

Abstract
I study the reorganization of the yeast transcriptional regulatory network after whole-genome duplication (WGD). Individual transcription factors (TFs) were computationally removed from the regulatory network, and the resulting networks were analysed. TF gene pairs that survive in duplicate from WGD show detectable redundancy as a result of that duplication. However, in most other respects, these duplicated TFs are indistinguishable from other TFs in the genome, suggesting that the duplicate TFs produced by WGD were rapidly diverted to distinct functional roles in the regulatory network. Separately, I find that genes targeted by many TFs appear to be preferentially retained in duplicate after WGD, an effect I attribute to selection to maintain dosage balance in the regulatory network after WGD.
Keywords: whole-genome duplication, network evolution, regulatory evolution
1. Introduction
The importance of changes in gene regulation in generating evolutionary novelty has long been appreciated (e.g. Wray et al. 2003). However, some questions about regulatory evolution could only be answered with the advent of genome sequencing and data such as that from chromatin-immunoprecipitation studies (Lee et al. 2002; Harbison et al. 2004). Such data have allowed the inference of reasonably complete transcriptional regulatory networks from organisms including the yeast Saccharomyces cerevisiae. As a result, we now know that the number of targets of a given transcription factor (TF) follows a power law (Guelzim et al. 2002; Luscombe et al. 2004), that the regulatory networks possess an overabundance of specific small ‘circuit’ motifs (Lee et al. 2002; Milo et al. 2002; Shen-Orr et al. 2002) of functional significance (Conant & Wagner 2003; Mangan & Alon 2003; Klemm & Bornholdt 2005; Prill et al. 2005) and that the networks are structured in a manner that imparts robustness to signals (Li et al. 2004; Klemm & Bornholdt 2005). Of course, transcriptional regulation is a dynamic process. Luscombe et al. (2004) have shown that gene regulation in response to exogenous stimuli tends to involve fewer TFs per target and shorter regulatory cascades than do endogenous regulatory controls (such as the cell cycle). Additionally, Jothi et al. (2009) found that differences in TF half-life were associated with a TF's position in the hierarchically organized regulatory network, such that TFs associated with environmental stimuli tended to be long-lived, while those TFs involved in computing the appropriate response to those stimuli had shorter half-lives.
Features such as network motifs imply that natural selection has shaped the regulatory network to specific functional requirements. But the network must also evolve according to certain underlying rules. One useful analogy is protein evolution, where natural selection preserves proteins of useful function, but the generation of the protein sequences themselves must also obey the rules of mutation and population genetics. Similar rules governing network evolution remain to be completely elucidated, but an important first step was provided by Teichmann & Babu (2004), who showed that much of the structure of the network in both S. cerevisiae and Escherichia coli was created by gene duplication. Another notable feature of network evolution is the rapid (and asymmetric) evolution of gene expression and regulatory interactions after such duplications (Gu et al. 2002, 2005). Interestingly, rapid and asymmetric evolution are also features of protein interaction networks (Wagner 2001, 2002).
Here, I study network evolution after a particular type of duplication: the whole-genome duplication (WGD) that occurred in an ancestor of S. cerevisiae (Wolfe & Shields 1997; Dietrich et al. 2004; Dujon et al. 2004; Kellis et al. 2004). The goal is to understand post-duplication evolution in the face of one of the vexing issues in the analysis of biological networks: the fact that we are often able to study the network only as it exists in a single modern organism.
Being unable to compare multiple networks makes it very hard to identify historical changes in them. Sequence data can identify TFs or target genes that have been duplicated but cannot easily identify the loss or gain of regulatory interactions. However, such changes are interesting because they represent the network equivalents of neofunctionalization or subfunctionalization (Force et al. 1999; Stoltzfus 1999; Hughes 2005; Conant & Wolfe 2006, 2008). Distinguishing these possibilities is essentially the task of analysing the interactions of duplicate genes to see if they are ancestral or novel (figure 1a). This analysis is reasonably straightforward, given a second network with which to infer the ancestral state, but quite challenging failing that.
Figure 1.

Regulatory network evolution. (a) Conceptual view of regulatory network evolution after WGD. Immediately after WGD, each ancestral regulatory interaction exists in four copies. As time progresses, both interactions and the TFs and target genes may be lost. Note that the loss of a TF or target also eliminates the interactions that gene possessed. In this thought experiment, we are aware of the ancestral network, meaning that we can distinguish between new interactions (red) and redundant (green) and non-redundant (blue) interactions surviving from WGD. Grey circle, transcription factor; white circle, target gene. In real situations, the ancestral network is unknown, meaning that we cannot make this distinction (adapted from Conant & Wolfe 2008). (b) A simple example of calculating the input diversity of a network. The top-level TFs (red) have minimal input diversity, indicated by vectors with only one non-zero entry. We calculate the vectors for intermediate TFs (purple) and target nodes (blue) iteratively (see §2) until the values converge.
