

Abstract
Vibrational coherence spectroscopy is used to study the low frequency dynamics of the truncated dimer of human cystathionine β-synthase (CBS). CBS is a pyridoxal-5′-phosphate-dependent heme enzyme with cysteine and histidine axial ligands that catalyzes the condensation of serine and homocysteine to form cystathionine. A strong correlation between the “detuned” coherence spectrum (which probes higher frequencies) and the Raman spectrum is demonstrated and a rich pattern of modes below 200 cm−1 is revealed. Normal coordinate structural decomposition (NSD) of the ferric CBS crystal structure predicts the enhancement of normal modes with significant heme “doming”, “ruffling”, and “saddling” content and they are observed in the coherence spectra near ~40 cm−1, ~60 cm−1, and ~90 cm−1. When pH is varied, the relative intensities and frequencies of the low-frequency heme modes indicate the presence of a unique protein-induced heme structural perturbation near pH 7 that differs from what is observed at higher or lower pH. For ferric CBS, we observe a new mode near ~25 cm−1, possibly involving the response of the protein, which exhibits a phase jump of ~π for excitation on the blue and red side of the Soret band maximum. The low frequency vibrational coherence spectrum of ferrous CBS is also presented, along with our efforts to probe its NO-bound complex. The CBS-CO geminate rebinding kinetics are similar to the CO-bound form of the gene activator protein CooA, but with the appearance of a significant additional kinetic inhomogeneity. Analysis of this inhomogeneity suggests that it arises from the two subunits of CBS and leads to a factor of ~20 for the ratio of the average CO geminate rebinding rates of the two subunits.
Keywords: femtosecond pump-probe, vibrational coherence spectroscopy, cystathionine β-synthase, low frequency modes, geminate rebinding, heme proteins
Introduction
Recent developments in femtosecond laser technologies allow new applications in the study of ultrafast excitation and decay of non-equilibrium states in biological systems, especially in proteins1–4. Vibrational coherence spectroscopy1;5–7(VCS) provides significant new structural information because it can monitor previously inaccessible low frequency vibrational modes of biological systems that fall within the spectral bandwidth of the femtosecond laser pulse. These low frequency modes are thermally accessible (< 200 cm−1) and therefore are most likely to play important roles as reaction coordinates in various protein mediated processes. Here we investigate heme proteins, which perform a broad variety of functions in living systems and contain one of the most visible and versatile cofactors in the biology (iron protoporphyrin IX, FePPIX).
The heme protein under investigation is the mammalian cystathionine β-synthase (CBS), which catalyzes a pyridoxal phosphate (PLP)-dependent condensation of serine and homocysteine to form cystathionine8–10. CBS is a ~63 kDa modular protein with an N-terminal domain containing the heme and PLP cofactors in the catalytic core and a C-terminal regulatory domain associated with S-adenosylmethionine binding11;12. In the truncated form, which is studied here, the topic of this investigation, the C-terminal regulatory domain is removed and the resulting catalytic core forms an active dimeric species. Each monomer has one heme and one PLP. The heme in the truncated form is six-coordinate and low spin in both the ferrous and ferric states13, with neutral nitrogen (His65) and a thiolate (Cys52) as ligands to the heme14;15.
Figure 1 depicts the alignment of the heme and PLP cofactors obtained from the crystal structure (PDBID:1M54)16 of ferric CBS at pH 8.0. A Schiff’s base linkage at Lys 119 and hydrogen bonds between the phosphate group and Thr257 and 260 on helix 6 of the protein positions the PLP cofactor. A hydrogen bond between Cys 52, which is coordinated to the heme iron, and Arg 266 (on helix 6) has been suggested17;18 to be a key element in signaling between the two cofactors when CO binds and displaces the cysteine ligand. The CO adduct of ferrous CBS results in complete loss of the enzymatic activity and displays vibrational frequencies and absorption spectra characteristic of a histidine-heme-CO complex in a hydrophobic environment, demonstrating that the Fe–S (Cys) bond is labile19.
Figure 1.
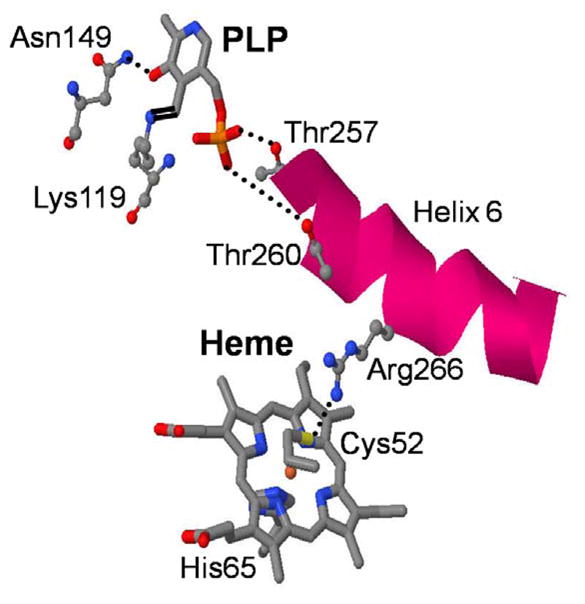
Crystal structures of ferric CBS at pH 8.0 generated from the PDBID:1M5416 is shown only with the heme and PLP cofactors. The hydrogen bonding between Cys 52 and Arg266, phosphate oxygen atoms of PLP to the Thr257 and Thr 260 and Asn 149 to the O3′ of the PLP are shown as dotted line. The formation of Schiff base between Lys 119 and PLP are also shown.
When NO binds to ferrous CBS it results in the formation of a five-coordinate nitrosyl species in which both endogenous ligands have been lost20. Like CO, NO inhibits CBS and demonstrates that the perturbation of the heme environment by these ligands is communicated to the PLP active site with concomitant inhibition of enzyme activity. Generally, this is consistent with a regulatory role for the heme group in the CBS reaction cycle. The investigation of bimolecular binding of CO to the truncated form of CBS suggested that CO binding is anticooperative17. Studies of CBS mutants have clarified that environmental changes at the heme are communicated to the active site of CBS through the involvement of the Arg266 residue21. Recently, Weeks et al. have investigated the CO inhibition mechanism with fluorescence and resonance Raman spectroscopies18. The loss of catalytic activity is proposed to arise from a tautomeric shift of PLP as CO binding displaces Cys52 and Arg 266, along with helix 6 and the PLP cofactor. If the Asn 149 H-bond to the oxygen (O′) of PLP is broken by this displacement, this alters the tautomeric state. (The tautomers of the internal aldimine of PLP in CBS are shown in Fig. S3 of the supporting material).
In this work VCS is used to probe the low frequency vibrational motions of the heme and how they depend on structural distortions that are induced by the CBS protein environment. Low frequency modes are “soft” (weak force constants) so that protein-induced distortions along these normal coordinates are the most likely to occur, allowing for protein control of the cofactor properties. For example, the heme doming coordinate, with frequency near 40 cm−1, has been found to be an important thermally accessible component of the CO binding reaction coordinate2;22. Other thermally accessible low-frequency heme modes, such as ruffling and saddling, that can mix iron and porphyrin orbitals are potentially important in governing redox processes. Frequency domain techniques (such as infrared and Raman) have difficulty probing the thermally accessible region below ~200 cm−1 in aqueous solutions. As a result, we utilize VCS in this study, in order to provide a first look at both the frequencies and the phases of the low frequency heme modes23;24.
Experimental
Sample preparation
Truncated human CBS lacking 143 amino acids at the C-terminus was purified from a recombinant expression system, as described previously13. The CBS protein was prepared in the pH range 6–9. For pH 8 and 9, 100 mM Tris buffer containing 250 mM KCl was used. For pH 6 to 7.5, 100 mM potassium phosphate buffer containing 250 mM KCl was used. For the vibrational coherence spectroscopy measurements, the concentration of the protein samples was adjusted so that the optical density of the sample at the pump wavelength was between 0.7 and 1.0 O.D. in a 1-mm pathlength spinning sample cell. The final concentration was between 100 and 150 μM. The PLP cofactor has a very low extinction coefficient ~5,000 M−1 cm−1 25 compared to heme in CBS ~81,000 M−1 cm−1 26, so its contribution to the coherence signals at equivalent concentration is negligible.
To prepare ferrous CBS, the protein was degassed for 20 min under an argon atmosphere and the protein concentration was adjusted so that the final absorbance of the reduced species was ~1 O.D./mm of path length at the pump wavelength. Subsequently, the samples were transferred into a glovebox and 3 μL of 1 M sodium dithionite (Na2S2O4) was added to 250 μL of buffered sample to obtain the ferrous species. To obtain the NO adducts, 2 μL of 1 M sodium nitrite (NaNO2) solution was added to the ferrous CBS sample. The reduced and NO-bound samples were then transferred into the air-tight sample cell.
