

Abstract
Statistical inference for mechanistic models of partially observed dynamic systems is an active area of research. Most existing inference methods place substantial restrictions upon the form of models that can be fitted and hence upon the nature of the scientific hypotheses that can be entertained and the data that can be used to evaluate them. In contrast, the so-called plug-and-play methods require only simulations from a model and are thus free of such restrictions. We show the utility of the plug-and-play approach in the context of an investigation of measles transmission dynamics. Our novel methodology enables us to ask and answer questions that previous analyses have been unable to address. Specifically, we demonstrate that plug-and-play methods permit the development of a modelling and inference framework applicable to data from both large and small populations. We thereby obtain novel insights into the nature of heterogeneity in mixing and comment on the importance of including extra-demographic stochasticity as a means of dealing with environmental stochasticity and model misspecification. Our approach is readily applicable to many other epidemiological and ecological systems.
Keywords: mechanistic model, iterated filtering, sequential Monte Carlo, measles, state-space model
1. Introduction
The ability to forecast, understand and control the spread of infectious diseases increasingly depends on the capacity to formulate and test mathematical models capturing key mechanisms. Accordingly, there has been a great deal of recent interest in techniques that allow one to fit mechanistic models to time-series data (Gibson & Renshaw 1998; Finkenstädt & Grenfell 2000; Bjørnstad & Grenfell 2001; Gibson & Renshaw 2001; Grenfell et al. 2001, 2002; Roberts & Stramer 2001; Bjørnstad et al. 2002; Finkenstädt et al. 2002; Grenfell et al. 2002; Keeling & Rohani 2002; Clark & Bjørnstad 2004; Koelle & Pascual 2004; Streftaris & Gibson 2004; Beskos et al. 2006; Cook et al. 2007; Cauchemez & Ferguson 2008; Cauchemez et al. 2008; Ferrari et al. 2008; Keeling & Ross 2008). Recent advances in statistical algorithms and computational hardware have made it possible to tailor statistical methodology directly to questions of scientific interest and have expanded the range of models that can be confronted with data. However, the nonlinear stochastic dynamical models arising in the study of infectious disease dynamics have proved relatively recalcitrant. Even the simplest useful models of disease transmission are highly nonlinear, and stochasticity cannot be ignored even in the most predictable of disease systems. Complicating the picture further is the ubiquitous presence of non-stationarity, measurement error and unobserved (hidden, or latent) variables. Finally, models of interest are typically formulated in continuous time, whereas data are sampled at discrete and sometimes irregular intervals. The large body of methodological literature devoted to the topic of inference for these partially observed stochastic dynamic systems is testament to its position as an important and unresolved challenge at the interface of the fields of ecology, epidemiology, mathematics and statistics.
As general approaches, all of the methods mentioned earlier face severe limitations. Some must modify the model, discretizing time to the scale of observations (Finkenstädt & Grenfell 2000; Grenfell et al. 2001, 2002; Bjørnstad et al. 2002; Finkenstädt et al. 2002; Clark & Bjørnstad 2004; Koelle & Pascual 2004). They rely upon fortuitous congruence between some time scale of the process under study (e.g. infection generation time) and that of the sampling interval. These methods are computationally cheap, but can lead to severely biased conclusions (Glass et al. 2003). Others rely on approximations that are valid only in restricted circumstances. For example, Cauchemez & Ferguson (2008) adopt a diffusion approximation to enable successful implementation of a Markov chain Monte Carlo approach; this approximation breaks down in small populations. On the other hand, straightforward Bayesian approaches have been demonstrated for small populations with purely demographic stochasticity (Gibson & Renshaw 1998, 2001); however, these methods break down for large populations (Roberts & Stramer 2001; Beskos et al. 2006) or when environmental stochasticity is non-negligible (Bretó et al. 2009). Finally, exact methods based on numerical solution of the so-called master equations (Keeling & Ross 2008) are available, but limited to low-dimensional models and small populations. In summary, each of these approaches makes demands upon the form of the model and/or the nature of the data and thus necessitates scientifically irrelevant or even inappropriate assumptions that can interfere with the hypotheses of interest.
In this paper, we use a relatively new approach with none of the aforementioned limitations (Ionides et al. 2006, 2007, 2009; King et al. 2008; Bretó et al. 2009). In particular, the method works on continuous time models, in large or small populations, with demographic or environmental stochasticity or both. It has the important advantage of operation based only on simulation from the dynamic model, without a need for explicit expressions for transition probabilities. The latter property has been called plug-and-play (Bretó et al. 2009). Plug-and-play inference methodology provides a powerful tool to enable carrying out data analysis via flexible, scientifically motivated classes of models. In particular, plug-and-play is extremely useful when one wishes to entertain multiple working hypotheses translated into multiple mechanistic models. Two properties analogous to plug-and-play have been considered in the contexts of optimization and complex system theory.
In optimization theory, methods requiring only evaluation of the objective function have been called gradient-free (Spall 2003). In such methods, computer code to evaluate the objective function can be plugged into the optimizer. This is particularly useful in situations in which the objective function is a complex, and possibly stochastic, function for which the analytic calculation of derivatives is difficult or impossible (Kocsis & Szepesvári 2006).
In the analysis of complex systems, methods that are based solely on simulations from computer codes representing a model have been called equation-free (Kevrekidis et al. 2004). A typical goal of such investigations is to determine the relationship between macroscopic phenomena (e.g. phase transitions) and microscopic phenomena (e.g. molecular interactions).
These analogies suggest that plug-and-play is a subject whose importance extends beyond our present topic of inference for partially observed stochastic dynamic systems. The conceptual commonality is that all these methodologies can be applied to models specified as ‘black-box’ computer codes. This enables flexible model development and therefore avoids confounding methodological issues with scientific hypotheses. It also facilitates both the comparison of alternative explanations and the transfer of methodological expertise between different systems. The price of plug-and-play is primarily in terms of computational effort: when properties of a model are available in closed form, computationally more efficient methods typically exist. As computational capabilities continue to grow, the ease with which new scientific questions can be asked and answered via plug-and-play methodology will make such techniques increasingly attractive.
We illustrate our new approach via a case study of measles transmission dynamics. A focus on this well-studied disease allows us to compare our conclusions with those derived previously by other means. Further, we demonstrate that our analysis leads to fresh insights not readily available via previous analyses. Measles has occupied a central place in mathematical epidemiology owing to its relatively straightforward diagnosis, the lifelong immunity following infection and the availability of high-quality case report data. Models that assume mass-action transmission on the scale of communities do remarkably well at reproducing the disease dynamics, particularly in large populations (Bailey 1955; Bartlett 1960; Fine & Clarkson 1982), although recent work has suggested that deviations from mass action are detectable in time-series data (Bjørnstad et al. 2002). Moreover, models incorporating purely demographic stochasticity have been deemed adequate to reproduce observed dynamical patterns. We revisit these questions by fitting a family of models that incorporate deviations from the mass-action assumption together with both environmental and demographic stochasticity. We fit these models to time-series data from both large and small populations. A comparison of results across community sizes indicates that there is little evidence in the data for deviations from mass-action transmission, at least as it has been modelled previously. On the other hand, our results do suggest a role for heterogeneity in transmission as a function of population size. With respect to the nature of stochasticity in this system, our results indicate a clear role for extra-demographic variability. Indeed, they suggest that failure to allow for extra-demographic variability may lead to severe biases in estimates of key parameters. These insights are possible because of the flexibility of the inference machinery, which also makes our approach more readily applicable to other, less exhaustively analysed, systems (e.g. King et al. 2008).