Lacking such an outgroup, I have approached the problem not by focusing on individual interactions, but rather by considering the differences between two sets of genes. In one set are those TFs that possess a paralogue (duplicate) created by WGD, whereas in the other are the remaining TFs, where the paralogue created by WGD has been lost along the lineage leading to S. cerevisiae (Scannell et al. 2007). I hypothesized that the duplicated TFs produced by WGD would retain some shared targets after WGD. It would seem to follow that such redundancy would also make these duplicated TFs less essential for gene regulation than other TFs.
2. Material and Methods
(a). Calculation of minimal path lengths and input diversity
For each network, I calculated the average minimum path length (the average over all nodes of the average for each node of the minimum number of edges needing to be traversed to reach any other node) with Dijkstra's algorithm (Yoon et al. 2006). This approach was then applied to all possible pruned networks.
As illustrated in figure 1b, to calculate input diversity, each node in the network was assigned a vector of length n, where n is the number of top-level TFs in the network. For a given gene x, I calculate the value of its vector element j given its input TFs 1, … , m as
| 2.1 |
All vectors except those of the top-level TFs are first initialized to zero. The calculation in equation (2.1) is then iterated for all target genes using the values from the last iteration from nodes 1, … , m until the values of the vectors for all nodes have converged. I then calculate the entropy of the target genes as
| 2.2 |
When calculating pruning statistics, the pruning of a top-level TF will necessarily reduce the input diversity (as n decreases). To control for this effect when calculating pruning effects, I used not the raw Shannon entropy but rather the scaled value of the observed entropy divided by the maximum possible entropy: −log2 (1/n′), where n′ is the number of top-level TFs in the pruned network.
(b). Comparison of the distribution of shared targets between the duplicated TF pairs and the remainder of the network
I assumed that the number of shared target genes for a pair of TFs follows a discrete power law distribution (visual inspection indicated that an exponential distribution was insufficiently ‘long tailed’ for these data; figure 2a). Thus, I assumed that the probability of observing a pair of genes with n shared targets is given by
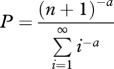 |
2.3 |
Because this distribution is defined over the interval 1 ≤ x < ∞, while these data are defined over 0 ≤ x < ∞, I fitted the number of shared targets plus one to this function (the n + 1 in the numerator above) using standard numerical techniques (Press et al. 1992). Using a likelihood ratio test (Sokal & Rohlf 1995), I compared a model where the probability of a shared target was allowed to differ between the pairs of WGD TFs and the other TF pairs with a constrained model where all pairs had the same probability of sharing a target. The same method was used for comparing the distribution of interaction degree between the WGD TFs and the other TFs, except that here I did not adjust the range of the input degrees.
Figure 2.
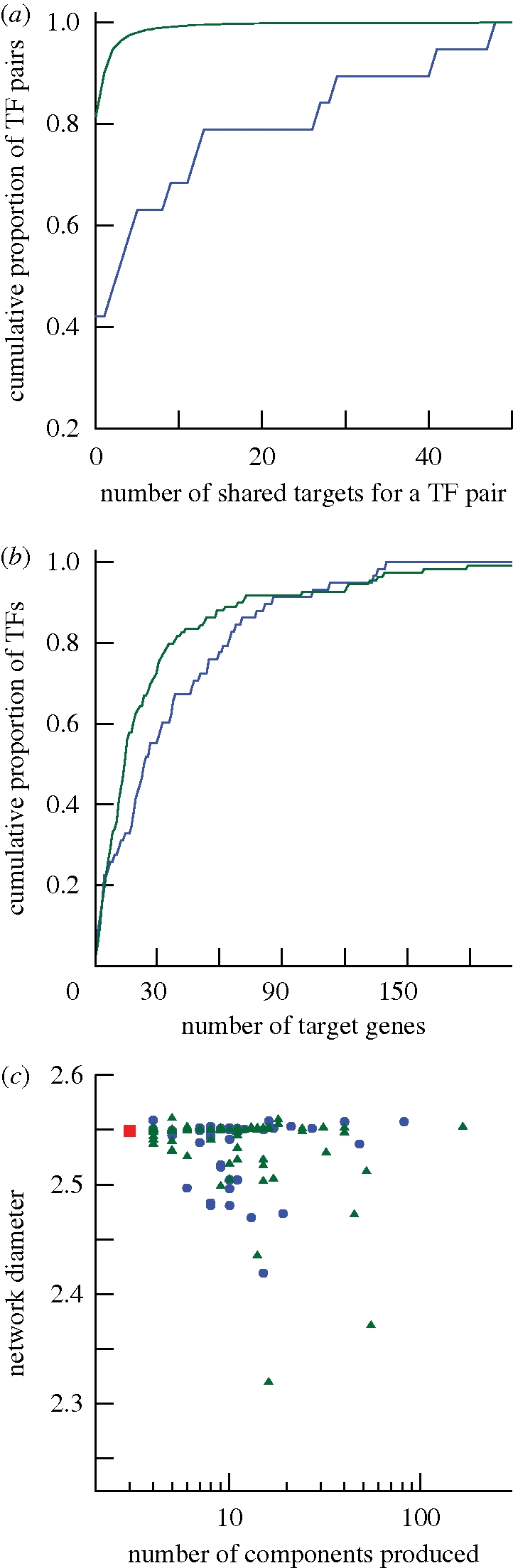
WGD-derived TFs possess an excess of shared targets but are not functionally redundant. Data from the network HR_03 for all panels. (a) Cumulative distribution of the number of shared targets for a pair of TFs: blue line, pairs of duplicated TFs created by WGD; green line, all other pairs of TFs. These distributions are significantly different (likelihood ratio test, p < 10−10). (b) Cumulative distribution of the number of target genes for a TF: blue line, TFs with a surviving duplicate from WGD; green line, all other TFs. There is no significant difference in these two distributions (likelihood ratio test, p > 0.05). (c) Comparison of TF knockout effects on two network statistics for TFs with (blue) and without (green) surviving duplicates from WGD. On the x-axis is the number of components produced by TF removal, on the y-axis is the resulting average path length. The values from the original network are shown in red. Blue circle, WGD TFs; green triangle, other TFs; red square, real network.
