

Abstract
Friedreich ataxia (FRDA) is a neurodegenerative disorder caused by an unstable GAA repeat expansion mutation within intron 1 of the FXN gene. However, the origins of the GAA repeat expansion, its unstable dynamics within different cells and tissues, and its effects on frataxin expression are not yet completely understood. Therefore, we have chosen to generate representative FRDA mouse models by using the human FXN GAA repeat expansion itself as the genetically modified mutation. We have previously reported the establishment of two lines of human FXN YAC transgenic mice that contain unstable GAA repeat expansions within the appropriate genomic context. We now describe the generation of FRDA mouse models by crossbreeding of both lines of human FXN YAC transgenic mice with heterozygous Fxn knockout mice. The resultant FRDA mice that express only human-derived frataxin show comparatively reduced levels of frataxin mRNA and protein expression, decreased aconitase activity, and oxidative stress, leading to progressive neurodegenerative and cardiac pathological phenotypes. Coordination deficits are present, as measured by accelerating rotarod analysis, together with a progressive decrease in locomotor activity and increase in weight. Large vacuoles are detected within neurons of the dorsal root ganglia (DRG), predominantly within the lumbar regions in 6-month-old mice, but spreading to the cervical regions after 1 year of age. Secondary demyelination of large axons is also detected within the lumbar roots of older mice. Lipofuscin deposition is increased in both DRG neurons and cardiomyocytes, and iron deposition is detected in cardiomyocytes after 1 year of age. These mice represent the first GAA repeat expansion-based FRDA mouse models that exhibit progressive FRDA-like pathology and thus will be of use in testing potential therapeutic strategies, particularly GAA repeat-based strategies.
Keywords: Friedreich ataxia, FRDA, FXN, Frataxin, GAA repeat, Mouse model
Friedreich ataxia (FRDA) is an autosomal recessive neurodegenerative disorder that is predominantly caused by a homozygous GAA repeat expansion mutation within intron 1 of the FXN gene (formerly FRDA) [1]. Normal individuals have 5 to 30 GAA repeat sequences, whereas affected individuals have from approximately 70 to more than 1000 GAA triplets [2]. The effect of the GAA expansion mutation is to reduce greatly the expression of frataxin [3], a mitochondrial protein that interacts with succinate dehydrogenase complex subunits [4] and acts both as an iron chaperone in iron–sulfur clusters and heme biosynthesis [5–8] and as an iron detoxifier [9]. A direct correlation has been identified between the size of the smaller of the two GAA repeat mutations (and hence the level of residual frataxin expression) and the age of onset and severity of FRDA disease phenotype [3,10–12]. Evidence suggests that the GAA repeat expansion may have its effect by adopting an abnormal triplex structure that interferes with FXN gene transcription [13,14], possibly by the formation of a sticky DNA complex that sequesters transcription factors [15]. It has also been suggested that GAA repeats may produce a heterochromatin-mediated gene silencing effect [16].
The generation of a representative mouse model of FRDA is considered important for the further understanding of disease pathology and the testing of potential therapeutic strategies. Thus far, strategies have been based on various gene-targeting approaches to modify the homologous mouse Fxn gene. Initially, Fxn knockout mice were shown to be embryonic lethal in the homozygous state [17]. Several Cre/loxP conditional knockout mice have subsequently been generated. These have each been shown to reproduce some pathological features of FRDA, including dysfunction of large sensory neurons, cardiodegeneration, diabetes, deficiency of Fe–S-containing enzymes, oxidative stress, and mitochondrial iron accumulation [18–21]. Despite these successes, conditional knockout models are by their nature selective in the tissues that contain loss or decreased levels of frataxin and they do not address the question of GAA repeat mutation dynamics. A different gene-targeting approach is to introduce a GAA repeat mutation into the mouse Fxn locus. However, this GAA repeat knock-in strategy is somewhat hampered by the fact that the Fxn intron 1 region of the mouse does not normally contain a GAA repeat sequence. Indeed, knock-in of a 230-GAA repeat sequence into the Fxn intron 1 has not reproduced triplet repeat instability in mice, although it did result in decreased frataxin production [22].
We hypothesized that a human genomic FXN transgene that contained a GAA repeat expansion at the correct intronic position would enable both GAA repeat instability and reduced frataxin expression to be obtained within the one single model. Our previous work demonstrated that a wild-type FXN YAC transgene could successfully rescue the embryonic lethal phenotype of the Fxn knockout mouse [17], indicating that the YAC contained the necessary regulatory elements for appropriate human frataxin expression in a frataxin null mouse [23]. We have subsequently generated two lines of human FXN YAC transgenic mice containing GAA repeat expansions derived from FRDA patient DNA that exhibit intergenerational and somatic instability of the GAA repeat [24]. We now describe the generation of GAA repeat expansion-based FRDA mouse models by crossbreeding of these FXN YAC transgenic mice with heterozygous Fxn knockout mice. Offspring are produced that express comparatively reduced levels of human-derived frataxin and rescue the homozygous Fxn knockout embryonic lethality. We detect FRDA-like biochemical, histopathological, and functional losses that will allow these mouse models to be used for testing effective FRDA therapeutic strategies.
Results
Human GAA expansion-containing FXN YAC transgenes rescue FRDA knockout embryonic lethality
Two lines of human FXN YAC transgenic mice that contain GAA repeat expansions of approximately 190 repeats or 190 + 90 repeats, designated YG22 and YG8, respectively, have previously been described [24]. To determine the viability of each GAA FXN transgene, we set up crosses between YG22 or YG8 mice (FXN+, Fxn+/+) and heterozygous Fxn knockout mice (Fxn+/−) [17]. The FXN+, Fxn+/− offspring from these crosses were further bred with Fxn+/− mice to generate FXN+, Fxn−/− “rescues.” Correct Mendelian ratios of rescue mice to overall offspring number were obtained from both YG22 and YG8 crosses (p > 0.25 in each case), indicating functional frataxin derived from both GAA repeat-containing transgenes. The rescue mice from both lines exhibit a normal life span, with mice surviving up to at least 2 years of age.