2. Methods: inference machinery, data and models
Consider a time series y1:N ≡ y1, … ,yN, consisting of N observations made at times t1, … ,tN. A stochastic model for y1:N implies a joint density f(y1:N|θ) given a vector of unknown parameters θ, where θ is in ℝnθ. Corresponding conditional densities for yn given y1:n–1 are written as 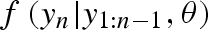 . Via the factorization
. Via the factorization 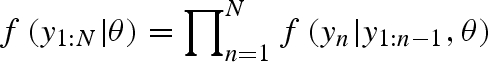 , the log-likelihood function is given by
, the log-likelihood function is given by 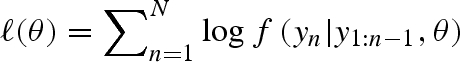 . Sequential Monte Carlo (Doucet et al. 2001; Arulampalam et al. 2002; Cappé et al. 2007) provides a standard method to obtain the log likelihood for stochastic dynamic systems. In our terminology, stochastic dynamic system is synonymous with partially observed Markov process (Ionides et al. 2006; Bretó et al. 2009); in the statistical literature, these systems are known as state-space models (Shumway & Stoffer 2006). Likelihood evaluation via sequential Monte Carlo has the feature that only simulations of sample paths are required; one need not have explicit forms for transition probabilities. That is, it has the plug-and-play property. Both Bayesian (Liu & West 2001; Toni et al. 2009) and frequentist (Ionides et al. 2006; Poyiadjis et al. 2006) approaches to plug-and-play likelihood-based inference via sequential Monte Carlo have been proposed. We adopt the maximum-likelihood approach of Ionides et al. (2006, 2009), which enables statistically efficient inference for general nonlinear stochastic dynamical systems. In contrast, the approximations developed by Liu & West (2001) are not statistically efficient, and lead to additional bias and variance in the resulting parameter estimates (Storvik 2002; Newman et al. 2008). The popularity of the artificial parameter evolution method of Liu & West (2001) may be due more to the convenience of its plug-and-play property than to its statistical properties. Various refinements and variations in the vein of the Bayesian methodology of Liu & West (2001) have been studied (Polson et al. 2008 and references therein); however, these typically do not posses the plug-and-play property.
. Sequential Monte Carlo (Doucet et al. 2001; Arulampalam et al. 2002; Cappé et al. 2007) provides a standard method to obtain the log likelihood for stochastic dynamic systems. In our terminology, stochastic dynamic system is synonymous with partially observed Markov process (Ionides et al. 2006; Bretó et al. 2009); in the statistical literature, these systems are known as state-space models (Shumway & Stoffer 2006). Likelihood evaluation via sequential Monte Carlo has the feature that only simulations of sample paths are required; one need not have explicit forms for transition probabilities. That is, it has the plug-and-play property. Both Bayesian (Liu & West 2001; Toni et al. 2009) and frequentist (Ionides et al. 2006; Poyiadjis et al. 2006) approaches to plug-and-play likelihood-based inference via sequential Monte Carlo have been proposed. We adopt the maximum-likelihood approach of Ionides et al. (2006, 2009), which enables statistically efficient inference for general nonlinear stochastic dynamical systems. In contrast, the approximations developed by Liu & West (2001) are not statistically efficient, and lead to additional bias and variance in the resulting parameter estimates (Storvik 2002; Newman et al. 2008). The popularity of the artificial parameter evolution method of Liu & West (2001) may be due more to the convenience of its plug-and-play property than to its statistical properties. Various refinements and variations in the vein of the Bayesian methodology of Liu & West (2001) have been studied (Polson et al. 2008 and references therein); however, these typically do not posses the plug-and-play property.
We used the implementation of iterated filtering contained in the software package Pomp (King et al. 2009) written for the R statistical computing environment (R Development Core Team 2006). This implementation follows the algorithm described by King et al. (2008, supplementary information therein). Heuristics and diagnostics, together with a discussion of computational issues, can be found in Ionides et al. (2006); a theoretical treatment is given by Ionides et al. (2009).
For complex dynamical systems, not all questions that one might wish to ask will be clearly answered by the available data. Typically, a question posed of the dynamic system is formalized as a hypothesis about a combination of parameters in a model for the system. If the likelihood function does not discriminate well between different values of some parameter combination, this parameter combination is said to be poorly identified. In such a case, one might proceed by making additional assumptions: adding constraints typically improves parameter identifiability at the cost of potential bias in the estimates of other parameters. In §3, we focus on questions the data are able to answer without further assumptions. A basic tool for this is the profile likelihood function, in which one parameter is varied over a range of values, while the likelihood is maximized over all remaining parameters (Barndorff-Nielsen & Cox 1994; Hilborn & Mangel 1997). In practice, we work with a smoothed estimate of the likelihood surface calculated via Monte Carlo (Ionides 2005).
The data we analyse consist of weekly measles case reports on 20 representative communities with population sizes ranging from 2000 to 3 000 000. The towns and cities were selected from 954 urban locations for which case reports have been compiled (Grenfell & Bolker 1998); specifically, we chose the largest 10 cities by population and sampled 10 more towns at random. Figure 1 shows the case reports and annual birth rates for two of these: London and Hastings. These data and their limitations have been discussed previously (Fine & Clarkson 1982; Bjørnstad et al. 2002). In particular, although measles became a notifiable disease in England and Wales in 1940, we follow Fine & Clarkson (1982) by starting our analysis in 1950 to avoid the disruptions caused by the second world war and the transition period following the introduction of the National Health Service in 1948. From 1950 onwards, it is believed that reporting was carried out fairly consistently, with between half and two-thirds of all cases being counted.
Figure 1.

Weekly reported measles cases (solid line) and annual birth rate (dotted line) for (a) London and (b) Hastings.
A basic susceptible–exposed–infectious–recovered (SEIR) model for measles divides the population into those individuals susceptible to infection, exposed (i.e. infected but not yet infectious), infectious and removed (i.e. quarantined or recovered and subsequently immune). The model is specified by describing the rates at which individuals move between compartments (Jacquez 1996; Matis & Kiffe 2000); figure 2 shows the transitions for the SEIR model of measles. A diagram such as figure 2 can be unambiguously interpreted as a deterministic system, a system with demographic stochasticity or a system with both demographic and extra-demographic stochasticity (Bretó et al. 2009). We give the equations for each of these interpretations in appendix A. Here, we will employ the stochastic interpretation, with both demographic and extra-demographic stochasticity, described in box 1 (in appendix A). The transition rates in a model such as figure 2 may, in general, depend on the state of the system and/or covariates. We specify the force of infection as
Figure 2.

Flow diagram for measles. The population is divided into four compartments: S, susceptible; E, exposed and infected but not yet infectious; I, infectious; R, recovered and immune. Births enter S at the rate μBS(t), and all individuals have a mortality rate μSD = μED = μID = μRD = m. The case reports, C, count infected individuals with probability ρ. Since diagnosed cases are treated with bed-rest and hence removed, infections are counted upon transition to R.
Box 1.
Euler scheme for a numerical solution of a Markov chain with stochastic rates. Here, (α, β) is the gamma distribution with shape parameter α and scale parameter is β, with corresponding mean αβ and variance αβ2. In the case σij = 0, Gamma(δ/σij2, σij2) is defined to take the non-random value δ. Further details on the gamma, multinomial and Poisson distributions can be found in introductory texts on probability and statistics (Casella & Berger 1990). In steps 5 and 7, Ri counts the number of individuals who remain in compartment i during the current Euler increment.
Set the initial value X(0) and time interval (0,T)
Set time increment δ; define tn = nδ and N = T/δ.
FOR n = 0 to N − 1
Generate independent noise increments, δΓij ∼ Gamma(δ/σij2,σij2).
- For i in 1, … ,k, generate process increments
where pij = pij({μij(tn, X(tn))},{δΓij}) is given by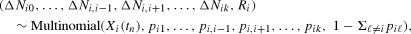
with μij = μij(t, x), defined in equation (A 2).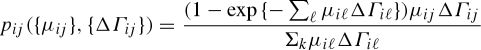
- Generate process increments (δN∅1, … ,δN∅k) via
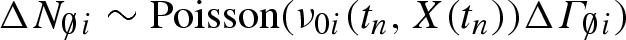
Set Xi(tn+1) = Ri + σj≠iδNji
END FOR
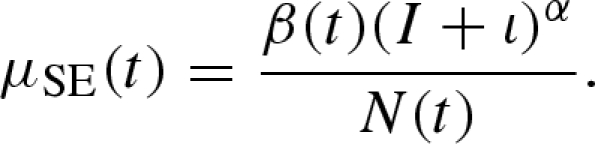 |
2.1 |
Here β(t) is the transmission rate; ι the mean number of infectives visiting the population at any given time; α a mixing parameter, with α = 1 corresponding to homogeneous mixing (Liu et al. 1986; Bjørnstad et al. 2002), and N(t) the population size, treated as known via interpolation from census data. Since transmission rates are closely linked to contact rates among children, which are higher during school terms (Schenzle 1984; Keeling & Grenfell 2002; Bauch & Earn 2003; Conlan & Grenfell 2007), we assume that β(t) reflects the pattern of British school terms and holidays. Specifically, we take
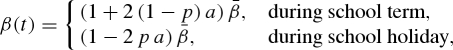 |
2.2 |
where p = 0.759 is the proportion of the year taken up by the school term, β̄ the mean transmission rate and a the relative effect of school holidays on transmission. For ease of interpretation, it is sometimes convenient to reparameterize β̄ in terms of the annual average basic reproductive ratio, R0, defined as the expected number of secondary infections engendered by an infective introduced into a fully susceptible population. When the duration of infection is much shorter than life expectancy, R0 ≈ β̄/μIR; here we employ a modification of this formula, given in appendix A.