(c). Identification of missing regulatory genes in three outgroup genomes
Using data from the Yeast Genome Order Browser (YGOB) project (Byrne & Wolfe 2005), I identified, for each TF, the syntenic locus in three non-WGD genomes: Ashbya gossypii (Dietrich et al. 2004), Kluyveromyces lactis (Dujon et al. 2004) and Kluyveromyces waltii (Kellis et al. 2004). The phylogenetic relationship between these three species and S. cerevisiae has been described by Kurtzman & Robnett (2003). For both the non-WGD species and S. cerevisiae, these syntenic loci may or may not have a gene present: the locus itself is identified by the shared flanking genes. Gene absence in S. cerevisiae will generally be the result of duplicate gene loss after WGD (meaning that one copy of the gene survives). In the other three species, YGOB also indicates whether a gene with sequence similarity exists elsewhere in the genome. Thus, the identification of gene absence in the three outgroup genomes is supported by both gene order and gene sequence data. I compared the effect of computational pruning from the network for genes where at least one of these species lacked an orthologue of the S. cerevisiae TF to that effect for TFs preserved across all three species.
(d). Comparison of WGD TF status and knockout fitness defect
Data on the fitness effects of gene knockouts were taken from Steinmetz et al. (2002). I averaged the knockout fitness on YPD (yeast extract, peptone, dextrose) media for the two time courses and omitted genes where these values differed by more than 0.05. Following Gu et al. (2003), I then normalized these measurements by the average value across all genes. Any gene annotated as essential by the Munich Information Center for Protein Sequences (MIPS; Mewes et al. 1999) was assigned a fitness value of zero.
3. Results
(a). Datasets
I used two datasets of TFs and their targets for these analyses. The first was the chromatin immunoprecipitation dataset of Harbison et al. (2004). These data consist of probabilities (p-values) of the binding of 203 TFs to the upstream regions of each gene in the yeast genome. I have used two different p-value thresholds for these analyses: p ≤ 10−3 (as used by Harbison et al. 2004; denoted HR_03 below) and p ≤ 10−4 (HR_04 below). Any TF lacking at least one interaction of the required stringency was omitted from the analysis. I have also used the published dataset of Luscombe et al. 2004 (LC), which consists of 142 TFs derived from both an earlier large-scale experiment and the literature.
(b). Genome duplication data
Data on which genes in the S. cerevisiae genome survive in duplicate from WGD were obtained from the YGOB project (Byrne & Wolfe 2005). For each dataset (HR_03, etc.), all TFs with surviving duplicates according to YGOB were assigned to the set of WGD TFs. All remaining TFs in each dataset are referred to as ‘other TFs’.
(c). Network statistics
A number of statistics have been proposed to evaluate the structure of biological networks, including the degree distribution (Jeong et al. 2001), clustering coefficient (Watts & Strogatz 1998; Wagner & Fell 2001) and network diameter (Jeong et al. 2000; Wagner & Fell 2001). To this list, I add a measure of information-processing capacity: the ‘input diversity’ (figure 1b). This statistic gives a scaled indication of the input a target gene receives from the set t of all TFs. It is calculated by first splitting t into two classes: the n top-level TFs that are not regulated by any other TFs (red in figure 1) and the t–n intermediate TFs that both regulate genes and are themselves regulated by other TFs (purple in figure 1). The regulatory influences on a given gene are represented as a vector with n elements (one for each top-level TF). Top-level TFs are described by discrete binary vectors with only a single non-zero entry corresponding to that TF's index number (e.g. Li et al. 2004). Other genes have non-zero entries from any top-level TFs that can be reached directly or indirectly from that gene. Values are calculated as described in §2 (figure 1b). Input diversity is defined as the Shannon entropy of this vector. Thus, a target gene with connections to only a single top-level TF would have minimal input diversity of 0, whereas a gene with direct inputs from all n top-level TFs would have an input diversity of −log2(1/n). One caveat to this approach is that some TFs will be misidentified as top-level regulators owing to missing regulatory interactions: in the present work, I do not attempt to gauge the magnitude of this problem. I also note that this approach has some similarities to that of Jothi et al. (2009), although it is less computationally complex and does not attempt to infer the hierarchical structure of the network.