Decreases in frataxin mRNA and protein expression
To determine the levels of frataxin mRNA expression in YG22 and YG8 rescue and transgenic mice we performed semiquantitative RT-PCR using primers that recognize both human and mouse frataxin cDNA, followed by digestion with human-specific and mouse-specific restriction enzymes. The DNA products were standardized by comparison with mouse Hprt RT-PCR controls and were then determined as a percentage of wild-type mouse values. The results confirmed the presence of human-only frataxin mRNA transcripts in both YG22 and YG8 rescue mice (Fig. 1A). The levels of transgenic FXN mRNA expression compared to endogenous mouse Fxn mRNA revealed decreases in all YG22 and YG8 rescue mouse tissues (Fig. 1B). The greatest, and statistically significant, decreases were detected in the cerebellum of both YG22 (62%) and YG8 (57%) rescues, together with the skeletal muscle of YG8 rescues (57%). This contrasted with YG22 and YG8 transgenic mouse samples, which all showed increases (101 to 145%) in overall frataxin mRNA expression, with approximately equal contributions from transgenic and endogenous frataxin mRNA.
Fig. 1.

mRNA expression analysis. (A) A representative RT-PCR image showing restriction-digested human frataxin products (112 and 29 bp) and mouse frataxin products (75 and 64 bp), together with mouse Hprt controls. mRNA samples were isolated from tissues of wild-type, YG22 rescue, YG8 rescue, YG22 transgenic, and YG8 transgenic mice (lanes 1, cerebrum; 2, brain stem; 3, cerebellum; 4, heart; 5, skeletal muscle). (B) Levels of frataxin mRNA expression as a percentage value of wild-type mouse expression. Values were generated by determining the means of six different RT-PCR experiments, each normalized to Hprt. Error bars indicate SEM. *p<0.05, **p<0.01.
To determine the levels of human frataxin expression in YG22 and YG8 rescue mice we used an anti-human recombinant mature frataxin antibody (α-hmfxn) in Western blot analysis of several different tissue lysates. Our initial analysis of equalized unaffected human and wild-type mouse skeletal muscle samples indicated that the α-hmfxn antibody reacts strongly with human frataxin to give a band of 18 kDa, but reacts only weakly with mouse frataxin to give a slightly larger band of 19 kDa (Fig. 2A). We determined the ratio of human to mouse signal intensities to be 2.3:1. Thus, although a direct comparison between the human frataxin levels in the YG22 and YG8 rescue mice and wild-type mouse frataxin levels was not possible, we were able to calculate the comparative levels of human frataxin indirectly. Analysis of our results (Fig. 2B–2D) revealed comparative frataxin levels of 67 and 86% in the cerebellum, 45 and 52% in the heart, and 23 and 23% in the skeletal muscle of YG22 and YG8 rescue mice, respectively, when standardized to an anti-porin mitochondrial antibody control (p < 0.01 for all, except YG8 cerebellum, which was not significant). These frataxin expression levels were further decreased to 67 and 42% in cerebellum, 37 and 25% in heart, and 10 and 9% in skeletal muscle of YG22 and YG8 rescue mice, respectively, when standardized to an anti-actin antibody control (p < 0.01 for all). This suggests that there is both a decrease in the relative amount of frataxin within the mitochondria and an overall decrease in mitochondria within the cells of these tissues, especially within the YG8 cerebellar and heart tissues compared with the corresponding YG22 tissues. Some confirmation of this latter point comes from our finding of variably decreased (60–90%) levels of porin in all YG22 and YG8 cerebellum, heart, and skeletal muscle tissues, compared with wild-type controls (data not shown). In contrast, the levels of frataxin in the cerebrum and brain stem from both YG22 and YG8 rescue mice are not similarly reduced, but show increases of between 1 and 2 times the endogenous mouse levels when standardized to porin and levels of 0.7 to 2.2 times when standardized to actin (Figs. 2B–2D). These data suggest an increased amount of frataxin within the mitochondria of the cerebrum and brain stem of both mice, with an approximately normal number of mitochondria in YG22, but a decrease in YG8. The fluctuating decreased or increased levels of frataxin protein that we determined in different rescue mouse tissues contrasts with our finding of constant decreased frataxin mRNA levels in all rescue mouse tissues. This discrepancy may be explained by tissue-dependent variations in posttranscriptional and posttranslational effects on transgenic frataxin protein production.
Fig. 2.

Frataxin and antioxidant enzyme expression levels in YG22 and YG8 rescue mice. (A) Western blot showing comparative levels of frataxin relative to actin in skeletal muscle samples from human (H), wild-type mouse (M), and YG22 transgenic mouse (TG). (B) Western blot of YG22 and YG8 rescue mouse tissue lysates (lanes 1, cerebrum; 2, brain stem; 3, cerebellum; 4, heart; 5, skeletal muscle) hybridized with antibodies against frataxin, actin, porin, MnSOD, and CuZnSOD. The very low levels of YG8 skeletal muscle frataxin, which appear to be negative in this image, were revealed upon longer exposure. (C, D) Levels of transgenic human frataxin expression as a percentage value of endogenous wild-type mouse frataxin expression, (C) relative to porin controls or (D) relative to actin controls (n=5–8). Tissues 1–5 are as described for (B). (E) Levels of CuZnSOD in rescue mice as a percentage value of wild-type mouse expression, normalized to actin (n=3–7). Tissues 1–5 are as described for (B). (F) Levels of MnSOD in rescue mice as a percentage value of wild-type mouse expression, normalized to porin (n=3–6). Tissues 1–5 are as described for (B). Error bars indicate SEM. *p<0.05, **p<0.01.
To investigate potential antioxidant changes, comparative levels of the antioxidant enzymes, CuZnSOD and MnSOD, were also determined from the same tissue lysates (Figs. 2B, 2E, and 2F). CuZnSOD showed increases of up to 166% in cerebrum, brain stem, cerebellum, and heart of both YG22 and YG8 rescue mice compared with wild-type controls and normalized with actin, although changes were not to a significant level. However, a significant decrease in CuZnSOD was detected in the skeletal muscle of both YG22 and YG8 rescue mice (40 and 22%, respectively, p < 0.01 for each). MnSOD showed increases of up to 146% in all tissues (with the exception of YG8 skeletal muscle, which was decreased to 80%) compared with wild-type controls and normalized with porin, although changes were not to a significant level.