A novel feature of our model is that we add white noise with intensity σSE to the transmission process. Some empirical support for the choice of white (i.e. temporally uncorrelated) noise is given by Lande et al. (2003). This variability in the rates allows the possibility of over-dispersion (McCullagh & Nelder 1989; Bretó et al. 2009). It is discussed at more length, together with a full specification of all the model equations, in appendix A.
The other transition rates are specified as follows: μEI is the rate at which exposed individuals become infectious, thus μEI−1 is the mean latent period; μIR is the recovery rate; μSD = μED = μID = μRD = m denotes a constant per-capita death rate, ignoring the negligible effect of disease-induced mortality; μBS(t) is the per-capita rate of recruitment of susceptibles, depending on the birth rate b(t), which is assumed to be known via interpolation from birth records. Births enter the susceptible class with a delay τ corresponding to the age at which children enter the high-risk school-age population. We also consider a cohort-entry effect that reflects the fact that a large cohort of first-year students—the majority of them serologically naive—enters the schools each autumn (Schenzle 1984; He 2006). Specifically, a fraction c of recruits into the susceptible class enter on the school admission day, and the remaining fraction (1–c) enter the susceptible class continuously. The cohort effect provides a parsimonious and mechanistically plausible alternative to previous suggestions that the transmission rate β(t) may increase following the start of the academic year (Fine & Clarkson 1982; Grenfell et al. 2002). The reporting process is taken to be on over-dispersed binomial, with reporting rate ρ and overdispersion parameter ψ (appendix A, equation (A 6)). Quantities appearing in the model are summarized in table 1.
Table 1.
List of symbols used in the paper.
| symbol | description | units | reference |
|---|---|---|---|
| μij | per-capita rate of transition from compartment i to j | yr−1 | figure 2, box 1 |
| σij | white-noise intensity on i → j transition rate | yr1/2 | box 1 |
| LP | latent period | day | equation (A 7) |
| IP | infectious period | day | equation (A 7) |
| ι | mean number of visiting infectives | — | equation (2.1) |
| α | mixing exponent | — | equation (2.1) |
| β(t) | transmission rate | yr−1 | equations (2.1) and (2.2) |
| N(t) | population size | — | equation 2.1 |
| a | amplitude of seasonality | — | equation 2.2 |
| b(t) | birth rate | yr−1 | equation (A 5) |
| c | cohort entry fraction | — | equation (A 5) |
| τ | recruitment delay | yr | equation (A 5) |
| m | mortality rate μjD for j ∈ {S, E, I, R} | yr−1 | figure 2 |
| ρ | reporting probability | — | equation (A 6) |
| ψ | reporting overdispersion | — | equation (A 6) |
| R0 | basic reproductive ratio | — | — |
3. Results
Maximum-likelihood parameter estimates for our sample of 20 communities are presented in table 2. These estimates are for the model in §2 with no constraints placed on the parameters. The variation in the parameter estimates between populations has two sources: it may represent differences in the population dynamics of measles between communities, or it may be due to weak identifiability of some combinations of parameters. We disentangle these two explanations via a detailed investigation of two of them, London and Hastings. However, table 2 does reveal some consistent patterns that are robust across the population samples and are therefore not strongly affected by identifiability issues. These patterns are valuable for indicating model features about which the data under investigation are informative without the incorporation of additional assumptions based on prior analyses.
Table 2.
Point estimates for 20 representative communities. N is 1950 population in thousands; LP and IP are the latent and infectious periods, respectively, in days, calculated from μEI and μIR as described in equation (A 7) of appendix A; other parameters and their units are as specified in table 1; the parameters τ = 4 yr and m = 0.02 yr−1 were not estimated. Maximized log-likelihood values are presented in the electronic supplementary material. The bottom row gives the Spearman rank correlation of the columns with N with * denoting significance at the 5 per cent level, ** the 1 per cent level and *** the 0.1 per cent level.
| N | α | σSE | R0 | LP | IP | a | ι | c | ρ | ψ | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Halesworth | 2 | 0.95 | 0.075 | 33 | 7.87 | 2.28 | 0.38 | 0.01 | 0.55 | 0.75 | 0.64 | |
| Lees | 4 | 0.97 | 0.060 | 28 | 7.28 | 1.93 | 0.16 | 0.03 | 0.62 | 0.60 | 0.69 | |
| Mold | 6 | 1.04 | 0.054 | 21 | 5.93 | 1.78 | 0.27 | 0.01 | 0.44 | 0.13 | 2.87 | |
| Dalton | 11 | 0.99 | 0.078 | 28 | 5.48 | 1.98 | 0.20 | 0.04 | 0.42 | 0.46 | 0.82 | |
| Oswestry | 11 | 1.04 | 0.070 | 53 | 10.29 | 2.72 | 0.34 | 0.03 | 0.26 | 0.63 | 0.48 | |
| Northwich | 18 | 0.93 | 0.069 | 23 | 8.07 | 2.55 | 0.30 | 0.05 | 0.39 | 0.78 | 0.40 | |
| Bedwellty | 29 | 0.94 | 0.061 | 25 | 6.82 | 3.03 | 0.16 | 0.04 | 0.35 | 0.31 | 0.95 | |
| Consett | 39 | 1.01 | 0.071 | 36 | 9.07 | 2.66 | 0.20 | 0.07 | 0.31 | 0.65 | 0.41 | |
| Hastings | 66 | 1.00 | 0.096 | 34 | 7.00 | 5.44 | 0.30 | 0.19 | 0.33 | 0.70 | 0.40 | |
| Cardiff | 245 | 1.00 | 0.054 | 34 | 9.86 | 3.09 | 0.22 | 0.14 | 0.27 | 0.60 | 0.27 | |
| Bradford | 294 | 1.00 | 0.053 | 36 | 10.80 | 3.22 | 0.25 | 0.27 | 0.30 | 0.60 | 0.19 | |
| Hull | 302 | 0.97 | 0.064 | 39 | 9.18 | 5.46 | 0.22 | 0.14 | 0.27 | 0.58 | 0.26 | |
| Nottingham | 307 | 0.98 | 0.038 | 23 | 5.72 | 3.69 | 0.16 | 0.17 | 0.34 | 0.61 | 0.26 | |
| Bristol | 443 | 1.01 | 0.039 | 27 | 6.19 | 4.94 | 0.20 | 0.44 | 0.34 | 0.63 | 0.20 | |
| Leeds | 510 | 1.00 | 0.078 | 48 | 9.48 | 10.92 | 0.27 | 1.25 | 0.59 | 0.67 | 0.17 | |
| Sheffield | 515 | 1.02 | 0.043 | 33 | 7.23 | 6.38 | 0.31 | 0.85 | 0.23 | 0.65 | 0.17 | |
| Manchester | 704 | 0.97 | 0.055 | 33 | 11.11 | 6.94 | 0.29 | 0.59 | 0.36 | 0.55 | 0.16 | |
| Liverpool | 802 | 0.98 | 0.053 | 48 | 7.90 | 9.80 | 0.30 | 0.26 | 0.19 | 0.49 | 0.14 | |
| Birmingham | 1118 | 1.02 | 0.056 | 35 | 11.23 | 7.90 | 0.31 | 1.09 | 0.61 | 0.56 | 0.19 | |
| London | 3390 | 0.98 | 0.088 | 57 | 13.14 | 12.51 | 0.55 | 2.90 | 0.56 | 0.49 | 0.12 | |
| r | 0.14 | −0.27 | 0.44 | 0.46* | 0.94*** | 0.26 | 0.94*** | −0.16 | −0.16 | −0.93*** |
In table 2, the homogeneity parameter α is consistently found to be close to 1. At first glance, this may suggest that there is little evidence for inhomogeneity of transmission in the data. Profile likelihood plots over α for London and Hastings are presented in figure 3. We see that the statistical uncertainty in the estimates for these two places is comparable to the geographic variability in the point estimates in table 2 (mean ± s.d. of 0.99 ± 0.03). This finding confirms the suggestion by Glass et al. (2003) that such exponents may be artefacts of the time discretization employed in earlier approaches (Bjørnstad et al. 2002; Grenfell et al. 2002). We can conclude that the phenomenon of inhomogeneous mixing—inasmuch as it plays a dynamic role—is best captured in some other way.
Figure 3.

Profile likelihood analysis of the mixing parameter, α, for (a) London and (b) Hastings. The solid black lines show the estimated profile log likelihood, derived from the Monte Carlo point estimates shown as circles. The dashed lines construct approximately 95 per cent CIs of (0.93, 1.03) for London and of (0.85, 1.03) for Hastings.