(d). TFs were more likely to be preserved in duplicate after WGD than genes in the genome at large
An overabundance of surviving duplicated TFs after WGD was observed in several species (Blanc & Wolfe 2004; Maere et al. 2005; Aury et al. 2006), but not initially in yeast (Seoighe & Wolfe 1999). However, more recent work has confirmed that the WGD-produced duplicates are indeed enriched for TFs (Chen et al. 2008; Conant & Wolfe 2008). Because of incomplete gene annotation and also because some duplicate genes appear to have lost their regulatory activity (e.g. Hittinger & Carroll 2007), there are a number of cases where only one member of such a duplicate gene pair is identified as a TF. Nonetheless, for all three datasets (HR_03, HR_04 and LC), there are significantly more WGD TFs than would be expected (p < 10−4, χ2-test).
(e). TF pairs surviving in duplicate from WGD share more target genes than would be expected by chance but do not differ in overall numbers of interactions
To test for redundancy produced by WGD, I compared the number of shared target genes for pairs of the WGD TFs to the number of shared targets for all other pairs of TFs (figure 2a). Unsurprisingly, for all three datasets, the pairs of WGD TFs share more targets than would be expected by chance (p < 10−10, likelihood ratio test; see §2). Interestingly, the WGD TFs have, on average, more regulatory interactions than other TFs (figure 2b), although this difference is significant only for the LC network (logistic regression, p = 0.028; Sokal & Rohlf 1995). Moreover, even if one removes all regulatory interactions shared between pairs of the WGD TFs, the WGD TFs still have no fewer targets than other TFs (logistic regression; p > 0.05). Likewise, genes that are targets of the WGD TFs have on average higher input diversity than do targets of other TFs, although again this difference is significant only for the LC dataset (logistic regression; p = 0.036).
(f). Duplicated TF genes may be slightly more dispensable than other TF genes
Given the excess of shared target genes among the WGD TFs, it is reasonable to ask if the WGD-produced redundancy in target genes reduces the importance of these duplicated TFs in the regulatory network. I tested whether the WGD TFs have less severe fitness defects when knocked out in S. cerevisiae than do other TFs (see §2). Average fitness is higher for all WGD TF knockouts: it is significantly higher for the LC network (logistic regression, p = 0.012).
(g). WGD status does not predict the effect of computational TF removal
To further test the prediction of increased dispensability among TFs duplicated at WGD, I created pruned networks that computationally simulate the effect of TF loss by removing each TF and its interactions from the network and then recalculating three network statistics: the number of components (how many pieces the network is broken into by the loss of a single TF), the average path length between two nodes and the average input diversity.
I asked whether the WGD TFs produced different network statistics after pruning compared to the other TFs. Using logistic regression, I found no differences between the WGD TFs and other TFs in the three statistics considered (p > 0.05; figure 2c). However, an alternative explanation for these results is that the network statistics considered are simply not useful measures of the effect of TF removal. To determine if this was the case, I carried out two analyses.
(h). Hypothetical ancestral TFs are more influential in the network than other TFs
First, I created pseudo-ancestral TFs for pairs of the WGD TFs by combining the interactions of the two TFs into a single ancestral gene, merging any redundant interactions. Note that the unrealistic assumption that no new regulatory circuits have evolved since the WGD is irrelevant when testing the value of these network statistics. For all three networks, the removal of pseudo-ancestral nodes was more likely to increase in the number of components than was the removal of other TFs (logistic regression, p < 0.02), indicating the increased centrality of these created TFs. For both datasets derived from the data of Harbison et al. (2004)—that is, HR_03 and HR_04—the loss of these ancestral nodes decreases the average path length more than the loss of other nodes (logistic regression, p < 0.005). All three networks also show a larger decrease in the input diversity when ancestral TFs are removed than is seen with other TFs (logistic regression, p < 0.04).
(i). Network parameters are correlated with phylogenetic dispersal
Second, I examined whether the network statistics used here were correlated with data on TF dispensability in three outgroup genomes that lack WGD. I thus used YGOB to determine whether each S. cerevisiae TF possessed an orthologue in these genomes (see §2). I compared the effect of TF removal for those TFs with orthologues in all the three species to that for TFs missing an orthologue in at least one species. In the HR_03 network, those TFs missing in at least one outgroup genome are less likely to break the network into a large number of components when removed (p < 0.03), whereas in both the HR_03 and HR_04 networks the removal of dispensable genes tends to reduce the input diversity less than the loss of other TFs (p < 0.05).
These analyses of both pseudo-ancestral and dispensable TFs suggest that statistics such as average path length, number of components and input diversity are real, if imperfect, measures of a TF's importance are imperfect in the regulatory network. This conclusion supports the contention that the lack of difference between the WGD TFs and other TFs is not simply an artefact of how these statistics measure importance.
(j). Duplicated TF genes were not more important prior to WGD
One explanation for the lack of difference between the WGD TFs and the other TFs in network measures is that the WGD TFs were actually more important prior to WGD and that they have subsequently become reduced in importance through the partitioning of ancestral functions (i.e. subfunctionalization). I thus asked whether the WGD TFs were less likely to be absent in the three non-WGD genomes than other TFs. Although the proportion of genes with orthologues is higher among the WGD TFs than for other TFs, this difference is not statistically significant in any network (table 1; p > 0.05). I therefore cannot conclude that these TFs were more important to the organism prior to WGD.