The absorption spectra of all samples were recorded after the preparation procedure to confirm that the desired chemical modifications were achieved and that they were complete. Absorption spectra were also taken at the conclusion of the experiments to check for possible photochemical damage and none was observed with the exception of the ferrous NO sample, which showed evidence of re-oxidation (see supporting information Fig. S1).
Optical System
The description of the optical system has been published7;24;27 and further details can be found in the supporting information. Two detection geometries for the VCS experiment allowed us to focus on different ranges of the coherent signal. An open-band detection scheme, which integrates over the full probe pulse bandwidth, improves the fidelity of the low frequency response in the range 20–100 cm−1 7;23;24;27. The detuned detection scheme selectively detects the higher frequency response in the coherent signal7;23;24;28 with improved reliability in the ~200–400 cm−1 region. The detuned detection is accomplished using a monochomator that detects 5 nm away from the carrier wavelength of the probe pulse with a bandwidth of 0.5 nm. The detuned results allow for direct comparison with frequency domain techniques like resonance Raman spectroscopy that probe modes above 200 cm−1. The CO binding kinetics were carried out using a two-color pump-probe setup working at 190 kHz as described previously29.
Data Analysis
The time domain optical response results from both population transfer and vibrational coherence. To obtain the residual coherence signal, the monotonic population decay components must be removed and digitization of the experimental signal is done using a lock-in amplifier (LIA) on a 24 bit scale so that it allows for a sufficient dynamic range to detect the low amplitude coherence signals. (The fractional change of transmittance, ΔT/T, for the open band oscillatory signals is on the order of 10−4–10−5).
The data analysis follows a sequential approach. Either linear predictive singular value decomposition (LPSVD) or a maximum entropy method (MEM) algorithm (which removes assumptions regarding the number of exponential decay processes) is used to retrieve the oscillatory signal. The oscillatory coherence signal arises from the field-induced superposition of the vibrational eigenstates that are coupled to the resonant electronic transition. Since both homogeneous and inhomogeneous decoherence processes damp the oscillatory signals as a function of time, the coherent superposition is lost after a few picoseconds. To quantify the coherent signals, the oscillatory signal left after the removal of the monotonic decay terms is fit to a sum of damped cosine functions,
| (1) |
using the LPSVD procedure. A standard FFT algorithim can also be employed 30 but the phase is not extracted without use of LPSVD. The low frequencies observed in the Raman spectrum can be used as a control to avoid over- or under-fitting the data. The detuned data (which optimizes the detection of the higher frequencies within the pulse bandwidth) are analyzed first to make sure that the global fitting parameters (such as the number of oscillators) are properly correlated with the frequencies determined independently from the Raman spectrum. The power spectra obtained from the detuned measurements generally extend below 200 cm−1, which represents the practical lower limit cutoff for heme protein resonance Raman measurements. After analysis of the detuned data, comparisons are made with the open band frequencies in the regions of overlap. The open band analysis starts by selecting a low number of oscillations and then gradually increasing that number until the higher frequencies are consistent with the detuned measurements and Raman spectroscopy.
The relative intensities of the coherences depend on the laser pulse bandwidth and detuning conditions. The LPSVD spectra, as presented, are not corrected for the Gaussian-distributed relative pumping efficiencies within the bandwidth of the laser pulse. In order to selectively enhance the lower frequency modes, the auto-correlation pulse widths are typically kept near 70 ± 5 fs. Examination of the data from numerous independent samples has generally produced very good reproducibility and strong correlations between the frequencies extracted using the three experimental conditions (Raman, detuned coherence, and open band coherence). We note that the phase (ϕi) cannot be determined from Raman measurements. We estimate that, for mode frequencies below 100 cm−1, the phases determined to be near (~) a particular value will have an error at or below ± π/6 (± 30°).
Resonance Raman spectra
Resonance Raman spectra were recorded using a standard setup with a 90° light-collection geometry and a single grating monochromator (model No. 1870B; Spex Industries, Edison, NJ). An optical polarization scrambler was inserted in front of the monochromator to obtain the intensity of the scattered light. The monochromator output was coupled to a liquid-nitrogen-cooled charge-coupled detector (Princeton Instruments). In order to improve detection in the low-frequency domain of Raman shifts, an interferometric notch filter (Kaiser Optical Systems, Ann Arbor, MI) was used to extinguish the laser light. Samples were excited with a 413.1 nm laser line generated by a krypton laser (Innova 300, Coherent) with powers ranging from ~ 5 to 33 mW. A standard quartz cuvette (NSG Precision Cells, Farmingdale, NY) was used for the experimental Raman measurements.
Results
Excitation profile of the ferric form
The absorption spectrum of the ferric form of CBS at pH 8 is shown in Fig. 2A. The absorption maximum is at 428 nm (Soret peak) and another broad asymmetric absorption band is present at 550 nm (Q band). Reduction of the heme results in a large red shift of the Soret peak to 448 nm consistent with retention of the thiolate ligand in the six-coordinate ferrous heme (analogous to the CO bound, thiolate ligated, cytochrome P450s). The femtosecond time resolved optical transmittance of ferric CBS was obtained at the excitation wavelengths shown as circles in Fig. 2A. The normalized time resolved transmittance is illustrated in Fig. 2B and the corresponding rate constants and amplitudes for the population dynamics are given in Table S1 of the supporting information.
Figure 2.

(A) Electronic absorption spectra of ferric CBS dimer at pH 8. The Soret absorption maximum is at 428 nm. (B) Normalized time-resolved optical transmittance (ΔT) of CBS at different pump/probe excitation wavelengths (shown as color-coded circles in Fig 1A). Near the Soret maximum (excitation at 425 and 432 nm), the transmittance traces show a bleaching-recovery signal (ΔT>0), whereas for 412 and 443 nm excitation the signal involves an antibleaching component (ΔT<0).
It is interesting to note in Fig. 2B that the data taken on the red (443 nm) and blue (412 nm) sides of the Soret maximum show a transient absorption (or anti-bleach, ΔT < 0), while the data near the peak (425 and 432 nm) display bleaching signals (ΔT > 0). Such behavior is expected when there is vibrational heating, which leads to transient red-shifting and broadening of the Soret band (along with a decrease in peak intensity)31. As the non-equilibrium heme system cools, the Soret band undergoes spectral diffusion as it narrows and blue shifts back to the initial equilibrium absorption spectrum. In addition to the effect of the heating and cooling process, photolysis of axial ligands or photoreduction (or a combination of all) can potentially be involved in the content of the transient signals. Because there is no detectable heme fluorescence or hot luminescence from the Soret excited state, excited state population beyond the 100 fs timescale is taken to be negligible32.
Figure 3 shows the open-band vibrational coherence spectra of the ferric state of CBS at pH 8 using the same excitation wavelengths as in Fig. 2B. The experimental data were analyzed using LPSVD. The left panels show the oscillatory data (small circles) and the LPSVD fits (solid red line). The LPSVD components corresponding to the dominant modes and their phases are also shown as the blue (~40 cm−1) or green (~25 cm−1) solid lines. The right panels show the corresponding amplitudes of the power spectra. The analysis of the experimental data shows that excitation near the equilibrium absorption maximum yields dominant modes at ~40 cm−1 and at ~90 cm−1. The phases of the low frequency modes for different excitations are given in Table 1. The ~40 cm−1 mode (γa) is thought to contain a large component of heme doming and is especially active when ligand photo-dissociation is present7;24;30;33–36 The ~90 cm−1 mode (γc) has been previously suggested to contain a large component of heme saddling7 and this assignment is supported by the NSD analysis discussed below.
Figure 3.

The open-band coherence spectra of the ferric state of the CBS dimer at pH 8. The different pump/probe excitation wavelengths are given in the figure. The experimental data were analyzed by using LPSVD and the left panels show the oscillatory components (circle) and the LPSVD fits (solid red line). The LPSVD components corresponding to the dominant modes and their phases are also shown displaced for visibility (~40 cm−1 as blue and ~25 cm−1 as green solid lines). The right panel shows the corresponding power spectrum amplitudes. For excitation near the Soret maximum (425 and 432 nm), the ~40 cm−1 and ~90 cm−1 modes dominate, while excitation further to the blue (412 nm) and red (443 nm) side of the Soret maximum yields a dominate mode near ~25 cm−1. The oscillation amplitudes are extracted by lock-in detection in arbitrary units, but they are on the order of ΔT/T~10−4–10−5 in this and the other figures.
Table 1.
The low frequency modes (< 200 cm−1) of ferric CBS at pH 8†.
| λex (nm) | γ0 | γa | γb | γc |
|---|---|---|---|---|
| 412 | 25[s] 298° |
- | 57[w] | 84[w] |
| 425 | - | 37[s] 70° |
60[w] | 86[s] 16° |
| 432 | 25[w] | 39[m] 87° |
62[w] | 86[s] 28° |
| 443 | 23[s] 133° |
38[w] | 66[w] | - |
The mode frequencies are in cm−1 and their phases in degree are denoted below. The [s, m, w] denote [strong, medium, weak] intensity in the extracted power spectrum.