Neurobehavioral deficits
The coordination ability of the YG22 and YG8 rescue mice was shown to be impaired from the age of 3 months as determined by reduced performance on an accelerating rotarod treadmill compared with wild-type littermate controls (p < 0.01) (Fig. 3A). However, the degree of impairment did not extend to overt ataxia in either line of mice up to the age of 2 years. Muscle strength, assessed by a forelimb grip strength test, was decreased in YG22 rescues from 9 months of age (p < 0.01), but no significant changes were detected in the YG8 rescues (Fig. 3B). Locomotor activity, assessed by examining the unrestricted movement of mice in an open field, was decreased in both lines. The YG22 rescues showed a decreased trend in locomotor activity from 6 months of age, but no statistically significant difference was seen until 1 year (p < 0.05). YG8 rescues, on the other hand, showed a significant decrease in locomotor activity from 6 months of age (p < 0.05) (Fig. 3C). Both YG22 and YG8 lines demonstrated an increase in weight, with statistically significant differences detected in YG22 rescues from 6 months of age (p < 0.01) and in YG8 rescues from 9 months of age (p < 0.01) (Fig. 3D). One reason for the observed gain in weights may be the decreased locomotor activity identified in the mice.
Fig. 3.

Functional studies of 3-, 6-, 9-, and 12-month-old YG22 and YG8 rescue mice, compared with wild-type controls. (A) Rotarod analysis of YG22 and YG8 rescue mice compared with wild-type littermate controls shows a coordination deficit in both rescue mice from 3 months of age (n=10 and 9, respectively). However, a direct comparison between YG22 and YG8 experiments is not possible due to the use of different rotarod acceleration rate settings carried out at different periods of time. (B) Grip strength analysis shows a weakness in YG22 rescues only from 9 months of age (n=6–10). (C) Locomotor analysis identifies a progressive decrease in the mobility of both rescue mice (n=6–8 and 7–13, respectively). (D) Weight increases are detected in both rescue mice (n=6–16). Error bars indicate SEM. *p<0.05, **p<0.01.
Electrophysiological analysis
Motor and sensory nerve conduction studies were performed on the sciatic and caudal nerves of 9- to 14-month-old YG22 rescue and wild-type control mice and 20-month-old YG8 rescue and wild-type control mice. Decreases in both the sensory action potential and the sensory conduction velocity of the older YG8 rescue mice were detected (Fig. 4), although not to statistical significance. However, no sensory nerve conduction changes were detected in the younger YG22 rescues, and no motor nerve conduction changes were detected in either line of mice (data not shown). Taken together, these findings suggest a mild, progressive peripheral sensory neuropathy, which is consistent with an FRDA phenotype.
Fig. 4.
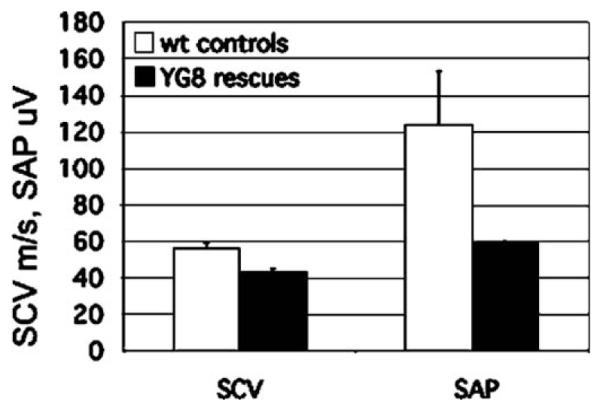
Sensory nerve conduction studies of 20-month-old YG8 rescue mice and wild-type controls showing a decrease in sensory conduction velocity (SCV) and sensory action potential (SAP), although not to significance (n=2).
Histological abnormalities in the dorsal root ganglia (DRG) and heart
To detect possible neuronal histopathology in the YG22 and YG8 rescue mice, we examined sections of brain, spinal cord, and DRG from eight YG22 rescues, six YG8 rescues, and three wild-type mice. By performing H&E staining and anti-calbindin immunohistology, we did not identify any of the cerebellar Purkinje cell abnormalities or granule cell losses that have previously been reported for frataxin conditional knockout mice [20]. Neither did we detect any abnormalities in the brain or spinal cord regions of either line of mice. However, we did identify pathological changes in the DRG. Prominent giant vacuoles were identified in the large sensory neuronal cell bodies of the DRG in both YG8 and YG22 rescue mice but not in wild-type controls (Fig. 5A). The vacuoles are round, single or multiple, and appear to be empty. We also detected peripheral margination of the nucleus in many large neuronal cell bodies, with or without vacuoles, suggestive of the process of central chromatolysis. These findings indicate specific degeneration of cells that also are a primary site of pathology in FRDA patients. Interestingly, the occurrence of DRG vacuoles shows a progressive position-dependent profile, which is similar in both lines of mice. Between the ages of 6 months to 1 year, vacuoles are detected only in the DRG of the lumbar region, but after 1 year of age, vacuoles also become apparent within DRG of the cervical region. Approximately 60% of the lumbar DRG sections from YG22 rescues and 70% of the lumbar DRG sections from YG8 rescues have cells containing vacuoles, with one to seven cells affected in each DRG section. In contrast, only 16% of the cervical DRG sections have vacuoles, and the vacuoles are detected in only one cell per DRG section. This profile resembles the distal-to-proximal “dying-back” phenomenon of neurodegeneration that is observed in FRDA patients.
Fig. 5.

Neuronal and cardiac histopathology. (A) H&E-stained section of lumbar DRG from a representative YG22 rescue mouse over 1 year of age, showing two neurons containing large vacuoles. Original magnification 400×. (B) Perl’s staining of a heart section from a representative YG22 rescue mouse over 1 year of age, showing characteristic blue staining indicating iron deposition. Original magnification 600×.
To determine potential hypertrophic cardiomyopathy, the heart weight/body weight ratio was determined and the heart examined for histopathology by H&E, trichrome Masson, and Perl’s staining. No increase in heart weight to body weight ratio was identified and no myofibril disarray or fibrosis was observed to suggest cardiomyopathy (data not shown). However, Perl’s staining identified iron deposition within the heart, but only from the oldest (14–18 months) YG22 rescue mice studied (Fig. 5B). In these cases, Perl’s staining of the cardiomyocytes was prominent but sparse, consistent with similar findings in FRDA patients.