The imported infection parameter ι is well identified in these data. There is a strong log-linear relationship between ι and N, consistent with the earlier findings of Bjørnstad et al. (2002) and with the results obtained by Xia et al. (2004), who analysed a much more extensive dataset using a gravity model. The evidence for such a relationship is stronger here than in a previous empirical investigation (Finkenstädt et al. 2002, fig. 6).
The extra-demographic stochasticity parameter σSE is consistently found to be around 0.06 (0.063 ± 0.015). The profile likelihood plots in figure 4 also indicate that this parameter is fairly well estimable, and the data strongly argue against the choice σSE = 0. Figure 4 suggests that a role for extra-demographic stochasticity is not only strongly indicated for large cities such as London but also clearly evidenced in smaller populations for which one might expect demographic stochasticity to dominate. One interpretation of this is that models with purely demographic stochasticity (i.e. all discrete-population continuous-time dynamic models that have been previously fitted to disease data) fail to capture an extra-demographic source of variability that has an important dynamic role. A priori exclusion of the possibility of such additional variability can lead to spurious confidence in the conclusions of the analysis (McCullagh & Nelder 1989) and to biased parameter estimates. Figure 4 shows that, in the absence of extra-demographic stochasticity, estimates of the latent and infectious periods are substantially lower when one allows for such stochasticity. This appears to arise because short transition times lead to fewer individuals in compartments E and I on average and therefore to a larger dynamic role for demographic stochasticity. In other words, the model compensates for the lack of extra-demographic stochasticity by attempting to increase the intensity of demographic stochasticity.
Figure 4.

Profile analysis of extrademographic variability, σSE, for (a) London and (b) Hastings. The solid black curve shows the estimated profile log likelihood, derived from the Monte Carlo point estimates shown as circles. The dashed lines construct approximately 95 per cent CIs for σSE. Corresponding profile estimates of the mean IP and mean LP are shown with dotted and dashed curves, respectively, with units given on the right-hand axis. The figure is calculated with the constraint α = 1. Dotted lines, IP; dashed lines, LP.
Each parameter of the model has an interpretation in terms of the basic biology of transmission and infection at the level of individual hosts. To the extent that the model is a faithful description of the population-level processes, one expects that parameter estimates should agree closely with estimates based on individual-level observations. When discrepancies occur, they are evidence for model misspecification or oversimplification. Strikingly, table 2 shows a strong log-linear relationship between population size and infectious period and, to a lesser extent, latent period. Since there is no evidence that the natural history of measles varies with population size, this relationship is indicative of such oversimplification. Figure 1 suggests that this simple model fails to account adequately for heterogeneity in transmission. Specifically, the model calls for short infectious periods and high transmissibility β to reproduce the explosive epidemics and interepidemic fadeouts typical of small communities. The data from large populations, in contrast, represent the aggregate of many local epidemics, in each of the many schools and local communities contained in a large conurbation. In these aggregated data, fadeouts are rare and the epidemic curve is generally shallower. The oversimple model can reproduce these features only by extending the infectious period and reducing transmissibility. Moreover, while the infectious period estimated in the small populations (2–4 days) is consistent with evidence from household studies (Hope Simpson 1948, 1952; Bailey 1956), the longer infectious periods estimated in the large populations are not. Previous studies have noted that deterministic versions of the mass-action model analysed here do a remarkably good job of qualitatively reproducing the dynamics of measles in large communities (Earn et al. 2000). Our analysis shows, however, that when both continuous time dynamics and stochasticity are taken into account, data from small populations afford a less-distorted view of the disease dynamics, at least when viewed through the lens of relatively simple models.
The basic reproductive ratio of measles is conventionally held to lie between 14 and 18 (Anderson & May 1991). The values of R0 in table 2 are variable but consistently high relative to previous estimates (mean ± s.d. of 34.7 ± 10.1 over 20 communities). Profile likelihoods over R0 for London and Hastings (see the electronic supplementary material) yield approximately 95 per cent confidence intervals (CIs) of (37, 60) and (28, 74), respectively. Earlier empirical work on transmission dynamics (Bjørnstad et al. 2002) also led to a relatively high value of R0 = 29.9 for London. Bjørnstad et al. (2002) were cautious about the interpretation of this result because of concerns about the effects of the discrete-time methods employed. We can more confidently say that these higher values of R0 are a genuine feature of the continuous-time dynamics, at least under the assumption of homogeneous mixing and exponentially distributed latent and infectious periods. Changing the distribution of the latter to a more general gamma distribution (Keeling & Grenfell 1997; Lloyd 2001b; Wearing et al. 2005) decreases the estimated 95 per cent CI of R0 to (25, 41) for London (see the electronic supplementary material, figure S6). Though this is a substantial quantitative change, it does not alter the qualitative conclusion that homogeneous population models require high values of R0 in order to be consistent with the data.
4. Discussion and conclusion
A major goal of biomathematical modelling is to seek conceptual simplicity in the face of biological complexity. In general, the questions of interest should dictate the form and complexity of the model used to address them. In the present context in particular, the appropriate degree of aggregation over population inhomogeneities (such as spatial, socio-economic, genetic and age variation) may depend on the goals of the investigation (Levin et al. 1997; May 2004). For measles, analyses based on models of homogeneous mixing populations have proved useful in the study of seasonality (Fine & Clarkson 1982; Bjørnstad et al. 2002; Ferrari et al. 2008) and effects of climate drivers (Lima 2009). Metapopulations of weakly coupled homogeneously mixing populations are central to current understanding of spatio-temporal disease dynamics (Grenfell et al. 2001; Xia et al. 2004). In addition, such models can yield insight into the fundamental predictability of disease outbreaks (Stone et al. 2007) and have been found adequate to predict the effects of variation in birth rate and some dynamical features associated with the institution of vaccination programmes (Earn et al. 2000). Homogeneous mixing models were also the basis of early work on local extinctions and vaccination strategies (Bartlett 1957), though age structure and infectious period distribution may have substantial roles to play in these questions (Schenzle 1984; Keeling & Grenfell 1997; Lloyd 2001b; Conlan et al. submitted). In particular, estimates of the parameter R0 are known to be highly sensitive to assumptions on age structure (Wallinga et al. 2001). Interpretations of homogeneous mixing models must be made in the context of their limitations.
Clinical and household studies of infectious diseases yield information regarding the transmission and progression of infection on the scale of individuals or families (Hope Simpson 1948, 1952; Bailey 1956). Such studies are complemented by data on disease prevalence or incidence on larger spatial scales. The recent development of likelihood-based approaches capable of dealing with the full spectrum of unavoidable complexities (Cauchemez & Ferguson 2008; Cauchemez et al. 2008; King et al. 2008)—unobserved variables, measurement error, process noise, nonlinearity, non-stationarity and covariates—means that we can now view a disease from the population point of view with something like the same clarity that we have for many years been able to view individual infections. Specifically, although we have long understood how to describe the population dynamics of infectious diseases using mathematical models, we have only recently gained the ability to fit these models to data using statistically sound methods. When biological quantities estimated in this way agree with those estimated from clinical or household studies, it can be interpreted as confirmation of the assumptions embodied in the model. Disagreement between the small- and large-scale points of view, however, raises interesting scientific questions. In the present study, we find broad agreement with previous studies concerning many of the model's parameters. On the other hand, we find significant departures with respect to three important quantities—R0, the infectious period and the intensity of extra-demographic stochasticity.
The quantity R0 is central in epidemiological theory because it has interpretations in terms of so many quantities of interest, including mean age of first infection, mean susceptible fraction, exponential-phase epidemic growth rate and vaccination coverage required for eradication. It is important to realize that the conjunction of these interpretations occurs only in the context of very simple models. Simple models necessarily lack flexibility. In reality, these interpretations diverge owing to heterogeneities in age, spatial location, host genetics, etc. It is therefore unrealistic to expect that estimates of R0 (or any other single quantity) derived from fitting a simple model to one sort of data should agree with estimates derived from other data sources. Rather, the key question should be: which biological interpretations are relevant in the context of the data used to inform the model? From a statistical point of view, this corresponds to the question: to what features of the data are the parameter estimates sensitive? Our estimate of the basic reproductive ratio, R0, like those of earlier studies focusing on aggregated case-count time series, is somewhat greater than estimates based on serological surveys and age-stratified incidence data. It is possible that this reflects the sensitivity of the time-series-derived estimates of R0 to peak incidence. Specifically, peak incidence strongly influences the estimates of both R0 and reporting rate ρ, but since ρ is well identified by other features of the data (namely, the long-run cumulative incidence), we suspect that the model requires a high R0 to match the observed peak epidemic case counts. Alternatively, the high R0 estimates may reflect the sensitivity of estimated R0 to early phase epidemic growth. If this is in fact the case, our estimates of R0 may more accurately reflect contact rates among the core group of school-age children than they do those of the population at large. In this case, the model is effectively extrapolating these rates to the adult population, about which these data have little direct information since so few adult cases occur. In contrast, from this point of view, the interpretation of R0 in terms of its definition as mean number of secondary infections in a fully susceptible population is hopelessly extrapolated. Likewise, the interpretation of R0 in terms of mean age of first infection is unjustified, since it necessarily depends on the age structure of transmission, which is not part of the model. Perhaps the most direct information pertaining to mean age at first infection in these data comes from the lag between changes in birth rate and their subsequent effects on incidence. This lag is explicitly captured in our model by the delay, τ, between birth and recruitment into the susceptible pool.