Although the γa and γc modes are enhanced near the peak of the Soret band, for excitation on the blue (412 nm) and red (443 nm) sides of the Soret maximum there is less apparent activity of these modes because a new mode (γ0) appears near ~25 cm−1 and dominates the coherence spectrum. As can be seen from Fig. 3, the phase of the ~25 cm−1 mode changes by ~ π as the wavelength is tuned between 412 nm and 443 nm. The possibility that the feature near 25 cm−1 could arise due to over-fitting or under-fitting of the data by the LPSVD algorithm was explored in some detail. When the power spectrum, obtained using LPSVD, was compared with that obtained using a maximum entropy analysis to extract the oscillatory components, it was found that both methods generated a spectrum with a strong 25 cm−1 component. As a result of this, and its π phase shift, we conclude that this feature is a genuine response of the material system and is not due to fitting artifacts, which can sometimes appear at very low frequencies.
To optimize the time-domain detection of the higher frequencies, we employed a detuned detection scheme, as outlined in the experimental section. The detuned experimental configuration selects for higher frequency vibrational components, while the open-band condition probes the lowest frequencies with the best resolution and fidelity and it also eliminates the non-resonant (birefringent) signals due to the solvent23;28;37. Figure 4 shows the correlation between the Raman and coherence spectra for CBS at pH 8. The coherence data were collected with both open-band and detuned detection, using pulses with a center (i.e., carrier) wavelength of 432 nm. The spontaneous resonance Raman experiments are unable to probe effectively below 150 cm−1 due to cancellation of the oppositely signed Raman Franck-Condon amplitudes38 as well as to limitations brought on by Rayleigh and quasielastic light scattering and the nonlinearities associated with the low frequency cutoff filters used to attenuate the large light flux in the region of laser excitation. In Fig. 4, the inset shows the oscillatory components of the experimental coherence data (detuned, blue upper curve and open-band, green lower curve). The detuned measurements utilize a 0.5 nm spectral window, detuned 5 nm to the blue of the carrier frequency maximum (λpr = 427 nm). Figure 4 shows good correlation between the open band and detuned detection techniques in both the low and higher frequency regions. One exception is the 242 cm−1 mode, which is absent from the open band measurement, presumably due to the loss of higher frequency resolution and (relative) intensity that is expected under open band detection conditions. It is likely that the 242 cm−1 mode has merged with the strong mode at 278 cm−1, yielding the feature at 266 cm−1. There is also good correlation (~ ±5 cm−1) between the power spectrum obtained with detuned detection (middle blue curve) and the resonance Raman measurements (top red curve), especially for the relatively strong 370 cm−1 mode and the mode at 180 cm−1. It is noteworthy that the feature assigned to the Fe-S vibration14 near 316 cm−1 is unresolved in the coherence spectra, possibly due to the differing resonance conditions (413 vs. 432 nm) or to photolysis of the Fe-S bond. In order to test for possible loss of the Fe-S bond via long-lived photoproducts in CBS, resonance Raman spectra of stationary samples were measured with varying power from 5 mW to 33 mW, but no changes were observed.
Figure 4.

The correlation between the Raman and coherence spectra for ferric CBS at pH 8. The Raman spectrum (red) was measured with excitation at 413.1 nm, whereas open band (green) and detuned (blue) coherence spectra were measured at a carrier wavelength of 432 nm. The detuned coherence data were collected with a 0.5 nm spectral window, detuned 5 nm to the blue of the carrier wavelength. The time domain oscillation data are shown in the inset as small circles and the LPSVD fits are the solid lines through the data. There is good correlation between the Raman and coherence spectral frequencies, with estimated errors of roughly ±5 cm−1.
As shown in Fig. 4 for ferric CBS, strong high frequency modes appear at 278 and 338 cm−1 in the detuned measurement, which correlate with Raman modes at 271 and 347 cm−1 (the latter has been assigned to ν839). It is interesting to note that the mode at 370 cm−1, which is assigned as δ(CβCcCd), is correlated well in all three measurements, due to its relatively long damping time.
Ferrous and NO bound CBS
The perturbation of the heme environment by NO binding is communicated to the active site PLP cofactor of CBS and inhibits enzymatic activity20 although this effect is much weaker than for CO19. Thus, studies of the NO bound form may help elucidate the regulatory role of the heme group in CBS. The electronic absorption spectra of the ferric and ferrous states, along with the ferrous NO bound form of CBS at pH 8 are shown in Fig. 5A. Figure 5B shows the femtosecond time resolved transmittance (ΔT) of the above samples, obtained by exciting at 425 nm. The lifetimes and amplitudes of the corresponding species obtained by exponential fitting are given in Table S2 of the supporting information.
Figure 5.

(A) Electronic absorption spectra of the ferric, ferrous, and ferrous NO bound forms of the CBS dimer at pH 8. The Soret absorption maxima are 428, 448 and 378 nm for the respective states. (B) Normalized time resolved transmittance (ΔT) of the above complexes obtained with pump/probe excitation at 425 nm. The kinetic traces show a bleaching recovery signal (ΔT>0) for all the three complexes.
The open-band coherence spectra of the ferric, ferrous, and the ferrous NO-bound forms of CBS at pH 8, are compared in Fig. 6. The pump/probe excitation wavelength is at 425 nm. The left panels show the oscillatory data and the LPSVD fits (solid red line). The LPSVD component corresponding to the dominant doming mode, γa, near 35–40 cm−1 is also shown (blue solid line) along with its extracted phase. The right panels show the corresponding amplitudes of the power spectra. When compared to the ferric form, the ferrous form has a much stronger γb mode found between ~60–70 cm−1 and a much weaker γc mode, which usually occurs in the vicinity of ~90 cm−1. The γb mode at ~60–70 cm−1 in nitrophorin was previously correlated with a ruffling distortion of the heme, based on x-ray structures and DFT calculations 40;41.
Figure 6.

The open-band coherence spectra of the ferric, ferrous, and ferrous NO bound forms of CBS at pH 8. The pump/probe excitation wavelength is at 425 nm. The left panels show the oscillatory components (circle) and the LPSVD fits (solid red line). The LPSVD components corresponding to the strong γa mode near ~40 cm−1 are also shown (blue solid line), along with the extracted phase. The right panels show the corresponding power spectrum amplitudes. When compared to the ferric state, the strong activity at ~90 cm−1 (γc) is diminished and replaced by a mode near ~60–70 cm−1 (γb) in the ferrous state. When NO is bound to the ferrous form, the ~90 cm−1 (γc) mode seems to reappear. We assign this spectrum to reoxidized material (see text).
When the ferrous NO bound sample is studied, the ~90 cm−1 mode reappears and the γb mode at ~60–70 cm−1 diminishes so that the power spectrum amplitudes of the ferric and ferrous-NO bound forms appear to be nearly the same. The absorption spectrum of the ferrous NO sample, following the coherence measurement suggests loss of NO and formation of an oxidized six-coordinate species (see supporting information). Because the ferric species is strongly resonant with the laser excitation at 425 nm, even a minority component could dominate the coherence spectrum and explain the similarity of the top and bottom panels of Fig. 6. This leads us to conclude that the lower panel in Fig. 6 depicts the VCS spectrum of oxidized material, rather than that of the ferrous NO complex.
pH dependence of ferric CBS
In order to investigate the tautomeric equilibrium of the PLP co-factor and potential protein induced structural perturbations of the heme, we measured the low-frequency coherence spectra of CBS as a function of pH. The ferric state was chosen for this study because it is stable in the pH regions studied.
The electronic absorption spectra of ferric CBS in the pH range 6 to 9 are shown in Fig. 7A. There is no shift of the Soret absorption maximum (428 nm) upon varying the pH. Figure 7B shows the pH dependence of the time resolved transmittance (ΔT) at 425 nm. The optical response involves a prompt bleaching-recovery signal (ΔT>0) at each pH. The trend, or appearance, of the kinetics is nearly the same for all pH samples. The exponential rate constants and amplitudes that fit the data at different pH values are given in Table S3 of the supporting information. The pH-independent average time constants and their respective amplitudes are 0.14 ps (49.1%), 0.62ps (22.6%) and 3.93 ps (28.3%). This latter time constant has larger uncertainty due to the limited time range of data set. However, it is consistently reproduced in a variety of experiments (c.f., Tables S1–S3 of supplementary information).
Figure 7.

(A) Electronic absorption spectra of the ferric state of the CBS dimer at pH 6 to 9. The Soret absorption maximum is located at 428 nm and is independent of sample pH. (B) Normalized time resolved transmittance (ΔT) of the ferric CBS samples as a function of pH was obtained using pump/probe excitation at 425 nm. The traces show a bleaching recovery signal (ΔT>0) that is independent of pH.