Ultrastructure pathology
Ultrastructure examination of lumbar DRG from 20-month-old YG8 rescue mice by electron microscopy confirmed the presence of large vacuoles within the large neuronal cell bodies (Fig. 6A), as previously detected by light microscopy. Chromatolysis was also confirmed by the finding of dispersion of the rough endoplasmic reticulum ribosomes, leaving regions of the cell devoid of any cytoplasmic organelles (Fig. 6B). Lipofuscin deposition, which represents a lysosomal protein polymer arising from iron-catalyzed oxidation, was pronounced in DRG neurons (Fig. 6C). Other changes within the lumbar DRG included the finding of swelling and secondary demyelination of large axons (Figs. 6D and 6E). These pathological changes are all in keeping with oxidative stress and mitochondrial dysfunction of degenerating large DRG neurons, which are then attempting to regenerate by activation of protein synthesis. Ultrastructure examination of the heart identified patches of lipofuscin deposition and lysosomes, together with an accumulation of dispersed free glycogen, disrupting the regular arrays of mitochondria within the cardiomyocytes (Fig. 6F). Lipofuscin deposition is a recognized feature within FRDA cardiomyocytes and dispersed free glycogen accumulation leading to myofibril distortion has been found in other forms of hypertrophic cardiomyopathy [25].
Fig. 6.

(A–E) Electron micrographs of lumbar DRG from 20-month-old YG8 rescue mice showing examples of: (A) giant vacuoles, (B) chromatolysis, and (C) lipofuscin deposits within the large neuronal cell bodies. Also detected within the lumbar DRG are instances of: (D) complete demyelination of a large axon with its associated Schwann cell and (E) large axonal swelling with reduced myelination. (F) Electron micrograph of cardiac muscle from a 20-month-old YG8 rescue mouse showing lipofuscin deposition and lysosomes disrupting an ordered array of mitochondria. Scale bars represent 10 μm.
Decreased aconitase activity and mitochondrial respiratory chain function
Previous studies of FRDA patient and mouse model tissues have demonstrated impaired activity of the iron–sulfur cluster (ISC)-containing aconitase enzyme and mitochondrial respiratory chain (MRC) complexes I, II, and III, but not the non-ISC-containing MRC complex IV [18,26]. Examination of heart tissue samples from 6-month-old YG8 rescue mice detected a decrease in aconitase activity to approximately 66% of wild-type mouse levels (p < 0.05) (Fig. 7). Decreases in MRC complex I and II/III activity were also detected (79 and 75% of controls, respectively), although not to significant levels. In contrast, isolated MRC complex II and MRC complex IV activities showed no change. These data are in general agreement with the previous reports of FRDA patient and mouse model samples. However, the isolated MRC complex II activity was not decreased in the YG8 rescue mice, as has previously been described in conditional knockout mouse models [18,20]. Therefore, it is possible that the level of frataxin reduction in these samples is only just sufficient to induce changes in the most sensitive enzyme, aconitase. The impaired aconitase activity may be due to either reduced ISC formation or oxidative damage.
Fig. 7.

Mitochondrial respiratory chain and aconitase activities in heart tissue from 6-month-old YG8 rescue mutant and control mice. (A) The specific activities of aconitase (Acon) and citrate synthase (CS). CS activities were divided by 10 (n=6). (B) The mitochondrial respiratory chain (MRC) activities expressed as a ratio with citrate synthase; MRC I, NADH coenzyme Q1 reductase (NQ1R); MRC II/III, succinate cytochrome c reductase (SCcR); MRC II, succinate coenzyme Q2 reductase (SQ2R); and MRC IV, cytochrome oxidase (COX). COX/CS ratio was multiplied by a factor of 10 (n=4). Activities are expressed as means ± SEM. *p=0.03.
Oxidative stress
To detect potential oxidative stress in our FRDA models, we determined the levels of oxidized proteins and malondialdehyde (MDA; a marker of lipoperoxides) in YG22 and YG8 rescue mouse cerebrum, cerebellum, heart, and skeletal muscle tissues compared with wild-type controls. Oxidized proteins were increased in all samples from both YG22 and YG8 rescue mice (Fig. 8A). The most prominent significant increases were detected in the cerebrum and cerebellum of both lines (p < 0.01), with lesser, but still significant, increases also detected in the heart and skeletal muscle of both YG8 (p < 0.01) and YG22 (p < 0.05) mice. The finding of increased oxidized proteins in the YG22 cerebrum is somewhat at odds with our previous finding of an increased level of frataxin protein in this tissue, suggesting reduced functional efficiency of the transgenic human frataxin protein. Increased lipid peroxidation, as determined by MDA levels, was also detected in cerebrum and heart samples from YG22 rescue mice, but only the heart showed a statistically significant increase (p < 0.05) (Fig. 8B).
Fig. 8.

Oxidative stress. (A) Oxyblot analysis detects protein oxidation in cerebrum, cerebellum, heart, and skeletal muscle tissues of 6- to 9-month old YG22 and YG8 rescue mice, compared with wild-type controls, measured in arbitrary units (a.u.) of densitometry (n =8–12). Error bars indicate SEM. *p < 0.05, **p < 0.01. (B) TBARS analysis shows levels of MDA (nmol/mg of protein) as a marker of lipoperoxidation in YG22 cerebrum and heart tissue compared with wild-type controls (n=6). Error bars indicate SEM. *p<0.05.
Discussion
Although the exact mechanisms that cause FRDA are still under investigation, several specific features of FRDA molecular pathology are now well established. Thus, the majority of FRDA patients are homozygous for GAA repeat expansion mutations within intron 1 of the FXN gene, leading to decreased levels of frataxin, subsequent oxidative stress, iron deposition, and ultimately neurodegeneration, primarily in the large sensory neurons of the DRG, and hypertrophic cardiomyopathy. We have been able to recapitulate many such features of FRDA disease in the mouse by establishing two lines of model mice that express frataxin only from GAA repeat expansion-containing FXN transgenes.