Table 1.
WGD TFs and phylogenetic dispersal.
| dataset | WGD TFsa | prop w/outgroup orthologuesb | other TFsc | prop w/outgroup orthologuesd | pe |
|---|---|---|---|---|---|
| HR_03 | 58 | 0.84 | 109 | 0.79 | 0.38 |
| HR_04 | 48 | 0.88 | 85 | 0.81 | 0.35 |
| LC | 48 | 0.85 | 93 | 0.81 | 0.48 |
aNumber of TFs with a surviving duplicate gene from WGD.
bProportion of the WGD TFs for which an orthologue exists in all three outgroup genomes examined (see §2).
cNumber of TFs lacking a surviving duplicate from WGD.
dProportion of other TFs for which an orthologue exists in all three outgroup genomes examined.
ep-value for the hypothesis test of different proportions of genes with three outgroup orthologues among the WGD TFs and among all other TFs. (χ2-test with one degree of freedom.)
(k). Network randomization
In addition to testing the usefulness of these measures of importance, the above analyses also suggest that purifying selection acts to retain certain features of the regulatory network. Thus, the fact that phylogenetic dispersal and input diversity are correlated implies that each of these two variables is probably associated with some underlying feature of the network that is being maintained by selection. To further explore this idea, I compared the measured statistics in the real networks to those seen in networks that had been randomly rewired while preserving the number of incoming and outgoing interactions for every TF and target gene. If we assume that the evolution of the real network has included the appearance of functionally important regulatory interactions, rewiring that network will disrupt the patterns of non-random attachment created by these interactions. Thus, if such patterns exist, we will see differences in summary statistics between the real network and the randomized ones, assuming that the statistics we have chosen are meaningful.
The HR_04 network shows significantly more components than randomized networks (p = 0.036), but no other networks showed this pattern, probably because the other two original networks had very few components (<4). The HR_04 network also shows significantly lower input diversity than do randomized networks (p = 0.006), a pattern also not seen in any other network (although I note that the average input diversity is higher for all three sets of randomized networks than for their respective real networks). The HR_03 network shows path lengths that are significantly shorter than random networks (p = 0.014). Both path length and input diversity are related to the degree with which the regulatory network is able to segregate signals to distinct sets of target genes. Thus, networks with longer path lengths will need to activate more intermediate TFs in order to propagate a signal to a target gene, while larger input diversity will imply that a given signal activates more target genes. One can argue that both of these properties might be undesirable under certain circumstances, because they make the cellular response to a stimulus less precise. One can therefore argue that natural selection acting to preserve efficient responses to stimuli might tend to produce networks that maintain lower values of input diversity and path length.
(l). WGD status and target gene characteristics
I also compared the regulation of target genes that possessed duplicates from WGD to other target genes to see if the number of TFs that acted upon a target gene or that gene's input diversity differed depending on WGD status. For networks HR_03 and LC, target genes with surviving duplicates from WGD were regulated by, on average, more TFs than other genes (p < 0.02). Similarly, in these two networks, targets with surviving duplicates had higher input diversity than did other genes (p < 0.003).
One explanation for this higher input degree among target genes with a duplicate from WGD is that it is caused by quartets of duplicated TFs and duplicated targets. However, the LC network actually shows fewer interactions between such quartets than would be expected by chance (p = 0.008), while the frequency of quartet interactions in HR_03 and HR_04 does not differ from the chance expectation (p > 0.05). It therefore does not appear that duplicate target genes owe their excess of regulatory interactions to surviving redundant interactions from WGD.
4. Discussion
Here, I have analysed the evolutionary patterns seen in the yeast transcriptional regulatory network following WGD. It is clear from the above results that the three networks considered do not always point to the same conclusions, and it is worth commenting on some possible explanations for this fact. The LC network, because it was partly drawn from literature data, may include some biases resulting from the TFs that researchers have chosen to study. Note that the WGD TFs in this network were the only ones to show statistically and significantly decreased essentiality and differences in network statistics from other TFs. The HR_03 and HR_04 datasets are more similar to each other, but the HR_04 network also shows some distinct patterns: its WGD-duplicated target genes show no increase in the number of TFs regulating them. I attribute this difference to the smaller number of genes and interactions in this network resulting from the increased stringency required to infer an interaction.
Genome duplication has long been thought to have an important role in reshaping regulatory networks (Ohno 1970; Freeling & Thomas 2006). The data above suggest that the new TFs created by WGD were relatively quickly incorporated into the regulatory network. This pattern is in contrast to my initial hypothesis that the WGD TFs would tend to be more generally dispensable than other TFs. Instead, although these TFs still show some features that trace to WGD (more shared interactions and possibly higher knockout fitness), in most respects they are indistinguishable from their non-duplicated counterparts: the WGD TFs have at least as many unique regulatory targets as do other TFs and do not show differences from other TFs in measures such as input diversity.