Figure 8 shows the results of vibrational coherence experiments at 425 nm with the CBS protein at different pH. The left panels show the oscillatory signals (circles) and the LPSVD fits (solid red line). The LPSVD components corresponding to the γa mode near ~40 cm−1 are also shown (blue solid lines). The right panels show the corresponding power spectrum amplitudes. At pH 8 and 9, the modes at ~40 cm−1 (γa), ~90 cm−1 (γc), and ~180 cm−1 are seen to dominate. When the pH is decreased to 7, the γc mode near 90 cm−1 disappears, or is strongly attenuated, and a new mode near ~75 cm−1 appears. At the same time, the γa mode near 40 cm−1 “softens” and shifts to 32 cm−1. Further decrease of the pH to 6 restores the ~90 cm−1 and 40 cm−1 modes, suggesting that a unique heme conformation is present near pH 7.
Figure 8.

The open-band coherence spectra of the ferric state of CBS at different pH (as in Fig. 6). The pump/probe excitation wavelength is at 425 nm. The left panels show the oscillatory components (circle) and the LPSVD fits (solid red line). The LPSVD component corresponding to the strong γa mode at ~40 cm−1 and the phase is also shown (blue solid line). The right panels show the corresponding amplitude of the power spectra. At pH 8 and 9, the modes at ~40 cm−1 (γa) and ~90 cm−1 (γc) are seen to dominate. When the pH is decreased to ~ 7, a mode at ~75 cm−1 (γb) appears and the mode at 90 cm−1 minimizes or disappears. A further decrease of pH to 6, reveals again the strong modes at ~40 cm−1 (γa) and ~90 cm−1 (γc).
NSD analysis
The normal coordinate structural decomposition (NSD) analysis for ferric CBS is shown in Fig. 9. The NSD analysis determines the geometric distortions of the heme when it is exposed to different protein environments41;42. The details of this type of analysis can be found in the supporting information. In short, we find the displacements along low frequency out-of-plane (OOP) modes of different symmetry (such as propellering, ruffling, saddling, waving(x), waving(y), doming and inverse doming) and use them to describe the observed x-ray structure of the heme.
Figure 9.

Crystal structures and NSD analysis of the heme in ferric CBS at pH 8.0. The displacements along each of the low frequency normal mode unit vectors is given in mass weighted coordinates (amu1/2 Å) of the Fe and 24 porphyrin (4N, 20C) atoms. The color coding for the modes is pro: propellering (blue), ruf: ruffling (green), sad: saddling (red), wav(x): wavingx (light blue), wav(y): wavingy (yellow), dom: doming (purple), invdom: inverse doming (gray). The minus sign of displacement is defined only for doming and inverse doming to indicate the direction of Fe displacement (+: proximal; −: distal). The crystal sctucture comes from the protein data bank (PDB) 1M5416. The NSD analysis shows a strong heme saddling distortion, which is consistent with the appearance of the ~90 cm−1(γc) mode in the coherence spectra of ferric CBS at pH 8.
The NSD analysis in Fig. 9 clearly shows a significant heme saddling distortion is present in the ferric CBS state. This is consistent with the appearance of the ~90 cm−1 (γc) mode in the coherence spectra of CBS at pH 8.0, which has a large component of saddling motion. The doming distortion in the resting state is roughly 40% that of saddling (Fig. 9) and we recall that the coherence intensities should scale as the square of the relative distortions41. Thus, the observation of a relatively strong ~40 cm−1 mode (Fig. 8) is somewhat surprising, although the relative pumping efficiency of the Gaussian laser pulse does enhance the 40 cm−1 mode relative to the 90 cm−1 mode to some degree. It is also possible that the x-ray structure-determined NSD analysis does not capture all aspects of the heme geometric distortions, since the crystal structure at pH 8.0 is taken at relatively low 2.9Å resolution16. Finally, we must also keep in mind that some degree of axial ligand photolysis is possible and this could be responsible for the enhancement of the doming mode.
Ligand binding kinetics
Because of its potential regulatory role as an inhibitor of CBS, we have also studied the CO rebinding kinetics of CBS using the two color pico/femto-second laser system29. We compare the CO geminate rebinding kinetics to CooA because this protein is known to be regulated by CO43. Fig. 10A shows a log-log plot comparing the CO geminate rebinding kinetics for CBS and CooA. The data were fit using the “SRC” model22 and the details are described in the discussion and in the supporting information. The fits in Fig. 10A represent the temporal evolution of the normalized survival population, N(t), of the five-coordinate heme photoproduct as it rebinds the CO ligand (Table 2). The kinetics of CO geminate rebinding in these two proteins take place on the 100 ps time scale and display large overall amplitudes (since there are 2 subunits, the Ig for CBS1,2 in Table 2 should be compared to 0.5 rather than 1.0). For CBS we found it necessary to take into account a level of kinetic inhomogeneity beyond a single SRC heme doming distribution2;22. We assign the additional inhomogeneity to differences in the CO rebinding barrier associated with the two distal pockets of the dimeric CBS structure. Figure S2 of the supporting information compares the CO bound Soret bands of Mb, CooA, and CBS, which independently indicates that significantly more inhomogeneity is present in the latter protein. Additional kinetic studies using multiple pump and probe wavelengths are needed to directly connect the Soret band and kinetic inhomogeneities in CBS.
Figure 10.

(A) Geminate rebinding kinetics of CO to CBS and CooA following excitation at 403 nm and probing at 440 nm. N(t) represents the temporal evolution of the normalized survival population of the five coordinate heme photoproduct as it rebinds the CO ligand. The red solid lines represent the fitting of the experimental data with the SRC model (The details are given in the supporting information). The kinetics of CO geminate rebinding in CBS take place on the 100ps-1ns timescale with a large (~80%) amplitude. (B). Comparison of CO rebinding kinetics in different heme proteins such as myoglobin, CBS and CooA. The geminate recombination of NO to Mb is also plotted. The qualitative similarity of the rebinding kinetics of CO to CooA and CBS with that of NO to Mb suggests that spin selection rules play a minor role in controlling the kinetics of ligand rebinding in heme proteins (see text).
Table 2.
SRC kinetic parametersa for heme-CO systems
| Sample | T | Ig | a0(Å) | σa(Å) | K(N/m) | k1(1011s−1) | k0(1011s−1) | H0(kJ/mol) |
|---|---|---|---|---|---|---|---|---|
| Mb | <180Kb | 1.0 | 0.35 | 0.11 | 13.8 | 1.7×10−3 | 2.8×10−2 | 7.0 |
| PPIX | <180Kc | 1.0 | 0.1 | 0.07 | 27.6d | 1.5 | 1.5 | 0 |
| CooA | 300K | 0.97 | 0.32 | 0.13 | 13.8 | 0.5 | 0.5 | 0e |
| CBS1 | 300K | 0.41 | 0.33 | 0.13 | 13.8 | 1.1 | 1.1 | 0e |
| CBS2 | 300K | 0.41 | 0.30 | 0.13 | 13.8 | 5.2×10−2 | 1.1f | 7.3f |
Temperature is given by T and the geminate amplitude by Ig. The heme doming distribution is specified by its mean value, a0, and width, σa; K is a harmonic approximation to the force constant for motion within the doming potential that is set by the 35–45 cm−1 doming frequency. The parameter k1 subsumes the Arrhenius prefactor, k0, and all “distal” contributions, H0, to the rebinding barrier. Thus, the non-exponential kinetics are fit using only 3 free parameters (a0, σa, and k1) in this model. The values of k0 and H0 can be extracted if measurements are done as a function of T (e.g., ref 64).
The geminate rebinding of MbCO must be studied at low temperature to prevent ligand escape.
The parameter values are fixed for PPIXCO below 180K and relax to larger values above this temperature (PPIX is Fe protoporphyrin IX; see ref 2 for details).
The value of K is increased by the square of the ratio of the low frequency modes determined from prior studies with and without a proximal histidine ligand (see ref 2 and 27 for details).
We assume H0 = 0 in order to keep the Arrhenius prefactor near ~1011s−1 as indicated by the PPIXCO results (temperature dependent studies are needed to verify this).
We tentatively assume that the Arrhenius prefactor of CBS2 is the same as found for CBS1.
The SRC kinetic parameters extracted for the two CBS rebinding components of the dimer (referred to as CBS1 and CBS2) are given in Table 2 where the large difference in k1 values should be noted. The fitting parameter k1 is the product of the normal Arrhenius prefactor k0 and the “distal” barrier, H0, contribution to the rebinding rate:
| (2) |
It is important to recognize that the last two columns of Table 2 are not fitting parameters. They are the fundamental quantities (k0, H0) that must be derived from temperature dependent kinetic measurements, along with application of Eq. 2. In Table 2 we have assumed that the prefactor, k0, is independent of subunit, which then suggests that the distal contribution to the rebinding barrier (H0) is significantly subunit-dependent.
