

Abstract
To ascertain the membrane topography of the multi-transmembrane spanning presenilin proteins PS-1 and PS-2, anti-peptide antibodies were raised to several specific amino acid sequences in the two proteins, and, after their specificity was ascertained, the anti-peptide antibodies were used in immunofluorescent labeling of live PS-transfected, cultured DAMI cells, which are impermeable to the antibodies, as well as of their fixed and permeabilized counterparts. In such experiments, antibodies that specifically stain the intact live cells must label epitopes of the PS proteins that are on the exterior face of the plasma membrane whereas those antibodies that do not stain the live cells but do stain the fixed and permeabilized cells must label epitopes that face the cytoplasmic side of the membrane. The results obtained were entirely in accord with the predictions of the seven-transmembrane spanning topography (like that of rhodopsin and the β-adrenergic receptor) and were totally inconsistent with the expectations for either the six- or eight-transmembrane topographies that have been proposed.
Keywords: anti-peptide antibodies, immunofluorescence microscopy, seven transmembrane-spanning protein family
Mutant forms of the two presenilin (PS) proteins PS-1 (1) and PS-2 (2, 3) have been identified as playing critical roles in early onset familial Alzheimer disease. This genetic information strongly suggests that the PS in their nonmutated forms are directly involved in the generation of the more common nonfamilial Alzheimer disease (AD), but their roles have not yet been elucidated. Some time ago, we published (4) an explicit hypothesis about how the PS proteins might function in AD, based on precedents in the literature (5, 6). We proposed that either PS-1 or PS-2, which are closely related integral membrane proteins, are expressed at, and in part protrude from, the plasma membranes of cells, where they serve as specific receptors for the β-amyloid precursor protein (β-APP) (for a review, see ref. 7), expressed at the cell surface of neighboring neurons in the brain. The intercellular specific molecular binding of either PS-1 or PS-2 on one cell to β-APP on the other, we proposed, creates a close adhesion between the two cells (8), which subsequently leads to the incorporation of contacting membrane elements into the interior of the β-APP-expressing neuron in the form of double-membrane-bounded vesicles (9). Once inside the neuron, these vesicles fuse with secondary lysosomes (5, 9), in which, we hypothesize, the internalized β-APP is broken down by specific proteases to the relevant forms of β-amyloid (Aβ), the oligopeptides that are the key components of the neuritic plaques in the brain that are thought to be responsible for AD (10).
In subsequent studies (11, 12), we proved by immunofluorescence microscopy experiments that the PS proteins were indeed expressed at the cell surfaces of untransfected and transfected cells in culture, including neurons; such surface expression had been missed or denied by others (13–18). We further demonstrated (11) that a specific cell–cell aggregation occurs upon mixing cells transfected with β-APP with cells transfected with either PS-1 or PS-2, thereby proving that β-APP binds specifically to PS-1 or PS-2 and can do so intercellularly, i.e., that the β-APP:PS-1 or β-APP:PS-2 binding is an example of a membrane-bound ligand–receptor transcellular interaction, as we originally proposed (4). That β-APP can bind specifically to the PS proteins has been confirmed recently in other types of studies with detergent-solubilized proteins (19, 20).
These findings emphasize the importance of the structure of the PS proteins and their topography in the plasma membranes of cells: The topography determines which regions of the PS molecules are accessible to, and involved in, the specific transcellular bonds they form with β-APP. The topography of the PS proteins has been contested (see Discussion), and various quite different versions involving six, seven, eight, and even nine transmembrane (TM)-spanning domains have been postulated. In this paper, by using a battery of anti-peptide antibodies generated to particular sequences in the PS proteins, which are characterized in the companion paper (21), we provide unequivocal evidence for a 7-TM spanning topography, as was proposed for these proteins when their amino acid sequences were deduced originally (1–3).
MATERIALS AND METHODS
Cell Culture and Transfections.
DAMI cells (American Type Culture Collection, CRL 9792) were cultured and transiently transfected with high efficiencies (60–80%) with pcDNA3 constructs of full length PS-1 and PS-2 as described (11).
Antibodies.