This result is not unexpected: duplication is often followed by rapid expression divergence (Gu et al. 2002, 2005). Indeed, this rapid divergence is one of the facts that supports the hypothesis that many phenotypic differences between organisms are due to changes in gene regulation (King & Wilson 1975; Jacob 1977; Wray et al. 2003). For the results described here, what remains unclear is the manner in which the regulatory divergence occurred. One hypothesis is that the extant WGD TFs have divided the ancestral regulatory functions between the two duplicate copies through subfunctionalization. While the interaction losses required for this pathway could indeed be rapid, a corollary of this hypothesis (in strict form) is that the pre-WGD TFs that gave rise to the duplicated TFs would have had more than twice as many regulatory targets as do the unduplicated TFs in modern S. cerevisiae. Were this not the case, I would not observe that the modern WGD TFs have the same number of regulatory targets as do other TFs. However, speaking against this hypothesis of the duplication and subfunctionalization of TFs of high importance, I find that the phylogenetic dispersal of the WGD TFs is not abnormally high, even though one would expect that putative ancestral TFs with such large numbers of interactions would tend to be selectively conserved and hence present in most yeast genomes. A second hypothesis to explain the similarity of the WGD TFs to the other TFs is that the former have gained regulatory targets since WGD. If one accepts the rapid turnover of regulatory interactions in the evolutionary time mentioned above, this hypothesis of interaction gain is consistent with the observation that different yeast species use different regulatory logic to produce the same phenotype (Tsong et al. 2006). Under this scenario, after WGD, neutral changes in regulation have occurred in both the WGD TFs and the other TFs, partially erasing the regulatory signature of WGD.
I would further suggest that the changes seen in these networks remind us that the regulatory network can actually be thought of as a computing device, taking inputs from the cellular surroundings and integrating them into cellular responses. The study of the hierarchical structure of the regulatory network by Jothi et al. (2009) is very intriguing in this respect as the structure that these authors deduce is reminiscent of a neural network (Flake 1998). Ironically, neural networks themselves originated as an analogy to an evolved computational engine: the brain.
Among the target genes in these regulatory networks, it appears that duplicated target genes surviving from WGD tend to have more regulatory interactions than would be expected. As this result is not due to the survival of the duplicated TF, target gene pairs, one explanation could be that these genes had more interactions at the time of duplication. This explanation actually has antecedents going back at least to Ohno (1970). The current formulation of the idea is the dosage balance hypothesis (Papp et al. 2003; Birchler & Veitia 2007; Edger & Pires 2009), which argues that single-gene duplications (i.e. non-WGD duplications) in dense parts of networks will tend to be selected against because they disrupt the stoichiometry of the network interactions. However, after WGD, these same densely connected genes will tend to be preserved in duplicate because all of their neighbouring genes are also duplicated. In those circumstances, natural selection will tend to disfavour the loss of a duplicate copy because that loss will introduce the same sorts of dosage imbalances produced by single-gene duplications. Freeling & Thomas (2006) pointed out that this preferential retention of highly connected gene duplicates can drive increased genetic complexity. I suggest that regulatory network divergence is not only intrinsically interesting, but also serves as a model for understanding the genesis of evolutionary novelty.
Acknowledgements
I would like to thank K. Byrne for assistance with the Yeast Gene Order Browser (http://wolfe.gen.tcd.ie/YGOB/), and P. Edger, J. Mower, S. Ó hÉigeartaigh, J. C. Pires and K. Wolfe for helpful discussions during the preparation of this manuscript. This work was supported by the Reproductive Biology Group of the Food for the Twenty-First Century programme at the University of Missouri and by a Science Foundation Ireland grant to K. Wolfe.