Figure 10B shows a log-log plot comparison of the CO and NO rebinding kinetics to myoglobin, measured over many decades in time, superimposed on the CO rebinding kinetics of CBS and CooA. In myoglobin only ~5% of the CO rebinds geminately44 and most of the photolyzed CO escapes to the solvent with a time constant of about 200 ns. In contrast, about 99% of NO rebinds to Mb geminately on a sub-nanosecond time scale. Prior work17 has demonstrated that on timescales longer than 10 ns, CO binding to CBS and CooA is bimolecular.
Discussion
Coherence spectroscopy selectively probes the low frequency modes in the range 10–200 cm−1. Since heme modes in this region are strongly excited by thermal fluctuations (kBT~200 cm−1 at 300 K), they are good candidates for reaction coordinates connected to protein function. Moreover, because these modes are “soft” and have weak force constants, they are the modes along which the protein can most easily distort the heme structure to modulate its electronic properties and reactivity. The changes in the phase and amplitude of the low frequency modes as the laser excitation wavelength is tuned across the resonant Soret absorption band yield information about the existence of underlying resonant electronic states and about the various mechanisms that are responsible for generating the coherent excitation of the heme.
Ferric CBS
Based on the appearance of the anti-bleaching (transient absorption, ΔT<0) signals on both sides of the Soret maximum shown in Fig. 2B, we have considered processes such as photoreduction, photolysis, and cooling dynamics as potentially contributing to the signals. Since the rate constants for the equilibrium recovery at 443 nm are ~3–4 ps or less, it is likely that they represent cooling dynamics associated with the spectral diffusion of the red-shifted and broadened vibrationally hot Soret band. However, kinetic data on longer timescales will be required in order to fully exclude the possibility of Fe-S bond photolysis45.
The vibrational coherence spectra show a strong mode (γ0) near 25 cm−1 that appears for excitation on both the blue and red sides of the ferric CBS Soret band (Fig. 3). The phase of this mode differs by ~π when the signals from the blue- and red-side Soret excitation are compared. In the absence of inhomogeneous broadening, the amplitude of a field driven coherence signal minimizes near the resonant absorption maximum whereas it maximizes for blue or red excitation23. In other words, a homogeneous vibrational coherence excitation profile is bilobed with a minimum at the resonant absorption peak. However, when the electronic spectra are inhomogeneously broadened, the bilobed feature is “washed out” 23. Hence, one explanation for the relatively weak intensity of the 25 cm−1 mode when ferric CBS is probed at 425 and 432 nm excitation is that this mode is coupled to a relatively homogeneous ferric 6-coordinate electronic resonance absorbing near 428 nm. The difference in phase (~π) that is observed for red and blue excitation is expected for degenerate pump-probe experiments when the coherent excitation creates a wavepacket with displaced coordinates that are independent of the pump excitation wavelength23;24. This can occur for wavepacket preparation on an excited electronic state surface or if a photoproduct ground state is formed during an ultrafast reaction. This condition is not met by a simple field driven preparation of a coherent wavepacket on the ground electronic state surface 23.
Since long-lived (>1ps) Soret-excited heme states are rare or non-existent32, one possibility to explain the 25 cm−1 mode phase and amplitude behavior involves the ultrafast non-radiative decay from the Soret excited state into the ground electronic state. Rapid non-radiative decay is generally associated with the iron containing heme group32;46 and indicates excited state admixtures of intermediate and ground electronic states are facilitating the ultrafast decay. The ultrafast return of the heme to its 6-coordinate electronic ground state (on timescales much faster than the ~1.3 ps period of the 25 cm−1 oscillation) suggests that the 25 cm−1 coherent wavepacket can be prepared by the pump fields with initial momentum and coordinate displacements that are independent of the excitation wavelength. Under such conditions, a coherent mode, coupled to a heme with a homogeneous Soret band absorbing near 428 nm, would exhibit an intensity dip near 428 nm and a phase difference of ~π for blue- and red-side excitation. The rapid exchange of energy between the heme and the protein could also potentially lead to a coherent protein response in this very low frequency region47–49. This possibility is of particular interest since calculations 50;51 have found a peak in the protein density-of-states near 20 cm−1. Since the heme chromophore in CBS is associated with regulation and signal transmission along the α-helix between Arg266 and the PLP co-factor17;21, it is also interesting to speculate that the 25 cm−1 mode may be connected to this signaling process. However, modes in the 20–25 cm−1 region have also been observed to some extent in heme model compounds27, suggesting that they may be inherent to the chromophore itself. For example, coherent signals in this region have been observed in other ferric 6-coordinated heme systems such as CooA, metMb, and bis Im-(Fe3+)PPIX material35.
Next we consider the appearance of the γa (doming) mode near 40 cm−1 in Fig. 3. The NSD analysis in Fig. 9 shows that distortions along the doming coordinate are present, but are relatively small in the 6-coordinate complex compared to saddling and ruffling. The fact that the coherence intensities should scale with the square of the distortion parameter41, suggests a small relative enhancement of the doming mode compared to ruffling and saddling. In contrast, Fig. 3 shows a relatively strong doming mode at 40 cm−1 (with phase near π/2) near the peak of the 6-coordinate Soret band. To some extent this could be due to the Gaussian distributed pump pulse efficiency. On the other hand, the relatively low (2.9Å) crystal structure resolution16 might lead to an underestimate of the heme doming distortion.
Another interpretation of the relatively strong enhancement of the 40 cm−1 mode at 425 and 432 nm is that ligand photolysis of some fraction of the sample takes place. This would generate a very short-lived transient five-coordinate ferric species with a blue-shifted Soret band and a strongly enhanced doming mode. As we know from our earlier work7;34–36, doming that is activated by photolysis corresponds to heme motion that changes both the local equilibrium position and momentum of the iron atom. Since the phase of the observed γa mode is near π/2, significant momentum must be transferred to the heme doming coordinate. This can occur either through recoil due to photolysis or via impulsive forces that are exerted on the heme iron during an ultrafast non-radiative decay process35.
In the event that one of the axial heme ligands is partially photolyzed (i.e., quantum yield less than unity), one would not see the otherwise expected phase jump of the 40 cm−1 mode from +π/2 to −π/2. This is because the excitations at 425 nm and 432 nm would both lie to the red of the putative transient 5-coordinate ferric Soret band maximum. Thus, the possibility of partial axial ligand photolysis in ferric CBS remains an open question. The optical response shown in Fig. 2B indicates that if such a process is taking place, axial ligand rebinding probably takes place on the same timescale as vibrational cooling, i.e., 3–4 ps. This would be somewhat faster than for ferrocytochrome c methionine photolysis, where rebinding is observed on the 6 ps timescale52, or for Dos, where the rebinding timescale is even longer45. However, it remains possible that longer timescale processes remain to be discovered in the CBS system, because a limited time range was employed in the present experiments.
Ferrous CBS
Next we turn to the ferrous forms of CBS and discuss the transmittance kinetics (Fig. 5) obtained by exciting at 425 nm, along with the corresponding coherence spectra (Fig. 6). The facile CO binding and displacement of Cys52 raises the possibility that the thiolate ligand might also be photolabile in the ferrous state. However, since we see a bleaching signal, indicating increased transmission at the photoproduct absorption band (~430 nm), it is not likely that significant cysteine photolysis takes place. The lack of significant cysteine photolysis is consistent with the formation of a relatively strong H-bond between Cys52 and Arg266, which presumably delocalizes the thiolate charge and stabilizes the 6-coordinate ferrous heme21. This charge delocalization may also be reflected by the absence (or diminution) of the “split-Soret” (or hyperporphyrin) absorption band that is often seen in cytochromes P45053. Generally, it appears that the transient absorption signals for ferrous CBS at pH 8 are primarily due to vibrational cooling following photon absorption and that it does not undergo significant ligand photolysis. As such, it would be the first 6-coordinate ferrous heme system we have studied that remains primarily 6-coordinate when photoexcited in the Soret band. As mentioned above, this is evidently the result of hydrogen bond stabilization of Cys52 by Arg26617.
We also note here that the mode at 68 cm−1, clearly seen for ferrous CBS in Fig. 6, is likely to contain a large contribution from heme ruffling 41. Strong enhancement of this mode suggests that heme ruffling distortions in the 6-coordinate reduced species at pH 8 that are larger than for the ferric state at the same pH. This indicates that heme conformational changes can be coupled to the iron redox state. Ruffling distortions are expected to stabilize the ferric heme by mixing the iron d-electron states with the porphyrin π orbitals40;54;55. A smaller ruffling distortion of the ferric heme would thus indicate a relative destabilization of the ferric state compared to the ferrous form, potentially leading to more facile heme reduction at pH 8. Since reduction of the heme is often a necessary condition for the binding of diatomic regulatory molecules, it offers another level of control to this complex protein system.