Eight polyclonal rabbit anti-peptide antibodies were raised to the following sequences. For PS-1: N2, SNGRPQGNSRQ (residues 51–61); I1, YTRKDGQL (residues 106–113); L1, SKNSKYNAE (residues 310–318); and L3, DSHLGPHRST (residues 345–354); and for PS-2: N1, ESPTPRSCQEGR (residues 24–35); N3, GVPGRPPGLE (residues 67–76); L2, SFGEPSY (residues 330–336); and C1, STDNLVRPF (residues 429–437). The oligopeptides for C1 and L3 antibodies were conjugated to keyhole limpet hemocyanin for immunization; the remaining oligopeptides as synthesized on a multiple antigen peptide were used as the immunogens. The synthesis and conjugations of the oligopeptides were performed by Research Genetics, Huntsville, AL; the preparation of the rabbit antisera was done by HTI Bio-products, Ramona, CA.
Immunofluorescent Microscopic Labeling.
Either live or fixed and permeabilized PS-1- or PS-2-transfected DAMI cells were first reacted in suspension with an appropriate one of the following six primary anti-peptide antisera at the indicated dilutions: N1 (1:100), I1 (1:200), L1 (1:50), L2 (1:350), L3 (1:200), and C1 (1:75), in PBS (pH 7.4) containing 3% BSA for 20 minutes at room temperature. The specificities of the antibodies N2 and N3 were suspect (21), and the results with these antibodies are therefore not included. Where indicated, inhibition of antibody labeling by the addition of an excess of the specific soluble oligopeptide conjugate was carried out as described (12). After centrifugal washing three times with PBS, the cells were resuspended in PBS containing 3% BSA and were then reacted with affinity-purified secondary antibodies, dichlorotriazinylamino fluorescein-conjugated donkey anti-rabbit IgG (1:100 dilution into PBS of a 1.5-mg/ml solution obtained from Jackson ImmunoResearch) at room temperature for 15 minutes. After washing, cells were examined in a solution of 1 mg/ml phenylene diamine in 100 mM Tris⋅HCl buffer (pH 8.5) containing 90% glycerol in a Zeiss Photoscope III epifluorescence instrument by using oil immersion with a ×60 objective lens and a fluorescein isothiocyanate filter or with Nomarski optics.
Fixation and Permeabilization of Cells.
The transfected DAMI cells were fixed for 5 minutes at room temperature in 3% paraformaldehyde in PBS, followed by a 5-minute treatment in 0.05% Nonidet P-40 and 0.05% Triton X-100 in 3% paraformaldehyde.
RESULTS
The eight different anti-peptide antibodies raised for these studies are characterized in a companion paper appearing in this issue of Proceedings (21). They were directed to amino acid sequences of PS-1 and PS-2 that best discriminated among the different proposed membrane topographies. Three (N1 and N3 for PS-2 and N2 for PS-1) were directed to epitopes within the NH2-terminal region preceding the first TM-spanning domain (Fig. 1, depicting the 7-TM spanning topography of PS-1 and PS-2); one (I1 for PS-1) directed to an epitope within the small hydrophilic loop region between TM stretches I and II; three (L1 and L3 for PS-1 and L2 for PS-2) directed to epitopes within the large loop region between the TM stretches VI and VII; and one (C1) directed to an epitope within the COOH-terminal region of PS-2, which also reacted with the same closely homologous region of PS-1. The antibodies N1 and L3 were those used in our earlier studies (11, 12). The antibodies N2 and N3 were of questionable specificity; the former was the only antibody that stained multiple bands in immunoblotting experiments of gently prepared DAMI cell extracts (see figure 1A, lane 13, in ref. 21) whereas the latter stained a main band that was, however, not PS-2 (see figure 1B, lane 10, in ref. 21). These two antibodies therefore were not used further in this paper. The remaining six antibodies satisfied the criteria that, in immunoblotting experiments, each mainly labeled the single band corresponding to the intact PS molecule and that this labeling was inhibited in the presence of an excess of the oligopeptide conjugate that was specific for that antibody (21).
Figure 1.
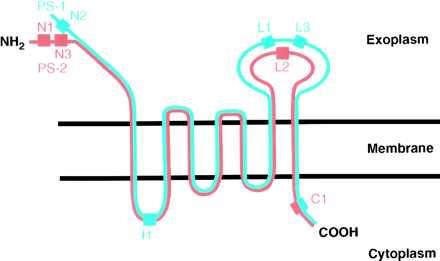
The proposed 7-TM spanning topography of the homologous PS-1 (blue) and PS-2 (red) proteins in the plasma membrane of a cell. PS-1 is a 467-aa polypeptide; PS-2 is a 448-aa. The colored blocks are situated at positions along the two polypeptides corresponding to the oligopeptide sequences (given in Materials and Methods) used to generate the several anti-peptide antibodies used in this study.