Since the comparatively high estimate of R0 does not appear to be a mere artefact of time discretization, the question remains as to why the population-level data suggest a more communicable disease than do the individual-level data. Existing estimates of R0 are sensitive to assumptions about the age structure of transmission that have not yet been fully resolved (Wallinga et al. 2001). Perhaps transmission in schools is relatively more effectual than within households. Alternatively, it may be that this discrepancy points to a need for a better description of infectious- and/or latent-period distribution, of the disease's age structure or both. In our results, the departure of the estimated latent and infectious periods from plausible values obtained from household studies grows with city size, and so the most likely explanation for this correlation is the spatial heterogeneity of transmission within towns and cities. The latter, we have shown, cannot usefully be captured via the simple device of an exponent in the transmission term. More detailed explorations of disaggregated data and/or models with explicit spatial structure have the potential to shed light on this question. Finally, the strong evidence in favour of extra-demographic stochasticity raises the question of precisely why such stochasticity aids in the explanation of the data. To what extent does this finding indicate the presence of genuine environmental stochasticity? To what extent does it indicate model misspecification? To address these questions, again, analysis based on more detailed models is called for. In the case of measles, there are sufficient data to entertain models featuring spatial inhomogeneity (Grenfell et al. 1995; Xia et al. 2004; Bjørnstad & Grenfell 2008), age structure inhomogeneity (Schenzle 1984; Bolker & Grenfell 1996; Keeling & Grenfell 1997) or both (Bolker & Grenfell 1995). Although it has been demonstrated that such models can do a good job of accounting for gross features of the data, there has been less emphasis on requiring these models to account for all features of the data.
Progress on inference methodology for parameter estimation from measles time series has emphasized SEIR-type models for homogeneous populations (Ellner et al. 1998; Bjørnstad et al. 2002; Cauchemez & Ferguson 2008; Keeling & Ross 2008). The study of age structure and spatial effects for measles has placed less emphasis on inferring parameters from data (one exception is Xia et al. 2004). This may be partly because of the additional difficulties of inference for such systems and partly because models based on homogeneous mixing do an impressive job of describing key dynamic features (Earn et al. 2000; Grenfell et al. 2002). Regardless of one's view of the importance of paying further attention to population inhomogeneities, there is a natural methodological question: how applicable are the techniques presented here for larger, more complex models? There is a computational price to pay for the convenience of plug-and-play statistical methods. For the results in table 2, one application of the plug-and-play-iterated filtering inference procedure (based on 50 iterations, with a Monte Carlo size of 104) implemented via the Pomp package in R (R Development Core Team 2006; King et al. 2009) took 5 h to run on a desktop machine. Computational effort scales roughly linearly with the number of parameters plus the number of state variables, so one can see that only a modest amount of additional structure could be included without hitting computational limitations. To increase computational efficiency, however, the iterated filtering algorithm can be implemented in a non-plug-and-play mode, in which the filter is either tailored to the particular model or takes advantage of available analytic properties of the transition densities. Such extensions, which would be required for much larger models, are a topic for future research. One can seek inspiration for future possibilities from numerical climate models, for which filtering operations (the computationally intensive step in the iterated filtering algorithm) have been carried out on systems with 5 × 106 state variables (Anderson & Collins 2007). Filtering in such high-dimensional situations requires the development and evaluation of appropriate approximations for the system under investigation.
In this article, we have carried out the first scientific investigation based on a new framework for continuous-time, discrete-state population dynamics with both demographic and extra-demographic noise (probabilistic and statistical properties of this model class were investigated by Bretó et al. (2009). Extra-demographic stochasticity (interpreted as noise in the rates of a discrete population Markov population model) is equivalent to the possibility of multiple individuals moving simultaneously between compartments (Bretó et al. 2009), and, as such, may be due to social events that affect the behaviour of many individuals (e.g. sporting events) or events that change disease transmissibility, such as variations in temperature and humidity. Variability in the rates gives the model additional flexibility that can also describe model misspecification. Analogously, when carrying out linear regression, it is customary to fit a line to data while understanding that the variation of the data around the line corresponds to unknown and unmodelled deterministic effects as well as to random fluctuations. For linear regression, one typically treats both these sources of uncertainty equally, and certainly all the usual standard errors and test statistics do not discriminate between them. We maintain that the same approach can be applied to dynamic models; in other words, the distinction between model misspecification and process stochasticity should be noted, though it will not usually affect the subsequent analysis.
The term extra-demographic stochasticity encompasses all sources of variability beyond the intrinsic demographic stochasticity that would be present in a homogeneous population. There are many circumstances in which such variability can be expected to be important. In particular, the variability of rates in our new framework offers an approach to modelling superspreading events (Lloyd-Smith et al. 2005). These events occur when variability between individuals, environmental effects or an interaction between the two results in a highly skewed distribution for the number of secondary cases caused by an index case. Superspreading has been documented in measles, but is of greater dynamic importance in other diseases such as severe acute respiratory syndrome (Lloyd-Smith et al. 2005). Conventional population models amenable to non-plug-and-play statistical analyses have been unable to include such effects readily. This is, therefore, one more example in which the flexibility of plug-and-play methodology holds the potential to encourage the development of scientifically appropriate models.
Acknowledgements
The authors thank Bryan Grenfell for valuable comments and for making the data available, Pejman Rohani for sharing his insight and Ottar Bjørnstad for useful conversations. We acknowledge the financial support of the National Science Foundation (grant EF nos 0430120 and 0545276 and DMS no. 0805533), the RAPIDD programme of the Science & Technology Directorate, Department of Homeland Security and the Fogarty International Center, National Institutes of Health. This work was conducted as a part of the Inference for Mechanistic Models Working Group supported by the National Center for Ecological Analysis and Synthesis, a Center funded by NSF (grant no. DEB-0553768), the University of California, Santa Barbara and the State of California.
Appendix A. compartment models with stochastic rates
Compartment models provide a unifying framework to describe population dynamics (Jacquez 1996; Matis & Kiffe 2000). Here, we write down a general compartment model to obtain formal representations of the transition rates presented in §2. Specifically, we present a framework in which these rates can be interpreted as a system of ordinary differential equations, a discrete population stochastic process with demographic stochasticity or a discrete population stochastic process with both demographic and extra-demographic stochasticity.
A compartment model is a vector-valued process X(t) = (X1(t), … , Xk(t)) denoting the (integer or real-valued) counts in each of k compartments labelled 1 to k. To keep track of individuals moving between compartments, we define flow processes Nij(t) such that Nij(t) – Nij(s) counts the number of transitions from compartment i to compartment j between times s and t. We introduce an extra compartment, labelled ∅, which provides a source and a sink to describe passages into and out of the system because of events such as birth, immigration, emigration and death. The basic characteristic of a compartment model is that X(t) can be written in terms of the flows Nij(t) from i to j, via a conservation of individuals' identity. Namely, for i ranging over 1, … ,k and j ranging over ∅, 1, … ,k, we write
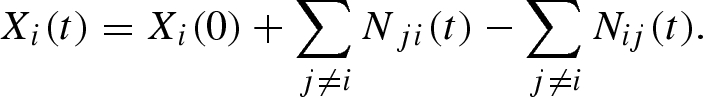 |
A 1 |
Each flow
Nij is associated with a rate function 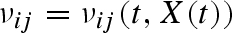 . The corresponding per capita transition rate
. The corresponding per capita transition rate 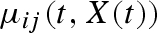 is defined, for
is defined, for 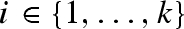 , by
, by
| A 2 |
There are many ways to develop concrete interpretations of such a compartment model (Bretó et al. 2009). For example, an ordinary differential equation interpretation is given by
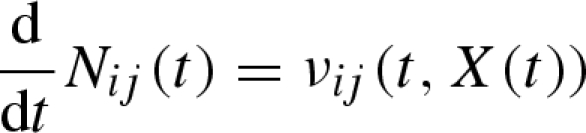 |
for i ≠ j with i and j ranging over ∅, 1, … ,k. In this case, equation (A 1) becomes
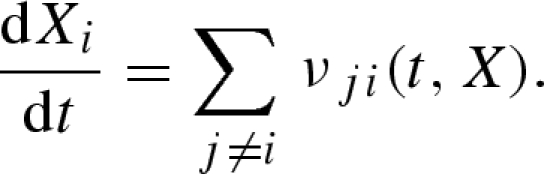 |
A 3 |
This deterministic, continuous-population system is known as the deterministic skeleton (Cushing et al. 1998; Coulson et al. 2004). Continuous-population stochastic generalizations include stochastic differential equation models (Ionides et al. 2006; King et al. 2008) and ordinary differential equations with slowly varying rates (Swishchuk & Wu 2003).