Dependence on pH
As we decrease the pH from 9 to 6 the following changes were observed in the low frequency coherence spectra of ferric CBS (Fig. 8). At pH 8 and 9 the spectra look nearly the same, suggesting that the heme structural distortions are effectively identical at these pH values. When we move to pH 7, we observe a new feature at 75 cm−1 that is possibly the γb mode, which was previously assigned to heme ruffling41, although this is an unusually high frequency. On the other hand, a γb mode near 68 cm−1 is seen to be relatively strongly enhanced in the ferrous enzyme (Fig. 6). In addition to the appearance of the feature at 75 cm−1, the γc mode near 87 cm−1 seems to disappear as pH 7 is approached. Given the broadness of the feature near 75 cm−1, particularly at pH 6.8, it is conceivable that it actually represents a superposition of both the γb (primarily ruffling content) and γc (primarily saddling content) modes that we expect to appear near 60 and 90 cm−1, respectively. Since both the frequencies and intensities of these modes can be affected by protein induced heme distortions, it is difficult to determine if the two peaks are changing intensity and merging or if the 87 cm−1 mode is disappearing and being replaced by a mode at 75 cm−1. In either case, a distinct pH dependence of what we believe to be primarily heme ruffling and saddling distortions is observed as the pH is lowered from 9 to 7. Since the ruffling distortion is thought to stabilize the ferric heme, this observation suggests the ferric heme is relatively more stabilized at pH 7 than at pH 8–9. This is consistent with a report56 that reduced CBS is more stable at higher pH. However, this report did not consider the pH dependence of the sodium dithionite reduction potential57, which might help to explain some of the pH-dependent stability observations.
A further decrease of the pH to 6 leads to loss of the 75 cm−1 feature and the return of the γc mode at 89 cm−1. The above observations can be explained if a protein conformation is stabilized near pH 7 where the heme is uniquely distorted along the ruffling and saddling coordinates41. We suggest that these changes are connected to the protonation state of PLP58;59 and/or the strength of the hydrogen bonding salt bridge between Cys 52 and Arg 266, which has been implicated in conveying regulatory signals between the heme and the PLP sites. At pH 8–9, the system is most reactive, while at pH 7 some sort of PLP tautomerizaton and/or heme conformational change has evidently taken place that may help poise the system to allow for better signaling and catalytic regulation at the expense of reactivity. Prior studies using resonance Raman spectroscopy56 have also noted subtle changes in the ferric CBS vibrational spectra, particularly in the Fe-S mode near 310–320 cm−1, that are consistent with a heme sensitivity to pH. As noted above, the heme ruffling mode has been previously implicated in the stabilization of the ferric heme state of nitroporphin40;41;54;55 and a pH sensitive ruffling distortion of the heme in CBS might alter its electronic properties and help tune the system for regulatory purposes.
CBS ligand binding kinetics
The CO geminate rebinding in CBS takes place on the 100 ps time scale and displays a large amplitude. Ultrafast geminate rebinding of CO has also been recently observed in other heme systems, e.g. CooA43, carboxymethyl cytochrome c (Cm-Cytc)60 and selected mutants61, DosH62, and BsTrHb (truncated hemoglobin from Bacillus subtilis)63. In CBS, only about 20% of the photolyzed CO material escapes the protein and rebinds in a bimolecular phase after the first 15 ns. This value is in reasonable agreement with the ~30% escape estimated by Puranik et al. using time resolved resonance Raman spectroscopy17. These authors also demonstrated that the rebinding rate of the displaced Cys52 is slow (4000 s−1) and that Cys52 binding in the first 200 ns is negligible. Thus the geminate recombination of CO in CBS, as shown in Fig. 10, is not competitive with the binding of Cys52.
Figure 10A compares CO binding in CBS with that of CooA. Both kinetic responses were fit using the “SRC” model2;22 and it is observed that CBS-CO rebinding has distinct sets of SRC parameters. The details of the fitting procedure are explained in the supporting information. The SRC model is based on a distribution of heme conformations along the doming coordinate, so that the enthalpic barrier for CO binding is separated into two parts
| (4) |
where Hp(a) represents the proximal barrier due to the heme doming and a is the generalized iron out-of-plane displacement. The quantity K is an effective force constant along the doming coordinate. H0 represents the remaining (mostly distal) contributions to the barrier and contains energies involving ligand docking sites and steric constraints associated with the distal pocket as well as a small a-independent term from the linearly coupled heme. The survival population at time t after photolysis can be written as2,
| (5) |
The parameter Ig represents the geminate amplitude and should be near unity for Eq. 5 to hold, which is the case for both CooA-CO and CBS-CO kinetics. The generalized force constant K is taken as 13.8 N/m, as found for other heme systems and from the frequency (~40 cm−1) of the heme doming mode. As a result, the relevant kinetic parameters {a0, σa}, that describe the heme out-of-plane distribution, P(a), and the distal pocket barrier (H0 in Eq. 2), can be found2 from the temperature dependent fitting parameters {A,C,k1} in Eq. 5 as,
| (6) |
Measurement of ultrafast kinetics as a function of temperature is difficult, but Eq. 6 has been used previously2 to evaluate the temperature dependence of {a0, σa} and to separately extract k0 and H0. For CBS we evaluated the data at a fixed temperature (293 K) found that we needed two terms like the one shown in Eq. 5 to describe the full kinetic inhomogeneity of the CO rebinding. We weighted them equally, one for each subunit of the dimer. The presence of additional inhomogeneity in CBS can also be inferred by comparing the Soret band linewidths of CO adducts as seen in the supporting information Fig. S2. It is unclear at this time if this is, or is not, a direct reflection of kinetically inequivalent dimers with different distal rebinding barriers. The distal pocket rebinding barrier, H0, usually associated with a CO docking site, is not expected to affect the Soret band inhomogeneity to the same degree as the heme doming distribution. Thus, it remains possible that the observed broadening of the CBS Soret band simply reflects a kinetically silent inhomogeneity in one or both of the dimers.
The extracted fitting parameters are discussed in more detail in the supporting information and are displayed in Table 2 (It should be noted that, in order to determine the last two columns of Table 2, a measurement of the temperature dependence of the fitting parameter k1 is necessary2 or else assumptions about k0 or H0 must be made as described in the footnotes to Table 2). A variety of other fitting approaches involving multiple exponentials and stretched exponentials were also examined and found to be inadequate as well as difficult to interpret. In order to reduce the number of fitting parameters, the fits to the data were carried out with some restrictions on the two subunit site distributions, Pi(a) (subscripts i=1,2 denote the different subunits). The distribution widths, σa, were restricted to be equal for the two subunits and they converged to 0.13Å. The centroids of the distributions were taken to be independent, but they converged to a0~0.3Å, very similar to what was found for CooA. The overall best fits for the geminate rebinding in the CBSCO dimer yielded two equally weighted heme distributions with nearly the same centroid and width, but very different values for k1. As shown in the supporting information, the fitting parameters lead to a ratio for the average geminate rates of the two subunits:
| (7) |
As emphasized in Table 2, the main difference between the two subunits is contained in the values for k1 (i.e., k11 and k12). In principle, this could indicate a difference in the underlying entropic barrier as reflected in the Arrhenius prefactors, k0, but a more likely reason is that the distal enthalpic barrier, H0, varies between the two subunits. If we assume that the prefactor k0 is the same for both binding sites, then by using Eq. 2 we find for T=300 K:
| (8) |
If H01 is taken to be > 0, the value for k0 would be unusually large, so we take H01~ 0 kJ/mol and find H02 ~7.2 kJ/mol. This is very similar to the value found for MbCO22, suggesting that a docking site with energetics similar to Mb is exposed in the second subunit of CBS upon CO binding to the first subunit. To be certain of this, measurements as a function of temperature are needed to separate k0 and H0 unambiguously, as demonstrated previously for the FePPIXCO system2.
The ratio of the average geminate CO rebinding rates falls within the error range of the ratio of the CO association constants ( ) previously found for the two components of the dimer17. The standard 3-state kinetic model64;65 as discussed in the supporting information shows that when the geminate amplitude is large (kBA≫kout), we have:
| (9) |
Where kout is the rate for the ligand to transition from the heme pocket to the solution and kBA and kAB signify the bond making and bond breaking step as CO binds to or dissociates from the heme iron within the protein distal pocket. Thus, the geminate rebinding results are consistent with the measured Kassoc if the difference in the CO association constants for the two subunits is primarily due to the difference in the geminate rates. A prior study17, using slower timescale kinetic probing, could detect no difference in the CO bimolecular kinetics, kon, because for large geminate amplitude systems this rate is either limited by the ligand entrance into the heme pocket, kin, or by the dissociation of Cys52, but not by the ultrafast geminate rate kBA. Since koff ~ kABkout/kBA depends inversely on kBA in the large geminate yield limit, the conclusion17 that the difference in the subunit association constants resides in the overall off rate is correct. Somewhat ironically, it is the geminate rebinding rate, kBA, that governs the difference in the subunit off-rates and therefore the overall association constants for CO binding.