We have proved (12) that, when live transfected DAMI cells are immunofluorescently labeled, only those epitopes of the PS proteins that are exposed at the cell surface are stained because such cells are not labeled with anti-tubulin antibodies, i.e., they are antibody-impermeable. Therefore, if the 7-TM spanning topography of the PS proteins (Fig. 1) is basically correct, we predicted the following immunofluorescent labeling results: Live DAMI cells transfected with PS-1 should be labeled with antibodies L1 and L3 but not with I1 or C1; and live PS-2-transfected DAMI cells should be labeled with antibodies N1 and L2 but not with C1. On the other hand, with the appropriate transfected cells that were first fixed and permeabilized before immunofluorescent labeling, the antibodies I1 and C1 now should have access to their specific epitopes located on the cytoplasmic side of the plasma membrane and on the luminal sides of the permeabilized endoplasmic reticulum (e.r.) and Golgi membranes and therefore should stain the cells. These predictions have been borne out entirely, as shown by the results in Fig. 2, which we describe next.
Figure 2.
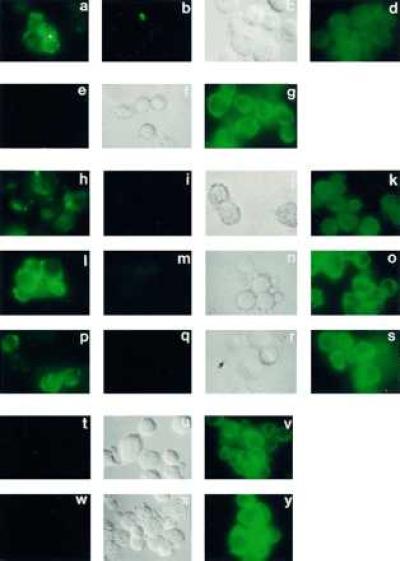
Immunofluorescent microscopic indirect labeling experiments with transfected DAMI cells, by using the several primary anti-peptide antibodies described in Materials and Methods and directed toward the epitopes shown in Fig. 1. [The N2 and N3 antibodies did not exhibit unique specificity for their epitope (21), unlike the other antibodies, and were therefore not included]. (a–d) DAMI cells transfected with PS-2 and immunolabeled with antibody N1: (a) live cells; (b) live cells labeled in the presence of excess specific soluble oligopeptide conjugate for N1; (c) the Nomarski image of b; (d) fixed and permeabilized cells. (e–g) DAMI cells transfected with PS-1 and immunolabeled with antibody I1: (e) live cells; (f) the Nomarski image of e; (g) fixed and permeabilized cells. (h–k) Same as a–d except that immunolabeling was of PS-1-transfected cells with antibody L3 and in i the specific soluble oligopeptide conjugate for L3 was used for inhibition. (l–o) DAMI cells transfected with PS-2 and immunolabeled with antibody L2: (l) live cells; (m) live cells labeled in the presence of excess specific soluble oligopeptide conjugate for L2; (n) the Nomarski image of m; (o) fixed and permeabilized cells. (p–s) DAMI cells transfected with PS-1 and immunolabeled with antibody L1: (p) live cells; (q) live cells labeled in the presence of excess specific soluble oligopeptide conjugate for L1; (r) the Nomarski image of q; (s) fixed and permeabilized cells. (t–v) DAMI cells transfected with PS-1 and immunolabeled with antibody C1: (t) live cells; (u) the Nomarski image of t; (v) fixed and permeabilized cells. (w–y) Same as t–v except with cells transfected with PS-2.