A Markov chain interpretation can be specified by the infinitesimal transition probabilities (Brémaud 1999)
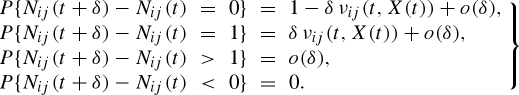 |
A 4 |
This Markov chain model includes only demographic stochasticity. Extra-demographic stochasticity is to be expected in data (Bjørnstad & Grenfell 2001); in particular, it arises because of variability in the rates of equation (A 4). We write σij(t, X(t)) for an intensity parameter corresponding to white noise in the rate νij(t, X(t)). Bretó et al. (2009) developed a general framework for adding white noise to the rates of Markov chain compartment models. The requirement that rates be non-negative rules out the use of Gaussian white noise; the simplest and most convenient parametric choice available is Gamma noise, which is employed in the Euler scheme of box 1. In box 1, δΓij has dimension of time, so δΓij/δ is an approximation to multiplicative noise over a Euler time increment of duration δ. Box 1 reduces to a multinomial Euler scheme (Cai & Xu 2007) if all the noise parameters are set to zero.
For measles, a well-studied compartment model corresponds to X(t) = (S(t), E(t), I(t), R(t)). The state ∅ models both a source (corresponding primarily to births) and a sink (corresponding primarily to deaths); thus in §2 we write N∅S = NBS, NS∅ = NSD, NE∅ = NED, NI∅ = NID and NR∅ = NRD. The per capita recruitment rate μBS(t) in §2 is given by
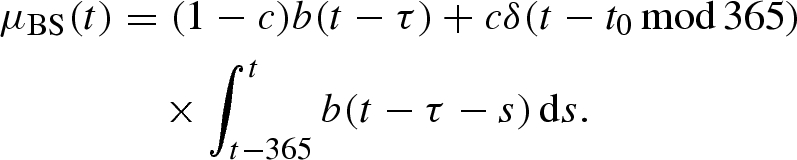 |
A 5 |
Here, b(t) is the birth rate, treated as known from annual birth data; δ(t − t0 mod 365) contributes a Dirac delta impulse to the recruitment rate when t falls on the same calendar day as t0, and we take t0 = 251 for the school admission day in England and Wales. The corresponding population recruitment rate is νBS = μBS(t)N(t), where N(t) is the population size, treated as known via smooth interpolation of census data. The force of infection, μSE(t), is given by equations (2.1) and (2.2). The calendar-day timings of the school holidays in equation (2.2) are as follows: Christmas, 356–365 and 0–6; Easter, 100–115; summer, 199–252; autumn half-term, 300–308.
To complete the model specification, we need to specify the measurement model. We write Zn = NIR(tn)–NIR(tn−1) for the complete case count in the nth reporting interval, supposing that cases are quarantined once they are identified. The corresponding case report yn is modelled via an overdispersed binomial distribution defined as a discretized normal random variable. Specifically, for y > 0, we take
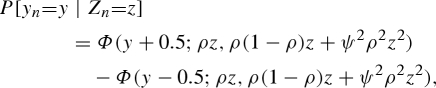 |
A 6 |
where Φ(·; μ, σ2) is normal(μ,σ2) cumulative distribution function. For y = 0, we replace y − 0.5 by −∞ in equation (A 6). Here, ρ is the reporting rate and ψ the overdispersion parameter. Equation (A 6) represents a normal approximation to the binomial distribution with extra variability added. For ψ2≪1, which is the case for all communities of population greater than 105 in the sample we investigated, the formulation in equation (A 6) approximates a binomial distribution for small z and a lognormal distribution for large z.
In the conventional SEIR Markov chain model given by equation (A 4), individuals move from E to I and then I to R at constant rates and so the latent and infectious periods are exponentially distributed. Gamma-distributed latent and infectious periods, which are believed to be more realistic, can be realized by dividing exposed and infectious classes into multiple stages (Anderson & Watson 1980; Lloyd 2001a,b). We implemented this and found a reduction in the estimates of R0 (as predicted by Wearing et al. 2005) but no substantial improvement in goodness of fit (figure S6, electronic supplementary material). In §2, we calculate R0 ignoring the negligible effects of mortality, taking R0 = β̄IP, where IP is the mean infectious period. Naïvely taking IP = μIR−1 in box 1 produces a significant distortion when μIR−1 is close to, or smaller than, the time discretization step δ. Throughout this work, we use δ = 1 day and we use the discrete-time formulas for IP and LP,
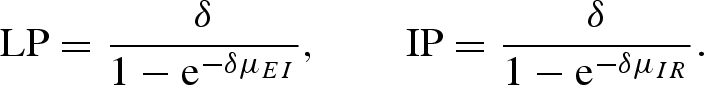 |
A 7 |
References
- Anderson D., Watson R. 1980. On the spread of a disease with gamma distributed latent and infectious periods. Biometrika 67, 191–198. ( 10.1093/biomet/67.1.191) [DOI] [Google Scholar]
- Anderson J. L., Collins N. 2007. Scalable implementations of ensemble filter algorithms for data assimilation. J. Atmos. Ocean. Technol. 24, 1452–1463. ( 10.1175/JTECH2049.1) [DOI] [Google Scholar]
- Anderson R. M., May R. M. 1991. Infectious diseases of Humans. Oxford, UK: Oxford University Press. [Google Scholar]
- Arulampalam M. S., Maskell S., Gordon N., Clapp T. 2002. A tutorial on particle filters for online nonlinear, non-Gaussian Bayesian tracking. IEEE Trans. Signal Process. 50, 174–188. ( 10.1109/78.978374) [DOI] [Google Scholar]
- Bailey N. T. J. 1955. Some problems in the statistical analysis of epidemic data. J. R. Stat. Soc. Ser. B 17, 35–68. [Google Scholar]
- Bailey N. T. J. 1956. On estimating the latent and infectious periods of measles: I. Families with two susceptibles only. Biometrika 43, 15–22. ( 10.2307/2333574) [DOI] [Google Scholar]
- Barndorff-Nielsen O. E., Cox D. R. 1994. Inference and asymptotics. London, UK: Chapman & Hall/CRC. [Google Scholar]
- Bartlett M. S. 1957. Measles periodicity and community size. J. R. Stat. Soc. Ser. A 120, 48–70. ( 10.2307/2342553) [DOI] [Google Scholar]
- Bartlett M. S. 1960. Stochastic population models in ecology and epidemiology, Methuen's monographs on applied probability and statistics, vol. 4 London, UK: Spottiswoode, Ballantyne & Co. Ltd. [Google Scholar]
- Bauch C. T., Earn D. J. D. 2003. Interepidemic intervals in forced and unforced SEIR models. In Dynamical systems and their applications in biology (eds Ruan S., Wolkowicz G., Wu J.). Fields Institute Communications, vol. 36, 33–44. Toronto, Canada: American Mathematical Society. [Google Scholar]
- Beskos A., Papaspiliopoulos O., Roberts G. O., Fearnhead P. 2006. Exact and computationally efficient likelihood-based estimation for discretely observed diffusion processes (with discussion). J. R. Stat. Soc. Ser. B 68, 333–382. ( 10.1111/j.1467-9868.2006.00552.x) [DOI] [Google Scholar]
- Bjørnstad O. N., Grenfell B. T. 2001. Noisy clockwork: time series analysis of population fluctuations in animals. Science 293, 638–643. ( 10.1126/science.1062226) [DOI] [PubMed] [Google Scholar]
- Bjørnstad O., Grenfell B. 2008. Hazards, spatial transmission and timing of outbreaks in epidemic metapopulations. Environ. Ecol. Stat. 15, 265–277. ( 10.1007/s10651-007-0059-3) [DOI] [Google Scholar]
- Bjørnstad O. N., Finkenstädt B. F., Grenfell B. T. 2002. Dynamics of measles epidemics: estimating scaling of transmission rates using a time series SIR model. Ecol. Monog. 72, 169–184. ( doi:10.1890/0012-9615(2002)072[0169:DOMEES]2.0.CO;2 ) ( 10.1890/0012-9615(2002)072[0169:DOMEES]2.0.CO;2) ( doi:10.2307/3100023 ) [DOI] [Google Scholar]
- Bolker B., Grenfell B. 1995. Space, persistence and dynamics of measles epidemics. Phil. Trans. R. Soc. Lond. B 348, 309–320. ( 10.1098/rstb.1995.0070) [DOI] [PubMed] [Google Scholar]