We have previously shown that the GAA repeat somatic instability pattern in our YG22 and YG8 transgenic mice closely resembles the GAA repeat dynamics in FRDA patients [24]. Our initial interpretation of the Western blot data from this study, which showed a more intense signal from the human frataxin compared to the mouse frataxin, led us to speculate on likely overexpression of human frataxin in YG22 or YG8 knockout rescue mice [24]. However, we have now demonstrated that both YG22 and YG8 rescue mice express comparatively decreased levels of human frataxin mRNA in all tissues and decreased levels of human frataxin protein in at least some tissues compared with endogenous mouse levels. Overall the YG8 rescue mice demonstrate slightly less frataxin mRNA and protein expression, combined with marginally higher amounts of protein oxidation, compared with the YG22 mice. Therefore, it is possible that the additional 90 GAA repeats in the YG8 line are responsible for producing a slightly more severe phenotype. Previous GAA expansion mutation knock-in studies have shown that mice expressing 25–36% endogenous mouse frataxin levels do not show an obvious pathological phenotype [22], although a clear microarray gene expression phenotype has been determined [27]. These mouse frataxin levels are similar to the human frataxin levels that we have determined in our heart samples. However, due to its slightly different amino acid composition, human frataxin may not function at 100% of endogenous mouse frataxin efficiency. Therefore, the levels of frataxin that we see here (67 and 42% in cerebellum, 37 and 25% in heart, and 10 and 9% in skeletal muscle of YG22 and YG8 rescue mice, respectively) may in fact represent an even more “functional” decrease, resulting in the pathological phenotype that we describe.
Age-related somatic increases in GAA repeats have previously been detected in the cerebrum, brain stem, and cerebellum of both YG22 and YG8 transgenic mice, but no such increases were detected in the heart or skeletal muscle [24]. Therefore, the pattern of frataxin expression in our YG22 and YG8 rescue mice (highest in cerebrum and brain stem, lowest in skeletal muscle) is not likely to be entirely due to somatic increases in GAA repeat expansions. A similar pattern of “high brain and low skeletal muscle” frataxin expression has been reported in lines of human FXN BAC transgenic mice that contain normal-sized GAA repeats [28]. Thus, unknown tissue-specific regulatory factors would appear to be acting on human FXN transgenes within the context of the mouse. As yet there are insufficient data available on human tissue samples to determine if a similar differential pattern of frataxin expression is present in FRDA and thus how representative our mouse models are in this regard.
That being said, the differentially reduced levels of frataxin in our mouse models are sufficient to induce a mild FRDA-like pathological phenotype. Impaired aconitase activity, oxidative stress, and functional deficits are apparent, although not severe, and neuronal histopathology and iron deposition within the heart are later progressive effects. This phenotype is indicative of the early effects of FRDA pathology that precede overt ataxia and the later development of hypertrophic cardiomyopathy. Indeed, these mild mouse models can be considered representative of the less severe, later onset cases of FRDA. The 190 and 190 + 90 GAA repeat expansion mutation sizes within our mouse models are also in keeping with the cases of later onset FRDA that have smaller GAA allele sizes of fewer than 200 repeats [29–33].
The vacuoles identified in the DRG of our mouse models appear similar to those in the “Cb” neuron-specific frataxin conditional knockout model reported by Simon et al. [20]. However, an additional finding within our models is the progression of vacuolar pathology from affected distal lumbar regions at only 6 months of age to more proximal cervical regions at 13–15 months. This resembles the dying-back phenomenon of neurodegeneration that is observed in FRDA patients. Although, to our knowledge, DRG vacuoles have not been described in FRDA patients, loss of large sensory DRG cell bodies is a hallmark of the disease. Thus, it is likely that we are identifying a milder or earlier effect in our mouse models, before the DRG neurons degenerate and are lost completely. The identification of lipofuscin deposition, chromatolysis, and swelling and demyelination of axons in the DRG can be explained by cells that are suffering from chronic oxidative damage. In this sense, our FRDA mouse models are similar to the mouse model of ataxia with vitamin E deficiency, which shows lipofuscin deposition in DRG of mice over 1 year of age, indicating chronic oxidative stress [34]. Although frataxin conditional knockout mouse models have reproduced many other features of FRDA pathology, there is currently some debate concerning oxidative stress status. Studies of neuronal and cardiac conditional knockout models indicate no, or insignificant, oxidative stress [35,36], whereas pancreatic and hepatic conditional knockout models demonstrate distinct oxidative stress [19,21]. Our own models demonstrate an obvious, but not severe, degree of oxidative stress. This may be due to the fact that our models have residual frataxin in all tissues, which may also be the case with the pancreatic and hepatic knockout models, but not the neuronal and cardiac knockout models, which have complete frataxin loss in specific tissues. Our mice also have a mitochondrial respiratory chain that shows only a slight overall functional deficit and, therefore, continues to produce damaging free radicals. The greater loss of aconitase activity than MRC complex activity deficits suggests that aconitase impairment is a prominent early feature, even in mild cases of FRDA. The increases that we found in CuZnSOD and MnSOD are generally consistent with a reaction to oxidative stress in our FRDA mouse model tissues. However, where there is the greatest decrease in frataxin (i.e., skeletal muscle) we actually see a decrease in both CuZnSOD and MnSOD. This indicates a different susceptibility or handling of this tissue to mild oxidative stress. The identification of iron deposition in the hearts of only our older mice confirms the later onset aspect of this pathology, as previously described for cardiac frataxin conditional knockout mouse mutants [18].
Understanding that FRDA is caused by an unstable GAA repeat sequence, leading to decreased levels of frataxin, has given rise to novel ideas for frataxin-increasing therapies. We believe that we have generated a GAA-repeat-based mouse model of FRDA that would be suitable for the investigation of many different therapeutic strategies. These include the possible use of iron chelators and antioxidants, targeted preferentially to the mitochondria [37,38], as well as novel drugs aimed at up-regulation of frataxin expression, such as hemin, butyric acid, and cisplatin [39,40]. However, the fact that our FRDA mouse models have been generated by the introduction of a GAA repeat expansion mutation also makes them amenable to novel therapeutic strategies aimed at interacting with and modifying the GAA repeat expansion itself.
Materials and methods
Production and genotyping of genetically modified mice
GAA repeat expansion-containing YG8 and YG22 transgenic mice [24] were first crossed with heterozygous Fxn knockout mice [17] to produce mice containing both the transgene and the knockout allele. A second cross with heterozygous Fxn knockout mice then produced viable FRDA model mice containing the human FXN transgene on an Fxn null background. Genotyping of GAA repeats and Fxn allele status was performed as previously described [24].
mRNA and protein expression analysis
mRNA was isolated and RT-PCR amplified for 25 cycles using human/mouse frataxin exon 3/4 primers FRT1 and RRTII, followed by digestion with mouse-specific BstXI and human-specific AflIII restriction enzymes as previously described [23]. Mouse Hprt RT-PCR was also carried out for 25 cycles using the primers and conditions described by Puccio et al. [18]. DNA products were resolved in 3% agarose gels and UV images were analyzed by UN-SCAN-IT (Silk Scientific Corp.) densitometry.