The antibodies N1 (Fig. 2a), L3 (Fig. 2h), L2 (Fig. 2l), and L1 (Fig. 2p) stained live DAMI cells transfected with the appropriate PS protein. This labeling was inhibited by an excess of the particular soluble oligopeptide conjugate specific for that antibody (Fig. 2 b, i, m, and q, respectively; the Nomarski images of these unstained cells are shown in Fig. 2 c, j, n, and r, respectively). This inhibition of antibody labeling by the soluble oligopeptide specific for that antibody attests to the specificity of each labeling shown in Fig. 2 a, h, l, and p. In additional experiments (not shown, but see ref. 12), we confirmed that these live transfected DAMI cells were not labeled with, and hence were impermeable to, anti-tubulin antibodies. Furthermore, the antibodies I1 with live PS-1 transfected cells (Fig. 2e) and C1 with either live PS-1-transfected (Fig. 2t) or PS-2-transfected (Fig. 2w) cells produced no labeling; their Nomarski images are displayed in Fig. 2 f, u, and x, respectively. On the other hand, if the cells were first fixed and permeabilized, they were intensely stained with these latter two antibodies (Fig. 2 g, v, and y, respectively). In addition, those four antibodies that labeled the appropriate live cells also intensely labeled their fixed and permeabilized counterparts (Fig. 2 d, k, o, and s), but the staining patterns, as expected for intracellular labeling, were more diffuse than for their respective surface labeling (Fig. 2 a, h, l, and p, respectively). The results therefore accord precisely with those predicted for the 7-TM spanning topography of the PS proteins in the plasma membrane (Fig. 1). In particular, the entirely similar results obtained with three independent antibodies (L1, L2, and L3) specifically directed to the large loop regions of either PS-1 or PS-2 strongly support the exoplasmic location of the large loop region characteristic of the 7-TM spanning topography.
DISCUSSION
Examination of the Kyte-Doolittle hydropathy plots (22) for the PS proteins, shown in Fig. 3A for PS-1 (1), reveals six sequences (I-VI) that are sufficiently hydrophopic to be strong candidates for TM spanning stretches, and all three topographic models, the 6-, 7-, and 8-TM spanners (ref. 23; refs. 1–3; and ref. 24, respectively), use them (Fig. 3B). However, how the models deploy these six TM spanning stretches is critically different. The 6- and 8-TM spanning models both start with the NH2-terminal domain positioned on the cytoplasmic side of the membrane, which then determines the alternating membrane orientations of the successive TM spanning stretches (and also, most important, the particular membrane sidedness of the successive hydrophilic loops between the TM spanning stretches) to be those shown in Fig. 3B. In the 7-TM model, however, the NH2-terminal domain is located in the exoplasm (or in the lumen of the intracellular e.r. or Golgi saccules). This determines that the orientation of each of the first six successive TM spanning stretches is the opposite of that in the 6- and 8-TM spanning models and likewise that each of the successive hydrophilic loops protrudes from the opposite face of the membrane in the 7-TM spanning model compared with its location in the 6- and 8-TM spanning models (Fig. 3B).
Figure 3.
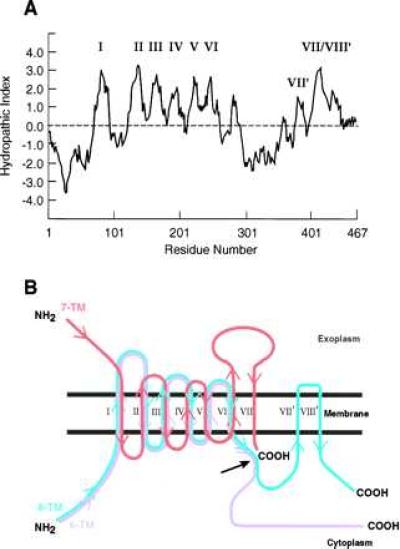
(A) The Kyte-Doolittle hydropathy plot for PS-1 (1) by using a window of 15 residues. The roman numerals indicate the hydrophobic sequences serving as TM spanning stretches in B (see text). (B) The topographies of the three proposed models of PS proteins, the 6- (magenta), 7- (red), and 8- (blue) TM spanning models. The correspondingly colored arrows indicate the polypeptide chain directions starting from the NH2 terminus (NH2) toward the COOH terminus (COOH) for each model. The long black arrow indicates the region of the proposed endoproteolytic cleavage of the PS proteins (see text).
The other topographic differences among the three models involve regions of the PS molecules that are carboxyl to TM VI. In the 6-TM spanning model, there are no further TM spanning stretches, and the rest of the sequence to the COOH terminus is located in the cytoplasm. In the 7-TM spanning model, only the highly hydrophobic stretch labeled VII/VIII′ in Fig. 3A spans the membrane (Fig. 3B) so that the COOH-terminal region is situated in the cytoplasm. In the 8-TM spanning model, the moderately hydrophobic stretch labeled VII′ in Fig. 3A, as well as the stretch labeled VII/VIII′, is threaded successively through the membrane. (All three models therefore locate the COOH terminus in the cytoplasm. Thus, labeling with antibody C1, although useful for other purposes, cannot discriminate among the topographies).