- Bolker B. M., Grenfell B. T. 1996. Impact of vaccination on the spatial correlation and persistence of measles dynamics. Proc. Natl Acad. Sci. USA 93, 12 648–12 653. ( 10.1073/pnas.93.22.12648) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brémaud P. 1999. Markov chains: Gibbs fields, Monte Carlo simulation, and queues. New York, NY: Springer. [Google Scholar]
- Bretó C., He D., Ionides E. L., King A. A. 2009. Time series analysis via mechanistic models. Ann. Appl. Stat. 3, 319–348. ( 10.1214/08-AOAS201) [DOI] [Google Scholar]
- Cai X., Xu Z. 2007. K-leap method for accelerating stochastic simulation of coupled chemical reactions. J. Chem. Phys. 126, 074 102 ( 10.1063/1.2436869) [DOI] [PubMed] [Google Scholar]
- Cappé O., Godsill S., Moulines E. 2007. An overview of existing methods and recent advances in sequential Monte Carlo. Proc. IEEE 95, 899–924. ( 10.1109/JPROC.2007.893250) [DOI] [Google Scholar]
- Casella G., Berger R. L. 1990. Statistical inference. Pacific Grove, CA: Wadsworth. [Google Scholar]
- Cauchemez S., Ferguson N. M. 2008. Likelihood-based estimation of continuous-time epidemic models from time-series data: application to measles transmission in London. J. R. Soc. Interface 5, 885–897. ( 10.1098/rsif.2007.1292) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauchemez S., Valleron A., Boëlle P., Flahault A., Ferguson N. M. 2008. Estimating the impact of school closure on influenza transmission from sentinel data. Nature 452, 750–754. ( 10.1038/nature06732) [DOI] [PubMed] [Google Scholar]
- Clark J. S., Bjørnstad O. N. 2004. Population time series: process variability, observation errors, missing values, lags, and hidden states. Ecology 85, 3140–3150. ( 10.1890/03-0520) [DOI] [Google Scholar]
- Conlan A., Grenfell B. 2007. Seasonality and the persistence and invasion of measles. Proc. R. Soc. B 274, 1133–1141. ( 10.1098/rspb.2006.0030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlan A. J. K., Rohani P., Lloyd A. L., Keeling M., Grenfell B. T. Submitted Resolving the impact of waiting time distributions on the persistence of measles. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook A. R., Otten W., Marion G., Gibson G. J., Gilligan C. A. 2007. Estimation of multiple transmission rates for epidemics in heterogeneous populations. Proc. Natl Acad. Sci. USA 104, 20 392–20 397. ( 10.1073/pnas.0706461104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson T., Rohani P., Pascual M. 2004. Skeletons, noise and population growth: the end of an old debate? Trends Ecol. Evol. 19, 359–364. ( 10.1016/j.tree.2004.05.008) [DOI] [PubMed] [Google Scholar]
- Cushing J. M., Costantino R. F., Dennis B., Desharnais R. A., Henson S. M. 1998. Nonlinear population dynamics: models, experiments, and data. J. Theor. Biol. 194, 1–9. ( 10.1006/jtbi.1998.0736) [DOI] [PubMed] [Google Scholar]
- Doucet A., de Freita N., Gordon N. 2001. Sequential Monte Carlo methods in practice. New York, NY: Springer. [Google Scholar]
- Earn D. J. D., Rohani P., Bolker B. M., Grenfell B. T. 2000. A simple model for complex dynamical transitions in epidemics. Science 287, 667–670. ( 10.1126/science.287.5453.667) [DOI] [PubMed] [Google Scholar]
- Ellner S. P., Bailey B. A., Bobashev G. V., Gallant A. R., Grenfell B. T., Nychka D. W. 1998. Noise and nonlinearity in measles epidemics: combining mechanistic and statistical approaches to population modeling. Am. Nat. 151, 425–440. ( 10.1086/286130) [DOI] [PubMed] [Google Scholar]
- Ferrari M. J., Grais R. F., Bharti N., Conlan A. J. K., Bjørnstad O. N., Wolfson L. J., Guerin P. J., Djibo A., Grenfell B. T. 2008. The dynamics of measles in sub-Saharan Africa. Nature 451, 679–684. ( 10.1038/nature06509) [DOI] [PubMed] [Google Scholar]
- Fine P. E. M., Clarkson J. A. 1982. Measles in England and Wales—I: an analysis of factors underlying seasonal patterns. Int. J. Epidemiol. 11, 5–14. ( 10.1093/ije/11.1.5) [DOI] [PubMed] [Google Scholar]
- Finkenstädt B. F., Grenfell B. T. 2000. Time series modelling of childhood diseases: a dynamical systems approach. J. R. Stat. Soc. Ser. C 49, 187–205. [Google Scholar]
- Finkenstädt B. F., Bjørnstad O. N., Grenfell B. T. 2002. A stochastic model for extinction and recurrence of epidemics: estimation and inference for measles outbreaks. Biostatistics 3, 493–510. ( 10.1093/biostatistics/3.4.493) [DOI] [PubMed] [Google Scholar]
- Gibson G. J., Renshaw E. 1998. Estimating parameters in stochastic compartmental models using Markov chain methods. IMA J. Math. Appl. Med. Biol. 15, 19–40. ( 10.1093/imammb/15.1.19) [DOI] [Google Scholar]
- Gibson G. J., Renshaw E. 2001. Likelihood estimation for stochastic compartmental models using Markov chain methods. Stat. Comput. 11, 347–358. ( 10.1023/A:1011973120681) [DOI] [Google Scholar]
- Glass K., Xia Y., Grenfell B. T. 2003. Interpreting time-series analyses for continuous-time biological models: measles as a case study. J. Theor. Biol. 223, 19–25. ( 10.1016/s0022-5193(03)00031-6) [DOI] [PubMed] [Google Scholar]
- Grenfell B. T., Bolker B. M. 1998. Cities and villages: infection hierarchies in a measles metapopulation. Ecol. Lett. 1, 63–70. ( 10.1046/j.1461-0248.1998.00016.x) [DOI] [Google Scholar]
- Grenfell B. T., Bolker B. M., Kleczkowski A. 1995. Seasonality and extinction in chaotic metapopulations. Proc. R. Soc. Lond. B 259, 97–103. ( 10.1098/rspb.1995.0015) [DOI] [Google Scholar]
- Grenfell B. T., Bjørnstad O. N., Kappey J. 2001. Travelling waves and spatial hierarchies in measles epidemics. Nature 414, 716–723. ( 10.1038/414716a) [DOI] [PubMed] [Google Scholar]
- Grenfell B. T., Bjørnstad O. N., Finkenstädt B. F. 2002. Dynamics of measles epidemics: Scaling noise, determinism, and predictability with the TSIR model. Ecol. Monogr. 72, 185–202. ( doi:10.2307/3100024 ) ( 10.2307/3100024) ( doi:10.1890/0012-9615(2002)072[0185:DOMESN]2.0.CO;2 ) [DOI] [Google Scholar]
- He D. 2006. Modeling of childhood infectious diseases. PhD thesis, McMaster University, Canada. [Google Scholar]
- Hilborn R., Mangel M. 1997. The ecological detective. Princeton, NJ: Princeton University Press. [Google Scholar]
- Hope Simpson R. E. 1948. The period of transmission in certain epidemic diseases: an observational method for its discovery. Lancet 252, 755–760. ( 10.1016/s0140-6736(48)91328-2) [DOI] [PubMed] [Google Scholar]
- Hope Simpson R. E. 1952. Infectiousness of communicable diseases in the household: measles, chickenpox, and mumps. Lancet 260, 549–554. ( 10.1016/s0140-6736(52)91357-3) [DOI] [PubMed] [Google Scholar]
- Ionides E. L. 2005. Maximum smoothed likelihood estimation. Stat. Sinica 15, 1003–1014. [Google Scholar]
- Ionides E. L., Bretó C., King A. A. 2006. Inference for nonlinear dynamical systems. Proc. Natl Acad. Sci. USA 103, 18 438–18 443. ( 10.1073/pnas.0603181103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionides E. L., Bretó C., King A. A. 2007. Modeling disease dynamics: cholera as a case study. In Statistical advances in the biomedical sciences (eds Biswas A., Datta S., Fine J., Segal M.), ch. 8. Hoboken, NJ: Wiley-Interscience. [Google Scholar]
- Ionides E. L., Bhadra A., King A. A. Iterated filtering. (http://arxiv.org/abs/0902.0347. ) 2009.