References
- Aury J. M., et al. 2006Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature 444, 171–178 (doi:10.1038/nature05230) [DOI] [PubMed] [Google Scholar]
- Birchler J. A., Veitia R. A.2007The gene balance hypothesis: from classical genetics to modern genomics. Plant Cell 19, 395–402 (doi:10.1105/tpc.106.049338) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G., Wolfe K. H.2004Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 16, 1679–1691 (doi:10.1105/tpc.021410) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne K. P., Wolfe K. H.2005The Yeast Gene Order Browser: combining curated homology and syntenic context reveals gene fate in polyploid species. Genome Res. 15, 1456–1461 (doi:10.1101/gr.3672305) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Xu L., Gu Z.2008Regulation dynamics of WGD genes during yeast metabolic oscillation. Mol. Biol. Evol. 25, 2513–2516 (doi:10.1093/molbev/msn212) [DOI] [PubMed] [Google Scholar]
- Conant G. C., Wagner A.2003Convergent evolution of gene circuits. Nat. Genet. 34, 264–266 (doi:10.1038/ng1181) [DOI] [PubMed] [Google Scholar]
- Conant G. C., Wolfe K. H.2006Functional partitioning of yeast co-expression networks after genome duplication. PLoS Biol. 4, e109 (doi:10.1371/journal.pbio.0040109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant G. C., Wolfe K. H.2008Turning a hobby into a job: how duplicated genes find new functions. Nat. Rev. Genet. 9, 938–950 (doi:10.1038/nrg2482) [DOI] [PubMed] [Google Scholar]
- Dietrich F. S., et al. 2004The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science 304, 304–307 (doi:10.1126/science.1095781) [DOI] [PubMed] [Google Scholar]
- Dujon B., et al. 2004Genome evolution in yeasts. Nature 430, 35–44 (doi:10.1038/nature02579) [DOI] [PubMed] [Google Scholar]
- Edger P. P., Pires J. C.2009Gene and genome duplications: the impact of dosage sensitivity on the fate of nuclear genes. Chromosome Res. 17, 699–717 (doi:10.1007/s10577-009-9055-9) [DOI] [PubMed] [Google Scholar]
- Flake G. W.1998The computational beauty of nature: computer explorations of fractals, chaos, complex systems, and adaptation Cambridge, MA: MIT Press [Google Scholar]
- Force A., Lynch M., Pickett F. B., Amores A., Yan Y., Postlethwait J.1999Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151, 1531–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M., Thomas B. C.2006Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome Res. 16, 805–814 (doi:10.1101/gr.3681406) [DOI] [PubMed] [Google Scholar]
- Gu Z., Nicolae D., Lu H. H.-S., Li S.2002Rapid divergence in expression between duplicate genes inferred from microarray data. Trends Genet. 18, 609–613 (doi:10.1016/S0168-9525(02)02837-8) [DOI] [PubMed] [Google Scholar]
- Gu Z., Steinmetz L. M., Gu X., Scharfe C., Davis R. W., Li H.2003Role of duplicate genes in genetic robustness against null mutations. Nature 421, 63–66 (doi:10.1038/nature01198) [DOI] [PubMed] [Google Scholar]
- Gu X., Zhang Z., Huang W.2005Rapid evolution of expression and regulatory divergences after yeast gene duplication. Proc. Natl Acad. Sci. USA 102, 707–712 (doi:10.1073/pnas.0409186102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelzim N., Bottani S., Bourgine P., Kepes F.2002Topological and causal structure of the yeast transcriptional regulatory network. Nat. Genet. 31, 60–63 (doi:10.1038/ng873) [DOI] [PubMed] [Google Scholar]
- Harbison C. T., et al. 2004Transcriptional regulatory code of a eukaryotic genome. Nature 431, 99–104 (doi:10.1038/nature02800) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger C. T., Carroll S. B.2007Gene duplication and the adaptive evolution of a classic genetic switch. Nature 449, 677–681 (doi:10.1038/nature06151) [DOI] [PubMed] [Google Scholar]
- Hughes A. L.2005Gene duplication and the origin of novel proteins. Proc. Natl Acad. Sci. USA 102, 8791–8792 (doi:10.1073/pnas.0503922102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F.1977Evolution and tinkering. Science 196, 1161–1166 (doi:10.1126/science.860134) [DOI] [PubMed] [Google Scholar]
- Jeong H., Tombor B., Albert R., Oltvai Z. N., Barabási L.2000The large-scale organization of metabolic networks. Nature 407, 651–654 (doi:10.1038/35036627) [DOI] [PubMed] [Google Scholar]
- Jeong H., Mason S. P., Barabási A.-L., Oltvai Z. N.2001Lethality and centrality in protein networks. Nature 411, 41–42 (doi:10.1038/35075138) [DOI] [PubMed] [Google Scholar]
- Jothi R., Balaji S., Wuster A., Grochow J. A., Gsponer J., Przytycka T. M., Aravind L., Babu M. M.2009Genomic analysis reveals a tight link between transcription factor dynamics and regulatory network architecture. Mol. Syst. Biol. 5, 294 (doi:10.1038/msb.2009.52) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M., Birren B. W., Lander E. S.2004Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 428, 617–624 (doi:10.1038/nature02424) [DOI] [PubMed] [Google Scholar]
- King M. C., Wilson A. C.1975Evolution at two levels in humans and chimpanzees. Science 188, 107–116 (doi:10.1126/science.1090005) [DOI] [PubMed] [Google Scholar]