The findings shown in Fig. 2 (which demonstrate that the NH2-terminal region as well as the long hydrophilic loop following transmembrane stretch VI are both located on the exoplasmic side of the plasma membrane whereas the short hydrophilic loop between TM stretches I and II and the COOH-terminal sequence are both located on the cytoplasmic side) strongly support the 7-TM spanning topography. They are, however, totally inconsistent with either the 6- or 8-TM spanning topographies (Fig. 3B), which if correct would require the exactly reversed results for every one of the antibodies except C1. We can think of no loopholes in these completely straightforward studies that might confute our conclusions that the 7-TM spanning topography for the PS proteins is correct and that either the 6- or 8-TM spanning one is not. Therefore, although ours are, at present, the only published experimental data leading to these conclusions, we have no hesitation in asserting them. Our experiments, by the way, were carried out by similar techniques of anti-peptide antibody and immunofluorescent microscopy and gave results closely analogous to those of Wang et al. (25) that experimentally confirmed the well known 7-TM spanning topography of the β-adrenergic receptor.
What, then, are the experimental results that have been adduced to support the 6- or 8-TM spanning topography? These have been of several kinds and originate from a number of laboratories; they cannot all be examined critically and in detail in this paper. Put briefly, however, the main evidence is of three types. One type involves a reported endoproteolytic cleavage of the PS proteins (for a recent review, see ref. 26) into an NH2-terminal ≈30-kDa fragment and a variety of COOH-terminal fragments, corresponding to cleavage in a region of the sequence following ≈30 residues after the TM stretch VI (represented by the long black arrow in Fig. 3B). It has been claimed that this is a normal physiological event occurring within cells by the action of a cytoplasmic protease on intact PS-1 and PS-2 molecules. If this were so, the proteolyzed region of the sequence would have to be located on the cytoplasmic side of the membrane as in the 6- or 8-, but opposite to, the 7-TM spanning topographies. However, in the accompanying paper (21), we provide evidence that this particular endoproteolytic event is most probably an artifact, which arises either during the prior treatment or extraction of the cells or tissues involved (including mouse brain); such proteolysis is not observed if sufficient precautions are taken to prevent it (12, 21). This endoproteolysis therefore is irrelevant to PS topography.
A second type of evidence involves experiments on the membrane intercalation of constructs of truncated forms of the PS proteins fused to various reporter sequences (23, 24). Such experiments rely on the basic assumption that every such construct, no matter how few TM stretches it retains or whatever the nature of the reporter and intervening sequences, on integration into a membrane will invariably reproduce the detailed membrane topography of the individual TM stretches that is characteristic of the native intact integral protein. Yet, in one of these PS studies (23), 7 of the 10 fusion proteins that were made gave discordant, unexplained, and ignored results. Furthermore, careful studies of the membrane integration of truncation mutants of the classic 7-TM spanning protein bovine rhodopsin (27) have shown that the “successful completion of different stages in the cellular processing of the protein (membrane insertion, N-linked glycosylation, stability to proteolytic degradation, and transport [beyond] the endoplasmic reticulum membrane) requires progressively longer lengths of the polypeptide chain” (27). In other words, the basic assumption of the fusion methods of topographical analysis does not always hold. These fusion-type experiments with PS (23, 24), therefore, do not warrant the high degree of confidence that has been attributed to them.