Frataxin Western blot analysis was carried out using a recombinant anti-mature frataxin antibody (G. Isaya, Mayo Clinic) as previously described [23]. Other immunodetection was carried on the same Western blots using antibodies against actin (Sigma), porin (Cambridge Bioscience), MnSOD (Abcam), and CuZnSOD (Abcam). Densitometry was carried out using UN-SCAN-IT software (Silk Scientific Corp.).
Functional studies
Rotarod analysis was performed using a Ugo-Basille 7650 accelerating rotarod treadmill apparatus. Mutant and control littermate mice ages 3, 6, 9, and 12 months (n=5–10) were placed on the rod and four trials were performed with the speed of the rotation gradually increasing from 4 to 40 rpm over a period of approximately 3–5 min. A period of 10 min rest was given between each trial. The time taken for each mouse to fall from the rod was recorded. Grip strength was assessed by holding mice by the base of the tail, while their forepaws gripped onto a horizontal bar that was attached to a tension spring. The mice were gently pulled backward until they let go of the bar and the distance pulled in millimeters was recorded. Locomotor activity was assessed by placing the mice in a gridded open-field Perspex box and the number of gridded squares entered by the mouse over a period of 20 s was recorded.
Electrophysiology
Electrophysiological recordings were carried out on anesthetized 20-month-old YG8 rescue mice, 9- to 14-month-old YG22 rescue mice, and wild-type controls. Motor nerve conduction was investigated by placing electrodes in the hind-paw muscle and stimulating the sciatic nerve. Measurements of amplitude, distal latency, and proximal latency were taken, and the motor conduction velocity was then determined. Sensory nerve conduction was investigated by placing electrodes at two positions in the tail skin and stimulating the caudal nerve. Measurements of the sensory action potential were taken, and the sensory conduction velocity was then determined.
Histology
For histological preparations, terminally anesthetized mice were fixed by intracardial perfusion with 4% paraformaldehyde in phosphate-buffered saline (PBS). The fixed mouse tissues were dissected, embedded in paraffin wax, and sectioned by standard methods. Sections were deparaffinized with Histoclear (National Diagnostics) and slides were stained with hematoxylin and eosin or trichrome Masson stains. Perl’s technique was used to detect ferric iron as previously described [17].
Electron microscopy
Mice of 20 months of age were perfused via a cannula in the heart with 1% glutaraldehyde and 1% paraformaldehyde plus 1% dextran, all in 0.1 M PIPES buffer. Tissues were dissected out, fixed for several hours in the same solution, and then postfixed overnight in 1% osmium tetroxide +1.5% potassium ferricyanide and 3% sodium iodate. Tissues were then dehydrated through increasing concentrations of ethanol and embedded in Durcupan via 1,2-epoxy propane. Thin sections for light microscopy were stained with thionin and acridine orange and ultrathin sections for electron microscopy were contrasted with uranyl acetate and lead citrate.
Biochemical measurements
Dissected mouse heart tissues were snap-frozen in liquid nitrogen. Mitochondrial respiratory chain and aconitase activities were determined as previously described [26].
Oxidative stress
Protein lysates were prepared according to Campuzano et al. [3]. For each assay, 10 μg of protein was used and the concentration was determined by using the BCA Protein Assay Reagent Kit (Pierce). Proteins were then modified by the use of the OxyBlot Protein Oxidation Detection Kit (Chemicon International). Duplicate samples were used for each assay and controls of unmodified samples were also included. After the assay was performed according to the manufacturer’s instructions, each sample was diluted to 100 μl in 1× Laemmli buffer [41], with 2-mercaptoethanol and no added glycerol. Samples were then applied to a slot-blot apparatus and washed with the same buffer, followed by electroblot transfer buffer. The filters were air-dried and treated with the antibody provided in the kit and subsequently visualized on X-OMAT X-ray film. Densitometry was performed using UN-SCAN-IT software (Silk Scientific Corp.) and the final values were calculated as ratios to the protein concentration used per assay.
TBARS (thiobarbituric acid-reactive substances) assay for MDA
Using a modification of the method described by Ledwozyw et al. [42], brain and heart tissues from wild-type and mutant mice were homogenized to 20% (w/v) in Dulbecco’s PBS. Samples were then centrifuged at 2000g for 10 min and supernatants were collected. Each supernatant (0.1 ml) was mixed with 20% (w/v) trichloroacetic acid in 0.6 M HCl and left at room temperature for 15 min. This was then mixed with thiobarbituric acid (TBA; prepared by dissolving 0.5 g of TBA in 6 ml of 1 M NaOH and then adding 69 ml of water). The mixture was then heated to 100°C for 30 min, cooled to room temperature for 10 min, and then extracted with 0.8 ml of n-butanol. Samples were vortexed and then centrifuged at 1500g for 10 min. The upper organic phase was collected and the absorbance of each sample was determined at 532 nm vs n-butanol. The concentration of MDA (which forms an adduct with TBA) was determined from a standard curve, obtained from using different concentrations of MDA (Aldrich). The protein concentration of each tissue homogenate supernatant was determined by the BCA Protein Assay (Pierce) and the final value of MDA in each tissue was presented as nmol/mg of protein used.
Statistical analysis
Mendelian ratios were determined using χ2 analysis, rotarod analysis was carried out using ANOVA with repeated measures, and all other functional, mRNA, Western blot, oxyblot, TBARS, and enzyme activities were analyzed using the Student t test.
Acknowledgments
We thank Michel Koenig and Helene Puccio (INSERM) for the Fxn knockout mice and helpful discussion, Grazia Isaya (Mayo Clinic and Foundation) for the kind gift of anti-human recombinant mature frataxin antibody, and Sebastian Brandner (UCL) for analysis of neuronal histological data. This work was supported by The Wellcome Trust, Friedreich’s Ataxia Research Alliance, Seek A Miracle/Muscular Dystrophy Association, and Ataxia, UK. S.A. is supported by The Wellcome Trust (Grant 070235).