Finally, we note that, if CO binding in the CBS system is “anti-cooperative” as suggested17, the CO binding must induce a quasi-static inequivalence in the two subunits that contrasts with the nearly identical subunits that appear in the ferric x ray structure16;66. If the subunits remained identical, we would not be able to observe the slower binding kinetic signal in these high repetition rate, low pump-pulse energy, measurements.
Figure 10B shows the CO and NO rebinding kinetics of myoglobin, where most of the photolyzed CO escapes to the solvent and about 99% of NO rebinds geminately on a subnanosecond time scale. This huge difference in the rebinding kinetics of the two ligands (and their temperature dependence) has been explained using a “harpoon” model2;67, where (as outlined above) the rebinding of CO depends upon the heme doming coordinate and a “distal pocket” barrier, but NO rebinding does not. It has also been suggested that NO binding to the heme iron has a spin allowed (ΔS=1) channel, whereas CO binding to the ferrous heme is a spin forbidden process (ΔS=2)68;69. However, after observing the ultrafast CO rebinding in CooA and CBS, and in heme model compounds2, the assumption that spin selection rules play a dominant role in controlling the kinetics of ligand geminate recombination is brought into question. Ligand rebinding studies of samples in high magnetic field are also consistent with the idea that spin selection rules do not play a key role in regulating CO binding to heme systems70.
CBS is one of several heme systems that rebind CO very rapidly. However, the specific reasons that underlie the phenomenon of ultrafast CO rebinding remain to be determined. One possibility is that the “distal pocket” enthalpic barriers are very small or take a relatively long time to develop. This can result in geminate recombination timescales that are faster than the entropy production timescales of the photolyzed protein-CO system. Such effects are synergistic and can dramatically reduce the entropic barrier for ligand binding2 leading to the enhanced prefactors for CO rebinding (k0 ~1011s−1) observed for CooA and CBS. Ultrafast temperature dependent kinetic studies are underway to help us better understand this interesting phenomenon.
Summary
In summary, we have studied CBS using vibrational coherence spectroscopy to access the thermally accessible spectral region below 200 cm−1. Comparisons of the vibrational coherence spectra with resonance Raman spectroscopy demonstrate very good correlation in the 150–400 cm−1 region where there is good frequency overlap and this helps to validate the modes extracted at lower frequencies. A mode near ~25 cm−1 is observed that can be explained by ultrafast non-radiative decay of the ferric six-coordinate species and coupling forces that activate either the protein material or a very low frequency mode of the heme. A heme mode with large doming content is observed in ferric CBS at ~40 cm−1 with a phase near π/2, indicating a significant momentum impulse is induced by the pump pulse via non-radiative decay, possibly involving ligand photolysis. The ferrous form of CBS shows a relatively strong γb mode that is thought to reflect a significant ruffling distortion compared to the ferric heme, affecting the reduction potential of the CBS system. We observe pH-dependent changes in the low frequency spectra that indicate a protein-induced alteration in heme structure near pH 7. This change could involve tautomeric states of the PLP co-factor and its interaction with the protein that are transmitted to the heme site. Ultrafast geminate rebinding of CO to CBS was also investigated for the first time and the kinetics displayed similarities with the CO regulatory protein CooA. The CBS-CO rebinding kinetics display distinct kinetic inhomogeneity, assigned to the dimer subunits, with underlying geminate rate ratios that are similar to the previously measured ratios for CO association.
Supplementary Material
Acknowledgments
This work is supported by grants from the NIH (DK35090 to P.M.C. and HL59984 to R.B.) and NSF (MCB-0744738 to P.M.C.) The authors thank Tom Poulos (University of California, Irvine) for providing the CooA sample and Minoru Kubo (University of Hyogo, Japan) for help during the initial stage of this project.
Footnotes
Supporting information available
The experimental details and preparation procedure for the ferrous NO bound form of CBS, the NSD and CO binding kinetic analysis using the SRC model. The absorption spectra of ferrous CBS-NO complex before and after the FCS measurement along with ferric CBS (Figure S1). The absorption spectra of the CO complexes of CBS, CooA and myoglobin and their full width half maximum (Figure S2). The scheme for ketoenamine and enolimine tautomers of the internal aldimine of PLP in CBS (Figure S3). The time constants and amplitudes for the optical response of the ferric, ferrous, and ferrous NO states of CBS at different pH and wavelength (Tables S1, S2, and S3). This material is available free of charge via the Internet at http://pubs.acs.org.
Reference List
- 1.Zhu L, Sage JT, Champion PM. Science. 1994;266:629–632. doi: 10.1126/science.7939716. [DOI] [PubMed] [Google Scholar]
- 2.Ye X, Ionascu D, Gruia F, Yu A, Benabbas A, Champion PM. Proc Natl Acad Sci US A. 2007;104:14682–14687. doi: 10.1073/pnas.0702622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vos MH. Biochim Biophys Acta. 2008;1777:15–31. doi: 10.1016/j.bbabio.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Cerullo G, Manzoni C, Luer L, Polli D. Photochemical & Photobiological Sciences. 2007;6:135–144. doi: 10.1039/b606949e. [DOI] [PubMed] [Google Scholar]
- 5.Vos MH, Lambry JC, Robles SJ, Youvan DC, Breton J, Martin JL. Proc Natl Acad Sci US A. 1991;88:8885–8889. doi: 10.1073/pnas.88.20.8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q, Schoenlein RW, Peteanu LA, Mathies RA, Shank CV. Science. 1994;266:422–424. doi: 10.1126/science.7939680. [DOI] [PubMed] [Google Scholar]
- 7.Gruia F, Kubo M, Ye X, Ionascu D, Lu C, Poole RK, Yeh SR, Champion PM. J Am Chem Soc. 2008;130:5231–5244. doi: 10.1021/ja7104027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banerjee R, Evande R, Kabil O, Ojha S, Taoka S. Biochim Biophys Acta. 2003;1647:30–35. doi: 10.1016/s1570-9639(03)00044-x. [DOI] [PubMed] [Google Scholar]
- 9.Miles EW, Kraus JP. J Biol Chem. 2004;279:29871–29874. doi: 10.1074/jbc.R400005200. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee R, Zou CG. Arch Biochem Biophys. 2005;433:144–156. doi: 10.1016/j.abb.2004.08.037. [DOI] [PubMed] [Google Scholar]
- 11.Evande R, Blom H, Boers GHJ, Banerjee R. Biochemistry. 2002;41:11832–11837. doi: 10.1021/bi026248d. [DOI] [PubMed] [Google Scholar]
- 12.Janosik M, Kery V, Gaustadnes M, Maclean KN, Kraus JP. Biochemistry. 2001;40:10625–10633. doi: 10.1021/bi010711p. [DOI] [PubMed] [Google Scholar]
- 13.Taoka S, Ohja S, Shan XY, Kruger WD, Banerjee R. J Biol Chem. 1998;273:25179–25184. doi: 10.1074/jbc.273.39.25179. [DOI] [PubMed] [Google Scholar]
- 14.Green EL, Taoka S, Banerjee R, Loehr TM. Biochemistry. 2001;40:459–463. doi: 10.1021/bi0010874. [DOI] [PubMed] [Google Scholar]
- 15.Ojha S, Hwang J, Kabil O, Penner-Hahn JE, Banerjee R. Biochemistry. 2000;39:10542–10547. doi: 10.1021/bi000831h. [DOI] [PubMed] [Google Scholar]
- 16.Taoka S, Lepore BW, Kabil O, Ojha S, Ringe D, Banerjee R. Biochemistry. 2002;41:10454–10461. doi: 10.1021/bi026052d. [DOI] [PubMed] [Google Scholar]