A third type of evidence (24) deals with the immunofluorescent labeling of the NH2-terminal domain of PS-1 inside PS-1-transfected Chinese hamster ovary cells that were permeabilized with streptolysin O. (Streptolysin O treatment renders the plasma membranes of cells permeable to antibodies whereas the e.r. membranes remain impermeable to them). The immunolabeling of the NH2-terminal domain of PS-1 in the streptolysin O-treated cells therefore suggested that this domain normally was located on the cytoplasmic side of the e.r. membrane, consistent with either the 6- or 8-TM spanning topography for PS-1 (Fig. 3B). There is, however, a possible alternative explanation for these findings, as follows. The integration of a multi-TM spanning protein like PS into the e.r. membrane is a cotranslational process that most likely occurs in a stepwise fashion (28, 29). As successive portions of a protein chain are synthesized starting at the NH2 terminus and are extruded from the membrane-bound ribosome over a period of several minutes, the successive hydrophobic domains are probably intercalated and folded into the membrane one after the other. It has been proposed, however, that, for the 7-TM spanning proteins (Type IIIb proteins, ref. 29), it is the second, and not the first, hydrophobic stretch that serves as part of the signal sequence to initiate the process and that the NH2 terminus and the first TM stretch are only translocated across the membrane in their final orientation at a considerably later time. In such circumstances, therefore, inside cells fixed at any one time, the e.r. membranes may contain significant amounts of as yet only partially integrated PS-1 molecules with their NH2 termini having not yet been translocated across the e.r. membrane, and therefore these termini are transiently accessible to immunolabeling on the cytoplasmic face of the membrane. By contrast to such streptolysin O-mediated intracellular immunolabeling experiments (24), in the studies reported herein with live intact cells, the immunolabeling was confined to the plasma membrane, where only those PS molecules that were fully integrated into the membrane were placed under observation.
In summary, we believe that our immunofluorescence evidence supporting the 7-TM spanning topography of the PS proteins carries greater weight than the evidence supporting either the 6- or 8-TM spanning topography.
The 7-TM spanning topography of the PS proteins places them among the very large family of 7-TM spanning, often G protein-coupled, receptors (for a review, see ref. 30), of which the bacterial and animal rhodopsins, and the β-adrenergic receptor, are well known members. Neither PS-1 nor PS-2, however, shows any significant amino acid sequence homologies with any of the other established family members. Furthermore, the PS proteins have no known oligosaccharide linkages to help assign their topography. This is why the present studies were essential to establish the 7-TM spanning topography experimentally.
Finally, a comment regarding the point mutational changes in the PS proteins (mainly in PS-1) that have been found with early onset familial AD families (for a recent review, see ref. 26). Approximately half of these changes arise within the several TM stretches of PS-1, and most of the remainder occur within the large hydrophilic loop between TM VI and TM VII in the 7-TM spanning topography. None has been found so far in the NH2-terminal domain. The sites within the large hydrophilic loop, which we have located on the exoplasmic face of the plasma membrane (Figs. 2 and 3B), therefore may be critically involved in the specific transcellular binding of either of the PS proteins to its cognate ligand, β-APP (11); the individual mutational changes may each alter either that binding or its consequences (4) and thereby serve to accelerate the onset of the disease.
Acknowledgments
This work was supported by the National Institutes of Health Grant 2RO1-NS27850 to N.N.D.
ABBREVIATIONS
- PS
presenilin
- AD
Alzheimer disease
- β-APP
β-amyloid precursor protein
- TM
transmembrane
- e.r.
endoplasmic reticulum
References
- 1.Sherrington R, Rogaev E I, Liang Y, Rogaeva E A, Levesque G, et al. Nature (London) 1995;375:754–760. [Google Scholar]
- 2.Levy-Lahad E, Wijsman E M, Nemens E, Andertson L, Goddard K A, Weber J L, Bird T D, Schellenberg G D. Science. 1995;269:970–973. doi: 10.1126/science.7638621. [DOI] [PubMed] [Google Scholar]