References
- [1].Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- [2].Pandolfo M. The molecular basis of Friedreich ataxia. Adv. Exp. Med. Biol. 2002;516:99–118. doi: 10.1007/978-1-4615-0117-6_5. [DOI] [PubMed] [Google Scholar]
- [3].Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, Trottier Y, Kish SJ, Faucheux B, Trouillas P, Authier FJ, Durr A, Mandel JL, Vescovi A, Pandolfo M, Koenig M. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 1997;6:1771–1780. doi: 10.1093/hmg/6.11.1771. [DOI] [PubMed] [Google Scholar]
- [4].Gonzalez-Cabo P, Vazquez-Manrique RP, Garcia-Gimeno MA, Sanz P, Palau F. Frataxin interacts functionally with mitochondrial electron transport chain proteins. Hum. Mol. Genet. 2005;14:2091–2098. doi: 10.1093/hmg/ddi214. [DOI] [PubMed] [Google Scholar]
- [5].Bulteau AL, O’Neill HA, Kennedy MC, Ikeda-Saito M, Isaya G, Szweda LI. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science. 2004;305:242–245. doi: 10.1126/science.1098991. [DOI] [PubMed] [Google Scholar]
- [6].Gerber J, Muhlenhoff U, Lill R. An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 2003;4:906–911. doi: 10.1038/sj.embor.embor918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yoon T, Cowan JA. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 2004;279:25943–25946. doi: 10.1074/jbc.C400107200. [DOI] [PubMed] [Google Scholar]
- [8].Schoenfeld RA, Napoli E, Wong A, Zhan S, Reutenauer L, Morin D, Buckpitt AR, Taroni F, Lonnerdal B, Ristow M, Puccio H, Cortopassi GA. Frataxin deficiency alters heme pathway transcripts and decreases mitochondrial heme metabolites in mammalian cells. Hum. Mol. Genet. 2005;14:3787–3799. doi: 10.1093/hmg/ddi393. [DOI] [PubMed] [Google Scholar]
- [9].Gakh O, Park S, Liu G, Macomber L, Imlay JA, Ferreira GC, Isaya G. Mitochondrial iron detoxification is a primary function of frataxin that limits oxidative damage and preserves cell longevity. Hum. Mol. Genet. 2005;15:467–479. doi: 10.1093/hmg/ddi461. [DOI] [PubMed] [Google Scholar]
- [10].Durr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel JL, Brice A, Koenig M. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N. Engl. J. Med. 1996;335:1169–1175. doi: 10.1056/NEJM199610173351601. [DOI] [PubMed] [Google Scholar]
- [11].Filla A, De Michele G, Cavalcanti F, Pianese L, Monticelli A, Campanella G, Cocozza S. The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am. J. Hum. Genet. 1996;59:554–560. [PMC free article] [PubMed] [Google Scholar]
- [12].Monros E, Molto MD, Martinez F, Canizares J, Blanca J, Vilchez JJ, Prieto F, de Frutos R, Palau F. Phenotype correlation and inter-generational dynamics of the Friedreich ataxia GAA trinucleotide repeat. Am. J. Hum. Genet. 1997;61:101–110. doi: 10.1086/513887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bidichandani SI, Ashizawa T, Patel PI. The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am. J. Hum. Genet. 1998;62:111–121. doi: 10.1086/301680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Grabczyk E, Usdin K. The GAA*TTC triplet repeat expanded in Friedreich’s ataxia impedes transcription elongation by T7 RNA polymerase in a length and supercoil dependent manner. Nucleic Acids Res. 2000;28:2815–2822. doi: 10.1093/nar/28.14.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sakamoto N, Ohshima K, Montermini L, Pandolfo M, Wells RD. Sticky DNA, a self-associated complex formed at long GAA*TTC repeats in intron 1 of the frataxin gene, inhibits transcription. J. Biol. Chem. 2001;276:27171–27177. doi: 10.1074/jbc.M101879200. [DOI] [PubMed] [Google Scholar]
- [16].Saveliev A, Everett C, Sharpe T, Webster Z, Festenstein R. DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature. 2003;422:909–913. doi: 10.1038/nature01596. [DOI] [PubMed] [Google Scholar]
- [17].Cossee M, Puccio H, Gansmuller A, Koutnikova H, Dierich A, LeMeur M, Fischbeck K, Dolle P, Koenig M. Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum. Mol. Genet. 2000;9:1219–1226. doi: 10.1093/hmg/9.8.1219. [DOI] [PubMed] [Google Scholar]
- [18].Puccio H, Simon D, Cossee M, Criqui-Filipe P, Tiziano F, Melki J, Hindelang C, Matyas R, Rustin P, Koenig M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe–S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001;27:181–186. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- [19].Ristow M, Mulder H, Pomplun D, Schulz TJ, Muller-Schmehl K, Krause A, Fex M, Puccio H, Muller J, Isken F, Spranger J, Muller-Wieland D, Magnuson MA, Mohlig M, Koenig M, Pfeiffer AF. Frataxin deficiency in pancreatic islets causes diabetes due to loss of beta cell mass. J. Clin. Invest. 2003;112:527–534. doi: 10.1172/JCI18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Simon D, Seznec H, Gansmuller A, Carelle N, Weber P, Metzger D, Rustin P, Koenig M, Puccio H. Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J. Neurosci. 2004;24:1987–1995. doi: 10.1523/JNEUROSCI.4549-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Thierbach R, Schulz TJ, Isken F, Voigt A, Mietzner B, Drewes G, von Kleist-Retzow JC, Wiesner RJ, Magnuson MA, Puccio H, Pfeiffer AF, Steinberg P, Ristow M. Targeted disruption of hepatic frataxin expression causes impaired mitochondrial function, decreased life span and tumor growth in mice. Hum. Mol. Genet. 2005;14:3857–3864. doi: 10.1093/hmg/ddi410. [DOI] [PubMed] [Google Scholar]