- 17.Puranik M, Weeks CL, Lahaye D, Kabil O, Taoka S, Nielsen SB, Groves JT, Banerjee R, Spiro TG. J Biol Chem. 2006;281:13433–13438. doi: 10.1074/jbc.M600246200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weeks CL, Singh S, Madzelan P, Banerjee R, Spiro TG. J Am Chem Soc. 2009;131:12809–12816. doi: 10.1021/ja904468w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taoka S, West M, Banerjee R. Biochemistry. 1999;38:2738–2744. doi: 10.1021/bi9826052. [DOI] [PubMed] [Google Scholar]
- 20.Taoka S, Banerjee R. J Inorg Biochem. 2001;87:245–251. doi: 10.1016/s0162-0134(01)00335-x. [DOI] [PubMed] [Google Scholar]
- 21.Singh S, Madzelan P, Stasser J, Weeks CL, Becker D, Spiro TG, Penner-Hahn J, Banerjee R. J Inorg Biochem. 2009;103:689–697. doi: 10.1016/j.jinorgbio.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srajer V, Reinisch L, Champion PM. J Am Chem Soc. 1988;110:6656–6670. [Google Scholar]
- 23.Kumar ATN, Rosca F, Widom A, Champion PM. J Chem Phys. 2001;114:701–724. [Google Scholar]
- 24.Rosca F, Kumar ATN, Ye X, Sjodin T, Demidov AA, Champion PM. J Phys Chem A. 2000;104:4280–4290. [Google Scholar]
- 25.Finlay TH. J Biol Chem. 1974;249:7476–7483. [PubMed] [Google Scholar]
- 26.Kery V, Poneleit L, Meyer JD, Manning MC, Kraus JP. Biochemistry. 1999;38:2716–2724. doi: 10.1021/bi981808n. [DOI] [PubMed] [Google Scholar]
- 27.Gruia F, Ye X, Ionascu D, Kubo M, Champion PM. Biophys J. 2007;93:4404–4413. doi: 10.1529/biophysj.107.114736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Constantine S, Zhou Y, Morais J, Ziegler LD. J Phys Chem A. 1997;101:5456–5462. [Google Scholar]
- 29.Yu AC, Ye X, Ionascu D, Cao WX, Champion PM. Rev Sci Instrum. 2005;76:114301. [Google Scholar]
- 30.Zhu L, Li P, Huang M, Sage JT, Champion PM. Phys Rev Lett. 1994;72:301–304. doi: 10.1103/PhysRevLett.72.301. [DOI] [PubMed] [Google Scholar]
- 31.Ye X, Demidov A, Rosca F, Wang W, Kumar A, Ionascu D, Zhu LY, Barrick D, Wharton D, Champion PM. J Phys Chem A. 2003;107:8156–8165. [Google Scholar]
- 32.Champion PM, Lange R. J Chem Phys. 1980;73:5947–5957. [Google Scholar]
- 33.Liebl U, Lipowski G, Negrerie M, Lambry JC, Martin JL, Vos MH. Nature. 1999;401:181–184. doi: 10.1038/43699. [DOI] [PubMed] [Google Scholar]
- 34.Rosca F, Kumar ATN, Ionascu D, Ye X, Demidov AA, Sjodin T, Wharton D, Barrick D, Sligar SG, Yonetani T, Champion PM. J Phys Chem A. 2002;106:3540–3552. [Google Scholar]
- 35.Gruia F, Kubo M, Ye X, Champion PM. Biophys J. 2008;94:2252–2268. doi: 10.1529/biophysj.107.122119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gruia F, Ionascu D, Kubo M, Ye X, Dawson J, Osborne RL, Sligar SG, Denisov I, Das A, Poulos TL, Terner J, Champion PM. Biochemistry. 2008;47:5156–5167. doi: 10.1021/bi7025485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ziegler LD, Fan R, Desrosiers AE, Scherer NF. J Chem Phys. 1994;100:1823–1839. [Google Scholar]
- 38.Schomacker KT, Bangcharoenpaurpong O, Champion PM. J Chem Phys. 1984;80:4701–4717. [Google Scholar]
- 39.Hu SZ, Morris IK, Singh JP, Smith KM, Spiro TG. J Am Chem Soc. 1993;115:12446–12458. [Google Scholar]
- 40.Roberts SA, Weichsel A, Qiu Y, Shelnutt JA, Walker FA, Montfort WR. Biochemistry. 2001;40:11327–11337. doi: 10.1021/bi0109257. [DOI] [PubMed] [Google Scholar]
- 41.Kubo M, Gruia F, Benabbas A, Barabanschikov A, Montfort WR, Maes EM, Champion PM. J Am Chem Soc. 2008;130:9800–9811. doi: 10.1021/ja800916d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shelnutt JA, Song XZ, Ma JG, Jia SL, Jentzen W, Medforth CJ. Chem Soc Rev. 1998;27:31–41. [Google Scholar]
- 43.Kumazaki S, Nakajima H, Sakaguchi T, Nakagawa E, Shinohara H, Yoshihara K, Aono S. J Biol Chem. 2000;275:38378–38383. doi: 10.1074/jbc.M005533200. [DOI] [PubMed] [Google Scholar]
- 44.Tian WD, Sage JT, Srajer VV, Champion PM. Phys Rev Lett. 1992;68:408–411. doi: 10.1103/PhysRevLett.68.408. [DOI] [PubMed] [Google Scholar]
- 45.Liebl U, Bouzhir-Sima L, Kiger L, Marden MC, Lambry JC, Negrerie M, Vos MH. Biochemistry. 2003;42:6527–6535. doi: 10.1021/bi027359f. [DOI] [PubMed] [Google Scholar]
- 46.Champion PM, Perreault GJ. J Chem Phys. 1981;75:490–491. [Google Scholar]
- 47.Armstrong MR, Ogilvie JP, Cowan ML, Nagy AM, Miller RJD. Proc Natl Acad Sci US A. 2003;100:4990–4994. doi: 10.1073/pnas.0936507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deak J, Chin HL, Lewis CM, Miller RJD. J Phys Chem B. 1998;102:6621–6634. [Google Scholar]
- 49.Miller RJD. Annu Rev Phys Chem. 1991;42:581–614. doi: 10.1146/annurev.pc.42.100191.003053. [DOI] [PubMed] [Google Scholar]
- 50.Go N. Biopolymers. 1978;17:1373–1379. [Google Scholar]
- 51.Go N, Noguti T, Nishikawa T. Proc Natl Acad Sci US A. 1983;80:3696–3700. doi: 10.1073/pnas.80.12.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang W, Ye X, Demidov AA, Rosca F, Sjodin T, Cao WX, Sheeran M, Champion PM. J Phys Chem B. 2000;104:10789–10801. [Google Scholar]
- 53.Hanson LK, Eaton WA, Sligar SG, Gunsalus IC, Gouterman M, Connell CR. J Am Chem Soc. 1976;98:2672–2674. doi: 10.1021/ja00425a050. [DOI] [PubMed] [Google Scholar]
- 54.Walker FA. Coord Chem Rev. 1999;186:471–534. [Google Scholar]
- 55.Walker FA. J Inorg Biochem. 2005;99:216–236. doi: 10.1016/j.jinorgbio.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 56.Pazicni S, Lukat-Rodgers GS, Oliveriusova J, Rees KA, Parks RB, Clark RW, Rodgers KR, Kraus JP, Burstyn JN. Biochemistry. 2004;43:14684–14695. doi: 10.1021/bi0488496. [DOI] [PubMed] [Google Scholar]
- 57.Mayhew SG. Eur J Biochem. 1978;85:535–547. doi: 10.1111/j.1432-1033.1978.tb12269.x. [DOI] [PubMed] [Google Scholar]
- 58.Echevarria GR, Catalan J, Blanco FG. Photochem Photobiol. 1997;66:810–816. [Google Scholar]
- 59.Metzler DE. J Am Chem Soc. 2002;79:485–490. [Google Scholar]
- 60.Silkstone G, Jasaitis A, Vos MH, Wilson MT. Dalton Trans. 2005:3489–3494. doi: 10.1039/b508183c. [DOI] [PubMed] [Google Scholar]
- 61.Silkstone G, Jasaitis A, Wilson MT, Vos MH. J Biol Chem. 2007;282:1638–1649. doi: 10.1074/jbc.M605760200. [DOI] [PubMed] [Google Scholar]
- 62.Yamashita T, Bouzhir-Sima L, Lambry JC, Liebl U, Vos MH. J Biol Chem. 2008;283:2344–2352. doi: 10.1074/jbc.M708123200. [DOI] [PubMed] [Google Scholar]
- 63.Feis A, Lapini A, Catacchio B, Brogioni S, Foggi P, Chiancone E, Boffi A, Smulevich G. Biochemistry. 2008;47:902–910. doi: 10.1021/bi701297f. [DOI] [PubMed] [Google Scholar]
- 64.Ye X, Yu AC, Georgiev GY, Gruia F, Ionascu D, Cao WX, Sage JT, Champion PM. J Am Chem Soc. 2005;127:5854–5861. doi: 10.1021/ja042365f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Henry ER, Sommer JH, Hofrichter J, Eaton WA. J Mol Biol. 1983;166:443–451. doi: 10.1016/s0022-2836(83)80094-1. [DOI] [PubMed] [Google Scholar]
- 66.Meier M, Janosik M, Kery V, Kraus JP, Burkhard P. Embo J. 2001;20:3910–3916. doi: 10.1093/emboj/20.15.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ionascu D, Gruia F, Ye X, Yu AC, Rosca F, Beck C, Demidov A, Olson JS, Champion PM. J Am Chem Soc. 2005;127:16921–16934. doi: 10.1021/ja054249y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Franzen S. Proc Natl Acad Sci US A. 2002;99:16754–16759. doi: 10.1073/pnas.252590999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Strickland N, Harvey JN. J Phys Chem B. 2007;111:841–852. doi: 10.1021/jp064091j. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Z, Benabbas A, Ye X, Yu A, Champion PM. J Phys Chem B. 2009;113:10923–10933. doi: 10.1021/jp9031805. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.