- Jacquez J. A. 1996. Compartmental analysis in biology and medicine, 3rd edn Ann Arbor, MI: BioMedware. [Google Scholar]
- Keeling M. J., Grenfell B. T. 1997. Disease extinction and community size: modeling the persistence of measles. Science 275, 65–67. ( 10.1126/science.275.5296.65) [DOI] [PubMed] [Google Scholar]
- Keeling M. J., Grenfell B. T. 2002. Understanding the persistence of measles: reconciling theory, simulation and observation. Proc. R. Soc. Lond. B 269, 335–343. ( 10.1098/rspb.2001.1898) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling M. J., Rohani P. 2002. Estimating spatial coupling in epidemiological systems: a mechanistic approach. Ecol. Lett. 5, 20–29. ( 10.1046/j.1461-0248.2002.00268.x) [DOI] [Google Scholar]
- Keeling M., Ross J. 2008. On methods for studying stochastic disease dynamics. J. R. Soc. Interface 5, 171–181. ( 10.1098/rsif.2007.1106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevrekidis I. G., Gear C. W., Hummer G. 2004. Equation-free: the computer-assisted analysis of complex, multiscale systems. AIChE J. 50, 1346–1354. ( 10.1002/aic.10106) [DOI] [Google Scholar]
- King A. A., Ionides E. L., Pascual M., Bouma M. J. 2008. Inapparent infections and cholera dynamics. Nature 454, 877–880. ( 10.1038/nature07084) [DOI] [PubMed] [Google Scholar]
- King A. A., Ionides E. L., Bretó C. M., Ellner S., Kendall B. 2009. pomp: Statistical inference for partially observed Markov processes (R package). (http://pomp.r.forge.r-project.org)
- Kocsis L., Szepesvári C. 2006. Universal parameter optimisation in games based on SPSA. Machine Learning 63, 249–286. ( 10.1007/s10994-006-6888-8) [DOI] [Google Scholar]
- Koelle K., Pascual M. 2004. Disentangling extrinsic from intrinsic factors in disease dynamics: a nonlinear time series approach with an application to cholera. Am. Nat. 163, 901–913. ( 10.1086/420798) [DOI] [PubMed] [Google Scholar]
- Lande R., Engen S., Sæther B.-E. 2003. Stochastic populated dynamics in ecology and conservation. New York, NY: Oxford University Press. [Google Scholar]
- Levin S. A., Grenfell B., Hastings A., Perelson A. S. 1997. Mathematical and computational challenges in population biology and ecosystems science. Science 275, 334–343. ( 10.1126/science.275.5298.334) [DOI] [PubMed] [Google Scholar]
- Lima M. 2009. A link between the North Atlantic Oscillation and measles dynamics during the vaccination period in England and Wales. Ecol. Lett. 12, 302–314. ( 10.1111/j.1461-0248.2009.01289.x) [DOI] [PubMed] [Google Scholar]
- Liu J., West M. 2001. Combining parameter and state estimation in simulation-based filtering. In Sequential Monte Carlo methods in practice (eds Doucet A., de Freitas N., Gordon N. J.), pp. 197–224. New York, NY: Springer. [Google Scholar]
- Liu W.-M., Levin S. A., Iwasa Y. 1986. Influence of nonlinear incidence rates upon the behavior of SIRS epidemiological models. J. Math. Biol. 23, 187–204. ( 10.1007/BF00276956) [DOI] [PubMed] [Google Scholar]
- Lloyd A. L. 2001a. Destabilization of epidemic models with the inclusion of realistic distributions of infectious periods. Proc. R. Soc. Lond. B 268, 985–993. ( 10.1098/rspb.2001.1598) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd A. L. 2001b. Realistic distributions of infectious periods in epidemic models: changing patterns of persistence and dynamics. Theor. Population Biol. 60, 59–71. ( 10.1006/tpbi.2001.1525) [DOI] [PubMed] [Google Scholar]
- Lloyd-Smith J. O., Schreiber S. J., Kopp P. E., Getz W. M. 2005. Superspreading and the effect of individual variation on disease emergence. Nature 438, 355–359. ( 10.1038/nature04153) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matis J. H., Kiffe T. R. 2000. Stochastic population models: a compartmental perspective. New York, NY: Springer. [Google Scholar]
- May R. M. 2004. Uses and abuses of mathematics in biology. Science 303, 790–793. ( 10.1126/science.1094442) [DOI] [PubMed] [Google Scholar]
- McCullagh P., Nelder J. A. 1989. Generalized linear models, 2nd edn London, UK: Chapman & Hall. [Google Scholar]
- Newman K. B., Fernandez C., Thomas L., Buckland S. T. 2008. Monte Carlo inference for state-space models of wild animal populations. Biometrics 63, 558–567. ( 10.1111/j.1541-0420.2008.01073.x) [DOI] [PubMed] [Google Scholar]
- Polson N. G., Stroud J. R., Muller P. 2008. Practical filtering with sequential parameter learning. J. R. Stat. Soc. B 70, 413–428. ( 10.1111/j.1467-9868.2007.00642.x) [DOI] [Google Scholar]
- Poyiadjis G., Singh S., Doucet A. 2006. Gradient-free maximum likelihood parameter estimation with particle filters. In Proc. Am. Control Conf. pp. 3062–3067. ( 10.1109/ACC.2006.1657187) [DOI] [Google Scholar]
- R Development Core Team 2006. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; ISBN 3-900051-07-0. [Google Scholar]
- Roberts G. O., Stramer O. 2001. On inference for partially observed nonlinear diffusion models using the Metropolis–Hastings algorithm. Biometrika 88, 603–621. ( 10.1093/biomet/88.3.603) [DOI] [Google Scholar]
- Schenzle D. 1984. An age-structured model of pre- and post-vaccination measles transmission. IMA J. Math. Appl. Med. Biol. 1, 169–191. ( 10.1093/imammb/1.2.169) [DOI] [PubMed] [Google Scholar]
- Shumway R. H., Stoffer D. S. 2006. Time series analysis and its applications, 2nd edn, New York, NY: Springer. [Google Scholar]
- Spall J. C. 2003. Introduction to stochastic search and optimization, Ch. 6. Hoboken, NJ: Wiley. [Google Scholar]
- Stone L., Olinky R., Huppert A. 2007. Seasonal dynamics of recurrent epidemics. Nature 446, 533–536. ( 10.1038/nature05638) [DOI] [PubMed] [Google Scholar]
- Storvik G. 2002. Particle filters for state-space models with the presence of unknown static parameters. IEEE Trans. Signal Process. 50, 281–289. ( 10.1109/78.978383) [DOI] [Google Scholar]
- Streftaris G., Gibson G. J. 2004. Bayesian analysis of experimental epidemics of foot-and-mouth disease. Proc. R. Soc. Lond. B 271, 1111–1117. ( 10.1098/rspb.2004.2715) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swishchuk A., Wu J. 2003. Evolution of biological systems in random media: limit theorems and stability. Dordrecht, The Netherlands: Kluwer Academic Publishers. [Google Scholar]
- Toni T., Welch D., Strelkowa N., Ipsen A., Stumpf M. P. 2009. Approximate Bayesian computation scheme for parameter inference and model selection in dynamical systems 6, 187–202. ( 10.1098/rsif.2008.0172) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallinga J., Levy-Bruhl D., Gay N. J., Wachmann C. H. 2001. Estimation of measles reproduction ratios and prospects for elimination of measles by vaccination in some western European countries. Epidemiol. Infect. 127, 281–295. ( 10.1017/s095026880100601x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wearing H. J., Rohani P., Keeling M. J. 2005. Appropriate models for the management of infectious diseases. PLoS Medicine 2, e174 ( 10.1371/journal.pmed.0020174) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y., Bjørnstad O. N., Grenfell B. T. 2004. Measles metapopulation dynamics: a gravity model for epidemiological coupling and dynamics. Am. Nat. 164, 267–281. ( 10.1086/422341) [DOI] [PubMed] [Google Scholar]