- 3.Rogaev E I, Sherrington R, Rogaeva E A, Levesque G, Ikeda M, et al. Nature (London) 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 4.Dewji N N, Singer S J. Science. 1996;271:159–160. doi: 10.1126/science.271.5246.159. [DOI] [PubMed] [Google Scholar]
- 5.Cagan R L, Kramer H, Hart A C, Zipursky S L. Cell. 1992;69:393–399. doi: 10.1016/0092-8674(92)90442-f. [DOI] [PubMed] [Google Scholar]
- 6.Levitan D, Greenwald I. Nature (London) 1995;377:351–354. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- 7.Selkoe D J. Annu Rev Cell Biol. 1994;10:373–403. doi: 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- 8.Singer S J. Science. 1992;255:1671–1677. doi: 10.1126/science.1313187. [DOI] [PubMed] [Google Scholar]
- 9.Bailey C H, Chen M, Keller F, Kandel E. Science. 1992;256:645–647. doi: 10.1126/science.1585177. [DOI] [PubMed] [Google Scholar]
- 10.Glenner G, Wong C. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 11.Dewji N N, Singer S J. Proc Natl Acad Sci USA. 1996;93:12575–12580. doi: 10.1073/pnas.93.22.12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dewji N N, Singer S J. Proc Natl Acad Sci USA. 1997;94:9926–9931. doi: 10.1073/pnas.94.18.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kovacs D M, Fausett H J, Page K J, Kim T-W, Moir R D, Merriam D E, Hollister R D, Hallmark O G, Mancini R, Felsenstein K M, Hyman B T, Tanzi R E, Wasco W. Nat Med. 1996;2:224–227. doi: 10.1038/nm0296-224. [DOI] [PubMed] [Google Scholar]
- 14.Cook D G, Sung J C, Golde T E, Felsenstein K M, Wojczyk B S, Tanzi R E, Trojanowski J Q, Lee V M-Y, Doms R W. Proc Natl Acad Sci USA. 1996;93:9223–9228. doi: 10.1073/pnas.93.17.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thinakaran G, Borchelt D R, Lee M K, Slunt H H, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey A I, Gandy S E, Jenkins N A, Copeland N G, Price D L, Sisodia S S. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 16.De Strooper B, Beullens M, Contreras B, Levesque L, Craessaerts K, Cordell B, Moechars D, Bollen M, Fraser P E, St. George-Hyslop P H, Van Leuven F. J Biol Chem. 1997;272:3590–3598. doi: 10.1074/jbc.272.6.3590. [DOI] [PubMed] [Google Scholar]
- 17.Kim T-W, Pettingill W H, Hallmark O G, Moir R D, Wasco W, Tanzi R E. J Biol Chem. 1997;272:11006–11010. doi: 10.1074/jbc.272.17.11006. [DOI] [PubMed] [Google Scholar]
- 18.Podlisny M B, Citron M, Amarante P, Sherrington R, Xia W, Zhang J, Diehl T, Levesque G, Fraser P E, Haass C, Koo E H M, Seubert P, St. George-Hyslop P H, Teplow D B, Selkoe D J. Neurobiol Dis. 1997;3:325–337. doi: 10.1006/nbdi.1997.0129. [DOI] [PubMed] [Google Scholar]
- 19.Weidemann A, Paliga K, Durrwang U, Czech C, Evin G, Masters C L, Beyreuther K. Nat Med. 1997;3:328–332. doi: 10.1038/nm0397-328. [DOI] [PubMed] [Google Scholar]
- 20.Xia W, Zhang J, Koo E H, Selkoe D J. Proc Natl Acad Sci USA. 1997;94:8208–8213. doi: 10.1073/pnas.94.15.8208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dewji N N, Do C, Singer S J. Proc Natl Acad Sci USA. 1997;94:14031–14036. doi: 10.1073/pnas.94.25.14031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kyte J, Doolittle R F. J Mol Biol. 1982;154:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 23.Lehmann S, Chiesa R, Harris D A. J Biol Chem. 1997;272:12047–12051. doi: 10.1074/jbc.272.18.12047. [DOI] [PubMed] [Google Scholar]
- 24.Doan A, Thinakaran G, Borchett D R, Slunt H H, Ratovitsky T, Podlisney M, Selkoe D J, Seeger M, Gandy S E, Price D L, Sisodia S S. Neuron. 1996;17:1023–1030. doi: 10.1016/s0896-6273(00)80232-9. [DOI] [PubMed] [Google Scholar]
- 25.Wang H-Y, Lipfert L, Malbon C C, Bahouth S. J Biol Chem. 1989;264:14424–14431. [PubMed] [Google Scholar]
- 26.Haass C. Neuron. 1977;18:687–690. doi: 10.1016/s0896-6273(00)80309-8. [DOI] [PubMed] [Google Scholar]
- 27.Heymann J A W, Subramaniam S. Proc Natl Acad Sci USA. 1997;94:4966–4971. doi: 10.1073/pnas.94.10.4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blobel G. Proc Natl Acad Sci USA. 1980;77:1496–1500. doi: 10.1073/pnas.77.3.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singer S J. Annu Rev Cell Biol. 1990;6:247–296. doi: 10.1146/annurev.cb.06.110190.001335. [DOI] [PubMed] [Google Scholar]
- 30.Spiegel A M. Annu Rev Physiol. 1996;58:143–170. doi: 10.1146/annurev.ph.58.030196.001043. [DOI] [PubMed] [Google Scholar]