:375–382. doi: 10.1167/iovs.04-0806. [DOI] [PubMed] [Google Scholar]
- 135.Wang W. Diamond SL. Does elevated nitric oxide production enhance the release of prostacyclin from shear stressed aortic endothelial cells? Biochem Biophys Res Commun. 1997;233:748–751. doi: 10.1006/bbrc.1997.6548. [DOI] [PubMed] [Google Scholar]
- 136.Warabi E. Takabe W. Minami T. Inoue K. Itoh K. Yamamoto M. Ishii T. Kodama T. Noguchi N. Shear stress stabilizes NF-E2-related factor 2 and induces antioxidant genes in endothelial cells: Role of reactive oxygen/nitrogen species. Free Radic Biol Med. 2007;42:260–269. doi: 10.1016/j.freeradbiomed.2006.10.043. [DOI] [PubMed] [Google Scholar]
- 137.Warabi E. Wada Y. Kajiwara H. Kobayashi M. Koshiba N. Hisada T. Shibata M. Ando J. Tsuchiya M. Kodama T. Noguchi N. Effect on endothelial cell gene expression of shear stress, oxygen concentration, and low-density lipoprotein as studied by a novel flow cell culture system. Free Radic Biol Med. 2004;37:682–694. doi: 10.1016/j.freeradbiomed.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 138.Won D. Zhu SN. Chen M. Teichert AM. Fish JE. Matouk CC. Bonert M. Ojha M. Marsden PA. Cybulsky MI. Relative reduction of endothelial nitric-oxide synthase expression and transcription in atherosclerosis-prone regions of the mouse aorta and in an in vitro model of disturbed flow. Am J Pathol. 2007;171:1691–1704. doi: 10.2353/ajpath.2007.060860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Woodman CR. Muller JM. Rush JW. Laughlin MH. Price EM. Flow regulation of ecNOS and Cu/Zn SOD mRNA expression in porcine coronary arterioles. Am J Physiol. 1999;276:H1058–1063. doi: 10.1152/ajpheart.1999.276.3.H1058. [DOI] [PubMed] [Google Scholar]
- 140.Yamawaki H. Haendeler J. Berk BC. Thioredoxin: A key regulator of cardiovascular homeostasis. Circ Res. 2003;93:1029–1033. doi: 10.1161/01.RES.0000102869.39150.23. [DOI] [PubMed] [Google Scholar]
- 141.Yamawaki H. Lehoux S. Berk BC. Chronic physiological shear stress inhibits tumor necrosis factor-induced proinflammatory responses in rabbit aorta perfused ex vivo. Circulation. 2003;108:1619–1625. doi: 10.1161/01.CIR.0000089373.49941.C4. [DOI] [PubMed] [Google Scholar]
- 142.Yamawaki H. Pan S. Lee RT. Berk BC. Fluid shear stress inhibits vascular inflammation by decreasing thioredoxin-interacting protein in endothelial cells. J Clin Invest. 2005;115:733–738. doi: 10.1172/JCI200523001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yao Y. Rabodzey A. Dewey CF., Jr Glycocalyx modulates the motility and proliferative response of vascular endothelium to fluid shear stress. Am J Physiol Heart Circ Physiol. 2007;293:H1023–1030. doi: 10.1152/ajpheart.00162.2007. [DOI] [PubMed] [Google Scholar]
- 144.Yeh JC. Otte LA. Frangos JA. Regulation of G protein-coupled receptor activities by the platelet-endothelial cell adhesion molecule, PECAM-1. Biochemistry. 2008;47:9029–9039. doi: 10.1021/bi8003846. [DOI] [PubMed] [Google Scholar]
- 145.Yoshizumi M. Fujita Y. Izawa Y. Suzaki Y. Kyaw M. Ali N. Tsuchiya K. Kagami S. Yano S. Sone S. Tamaki T. Ebselen inhibits tumor necrosis factor-alpha-induced c-Jun N-terminal kinase activation and adhesion molecule expression in endothelial cells. Exp Cell Res. 2004;292:1–10. doi: 10.1016/j.yexcr.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 146.Zakkar M. Chaudhury H. Sandvik G. Enesa K. Luong le A. Cuhlmann S. Mason JC. Krams R. Clark AR. Haskard DO. Evans PC. Increased endothelial mitogen-activated protein kinase phosphatase-1 expression suppresses proinflammatory activation at sites that are resistant to atherosclerosis. Circ Res. 2008;103:726–732. doi: 10.1161/CIRCRESAHA.108.183913. [DOI] [PubMed] [Google Scholar]