- [22].Miranda CJ, Santos MM, Ohshima K, Smith J, Li L, Bunting M, Cossee M, Koenig M, Sequeiros J, Kaplan J, Pandolfo M. Frataxin knockin mouse. FEBS Lett. 2002;512:291–297. doi: 10.1016/s0014-5793(02)02251-2. [DOI] [PubMed] [Google Scholar]
- [23].Pook MA, Al-Mahdawi S, Carroll CJ, Cossee M, Puccio H, Lawrence L, Clark P, Lowrie MB, Bradley JL, Cooper JM, Koenig M, Chamberlain S. Rescue of the Friedreich’s ataxia knockout mouse by human YAC transgenesis. Neurogenetics. 2001;3:185–193. doi: 10.1007/s100480100118. [DOI] [PubMed] [Google Scholar]
- [24].Al-Mahdawi S, Pinto RM, Ruddle P, Carroll C, Webster Z, Pook M. GAA repeat instability in Friedreich ataxia YAC transgenic mice. Genomics. 2004;84:301–310. doi: 10.1016/j.ygeno.2004.04.003. [DOI] [PubMed] [Google Scholar]
- [25].Verloes A, Massin M, Lombert J, Grattagliano B, Soyeur D, Rigo J, Koulischer L, van Hoof F. Nosology of lysosomal glycogen storage diseases without in vitro acid maltase deficiency: delineation of a neonatal form. Am. J. Med. Genet. 1997;72:135–142. [PubMed] [Google Scholar]
- [26].Bradley JL, Blake JC, Chamberlain S, Thomas PK, Cooper JM, Schapira AH. Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum. Mol. Genet. 2000;9:275–282. doi: 10.1093/hmg/9.2.275. [DOI] [PubMed] [Google Scholar]
- [27].Coppola G, Choi S-H, Santos MM, Miranda CJ, Tentler D, Wexler EM, Pandolfo M, Geschwind DH. Gene expression profiling in frataxin deficient mice: microarray evidence for significant expression changes without detectable neurodegeneration. Neurobiol. Dis. 2006;22:302–311. doi: 10.1016/j.nbd.2005.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sarsero JP, Li L, Holloway TP, Voullaire L, Gazeas S, Fowler KJ, Kirby DM, Thorburn DR, Galle A, Cheema S, Koenig M, Williamson R, Ioannou PA. Human BAC-mediated rescue of the Friedreich ataxia knockout mutation in transgenic mice. Mamm. Genome. 2004;15:370–382. doi: 10.1007/s00335-004-3019-3. [DOI] [PubMed] [Google Scholar]
- [29].Berciano J, Mateo I, De Pablos C, Polo JM, Combarros O. Friedreich ataxia with minimal GAA expansion presenting as adult-onset spastic ataxia. J. Neurol. Sci. 2002;194:75–82. doi: 10.1016/s0022-510x(01)00681-5. [DOI] [PubMed] [Google Scholar]
- [30].Gellera C, Pareyson D, Castellotti B, Mazzucchelli F, Zappacosta B, Pandolfo M, Di Donato S. Very late onset Friedreich’s ataxia without cardiomyopathy is associated with limited GAA expansion in the ×25 gene. Neurology. 1997;49:1153–1155. doi: 10.1212/wnl.49.4.1153. [DOI] [PubMed] [Google Scholar]
- [31].Lhatoo SD, Rao DG, Kane NM, Ormerod IE. Very late onset Friedreich’s presenting as spastic tetraparesis without ataxia or neuropathy. Neurology. 2001;56:1776–1777. doi: 10.1212/wnl.56.12.1776. [DOI] [PubMed] [Google Scholar]
- [32].McDaniel DO, Keats B, Vedanarayanan VV, Subramony SH. Sequence variation in GAA repeat expansions may cause differential phenotype display in Friedreich’s ataxia. Mov. Disord. 2001;16:1153–1158. doi: 10.1002/mds.1210. [DOI] [PubMed] [Google Scholar]
- [33].Sorbi S, Forleo P, Cellini E, Piacentini S, Serio A, Guarnieri B, Petruzzi C. Atypical Friedreich ataxia with a very late onset and an unusual limited GAA repeat. Arch. Neurol. 2000;57:1380–1382. doi: 10.1001/archneur.57.9.1380-a. [DOI] [PubMed] [Google Scholar]
- [34].Yokota T, Igarashi K, Uchihara T, Jishage K, Tomita H, Inaba A, Li Y, Suzuki H, Mizusawa H, Arai H. Delayed-onset ataxia in mice lacking α-tocopherol transfer protein: model for neuronal degeneration caused by chronic oxidative stress. Proc. Natl. Acad. Sci. USA. 2001;98:15185–15190. doi: 10.1073/pnas.261456098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Seznec H, Simon D, Bouton C, Reutenauer L, Hertzog A, Golik P, Procaccio V, Patel M, Drapier JC, Koenig M, Puccio H. Friedreich ataxia: the oxidative stress paradox. Hum. Mol. Genet. 2005;14:463–474. doi: 10.1093/hmg/ddi042. [DOI] [PubMed] [Google Scholar]
- [36].Seznec H, Simon D, Monassier L, Criqui-Filipe P, Gansmuller A, Rustin P, Koenig M, Puccio H. Idebenone delays the onset of cardiac functional alteration without correction of Fe–S enzymes deficit in a mouse model for Friedreich ataxia. Hum. Mol. Genet. 2004;13:1017–1024. doi: 10.1093/hmg/ddh114. [DOI] [PubMed] [Google Scholar]
- [37].Jauslin ML, Meier T, Smith RA, Murphy MP. Mitochondria-targeted antioxidants protect Friedreich ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003;17:1972–1974. doi: 10.1096/fj.03-0240fje. [DOI] [PubMed] [Google Scholar]
- [38].Richardson DR, Mouralian C, Ponka P, Becker E. Development of potential iron chelators for the treatment of Friedreich’s ataxia: ligands that mobilize mitochondrial iron. Biochim. Biophys. Acta. 2001;1536:133–140. doi: 10.1016/s0925-4439(01)00041-2. [DOI] [PubMed] [Google Scholar]
- [39].Sarsero JP, Li L, Wardan H, Sitte K, Williamson R, Ioannou PA. Upregulation of expression from the FRDA genomic locus for the therapy of Friedreich ataxia. J. Gene Med. 2003;5:72–81. doi: 10.1002/jgm.320. [DOI] [PubMed] [Google Scholar]
- [40].Voncken M, Ioannou P, Delatycki MB. Friedreich ataxia—update on pathogenesis and possible therapies. Neurogenetics. 2004;5:1–8. doi: 10.1007/s10048-003-0170-z. [DOI] [PubMed] [Google Scholar]
- [41].Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- [42].Ledwozyw AMJ, Stepien A, Kadziolka A. The relationship between plasma triglycerides, cholesterol, total lipids and lipid peroxidation products during human atherosclerosis. Clin. Chim. Acta. 1986;155:275–284. doi: 10.1016/0009-8981(86)90247-0. [DOI] [PubMed] [Google Scholar]