- Klemm K., Bornholdt S.2005Topology of biological networks and reliability of information processing. Proc. Natl Acad. Sci. USA 102, 18 414–18 419 (doi:10.1073/pnas.0509132102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtzman C. P., Robnett C. J.2003Phylogenetic relationships among yeasts of the ‘Saccharomyces complex' determined from multigene sequence analyses. FEMS Yeast Res. 3, 417–432 (doi:10.1016/S1567-1356(03)00012-6) [DOI] [PubMed] [Google Scholar]
- Lee T. I., et al. 2002Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298, 799–804 (doi:10.1126/science.1075090) [DOI] [PubMed] [Google Scholar]
- Li F., Long T., Lu Y., Ouyang Q., Tang C.2004The yeast cell-cycle network is robustly designed. Proc. Natl Acad. Sci. USA 101, 4781–4786 (doi:10.1073/pnas.0305937101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscombe N. M., Babu M. M., Yu H., Snyder M., Teichmann S. A., Gerstein M.2004Genomic analysis of regulatory network dynamics reveals large topological changes. Nature 431, 308–312 (doi:10.1038/nature02782) [DOI] [PubMed] [Google Scholar]
- Maere S., de Bodt S., Raes J., Casneuf T., van Montagu M., Kuiper M., van de Peer Y.2005Modeling gene and genome duplications in eukaryotes. Proc. Natl Acad. Sci. USA 102, 5454–5459 (doi:10.1073/pnas.0501102102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan S., Alon U.2003Structure and function of the feed-forward loop network motif. Proc. Natl Acad. Sci. USA 100, 11 980–11 985 (doi:10.1073/pnas.2133841100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewes H. W., Heumann K., Kaps A., Mayer K., Pfeiffer F., Stocker S., Frishman D.1999MIPS: a database for genomes and protein sequences. Nucleic Acids Res. 27, 44–48 (doi:10.1093/nar/27.1.44) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo R., Shen-Orr S., Itzkovitz S., Kashtan N., Chklovskii D., Alon U.2002Network motifs: simple building blocks of complex networks. Science 298, 824–827 (doi:10.1126/science.298.5594.824) [DOI] [PubMed] [Google Scholar]
- Ohno S.1970Evolution by gene duplication New York, NY: Springer [Google Scholar]
- Papp B., Pal C., Hurst L. D.2003Dosage sensitivity and the evolution of gene families in yeast. Nature 424, 194–197 (doi:10.1038/nature01771) [DOI] [PubMed] [Google Scholar]
- Press W. H., Teukolsky S. A., Vetterling W. A., Flannery B. P.1992Numerical recipes in C: the art of scientific computing New York, NY: Cambridge University Press [Google Scholar]
- Prill R. J., Iglesias P. A., Levchenko A.2005Dynamic properties of network motifs contribute to biological network organization. PLoS Biol. 3, e343 (doi:10.1371/journal.pbio.0030343) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannell D. R., Frank A. C., Conant G. C., Byrne K. P., Woolfit M., Wolfe K. H.2007Independent sorting-out of thousands of duplicated gene pairs in two yeast species descended from a whole-genome duplication. Proc. Natl Acad. Sci. USA 104, 8397–8402 (doi:10.1073/pnas.0608218104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoighe C., Wolfe K. H.1999Yeast genome evolution in the post-genome era. Curr. Opin. Microbiol. 2, 548–554 (doi:10.1016/S1369-5274(99)00015-6) [DOI] [PubMed] [Google Scholar]
- Shen-Orr S. S., Milo R., Mangan S., Alon U.2002Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 31, 64–68 (doi:10.1038/ng881) [DOI] [PubMed] [Google Scholar]
- Sokal R. R., Rohlf F. J.1995Biometry, 3rd edn New York, NY: W. H. Freeman and Company [Google Scholar]
- Steinmetz L. M., et al. 2002Systematic screen for human disease genes in yeast. Nat. Genet. 31, 400–404 [DOI] [PubMed] [Google Scholar]
- Stoltzfus A.1999On the possibility of constructive neutral evolution. J. Mol. Evol. 49, 169–181 (doi:10.1007/PL00006540) [DOI] [PubMed] [Google Scholar]
- Teichmann S., Babu M. M.2004Gene regulatory network growth by duplication. Nat. Genet. 36, 492–496 (doi:10.1038/ng1340) [DOI] [PubMed] [Google Scholar]
- Tsong A. E., Tuch B. B., Li H., Johnson A. D.2006Evolution of alternative transcriptional circuits with identical logic. Nature 443, 415–420 (doi:10.1038/nature05099) [DOI] [PubMed] [Google Scholar]
- Wagner A.2001The yeast protein interaction network evolves rapidly and contains few redundant duplicate genes. Mol. Biol. Evol. 18, 1283–1292 [DOI] [PubMed] [Google Scholar]
- Wagner A.2002Asymmetric functional divergence of duplicate genes in yeast. Mol. Biol. Evol. 19, 1760–1768 [DOI] [PubMed] [Google Scholar]
- Wagner A., Fell D. A.2001The small world inside large metabolic networks. Proc. R. Soc. Lond. B 268, 1803–1810 (doi:10.1098/rspb.2001.1711) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts D. J., Strogatz S. H.1998Collective dynamics of ‘small-world' networks. Nature 393, 440–442 (doi:10.1038/30918) [DOI] [PubMed] [Google Scholar]
- Wolfe K. H., Shields D. C.1997Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387, 708–713 (doi:10.1038/42711) [DOI] [PubMed] [Google Scholar]
- Wray G. A., Hahn M. W., Abouheif E., Balhoff J. P., Pizer M., Rockman M. V., Romano L. A.2003The evolution of transcriptional regulation in eukaryotes. Mol. Biol. Evol. 20, 1377–1419 (doi:10.1093/molbev/msg140) [DOI] [PubMed] [Google Scholar]
- Yoon J., Blumer A., Lee K.2006An algorithm for modularity analysis of directed and weighted biological networks based on edge-betweenness centrality. Bioinformatics 22, 3106–3108 (doi:10.1093/bioinformatics/btl533) [DOI] [PubMed] [Google Scholar]