

Abstract
Nitric oxide synthase-2 (NOS2) plays a critical role in reactive nitrogen species generation and cysteine modifications that influence mitochondrial function and signaling during inflammation. Here, we investigated the role of NOS2 in hepatic mitochondrial biogenesis during E. coli peritonitis in mice. NOS2-/- mice displayed smaller mitochondrial biogenesis responses than Wt mice during E. coli infection according to differences in mRNA levels for the PGC-1α co-activator, nuclear respiratory factor-1, mitochondrial transcription factor-A (Tfam), and mtDNA polymerase (Polγ). NOS2-/- mice did not significantly increase mitochondrial Tfam and Polγ protein levels during infection in conjunction with impaired mitochondrial DNA (mtDNA) transcription, loss of mtDNA copy number, and lower State 3 respiration rates. NOS2 blockade in mitochondrial-GFP reporter mice disrupted Hsp60 localization to mitochondria after E. coli exposure. Mechanistically, biotin-switch and immunoprecipitation studies demonstrated NOS2 binding to and S-nitros(yl)ation of Hsp60 and Hsp70. Specifically, NOS2 promoted Tfam accumulation in mitochondria by regulation of Hsp60-Tfam binding via S-nitros(yl)ation. In hepatocytes, site-directed mutagenesis identified 237Cys as a critical residue for Hsp60 S-nitros(yl)ation. Thus, the role of NOS2 in inflammation-induced mitochondrial biogenesis involves both optimal gene expression for nuclear-encoded mtDNA-binding proteins and functional regulation of the Hsp60 chaperone that enables their importation for mtDNA transcription and replication.
Keywords: heat shock proteins, mitochondria, mitochondrial biogenesis, nitric oxide, reactive oxygen species, sepsis
Introduction
Multiple organ failure in severe bacterial infections is a major cause of mortality [1], and nitric oxide (NO) produced by the inducible NO synthase (NOS2) is considered a damaging factor [2]. Among the deleterious effects of NO are mitochondrial damage and loss of electron transport function, generally coinciding with high rates of NOS2 activity, whereas physiological NO levels are usually deemed protective [3]. The calcium-independent NOS2 isoform has the highest capacity for L-arginine catalysis and is the principal source of NO-induced hypotension and vascular injury [4], but this requires enzyme induction during the host response, which in macrophages and other immune cells, yields bactericidal levels of NO [5, 6].
Quite apart from microbial killing, NOS2 is integral to host defense [7], but the enzyme's salutary effects are confounded by collateral cellular damage. NOS2-/- mice show variable susceptibility to infections, indicating a high specificity of action for NOS2 [8]. For instance, hepatic and pulmonary injury by LPS in NOS2-/- mice is comparable to that of wild-type (Wt) mice [7], and NOS2 contributes to hypotension in LPS-induced shock [9], but NOS2-/- mice do not consistently demonstrate improved survival [7, 10]. NOS2 also promotes tissue repair, for example, through angiogenesis [11], but these protective mechanisms, especially in the liver, which orchestrates critical early-phase cytokine production, are poorly understood.
NOS2 produces vasodilation and hypotension via guanylate cyclase [9], but many toxic effects are generated by peroxynitrite (ONOO-) and related products [12]. NO also produces post-translational protein effects via S-nitros(yl)ation (SNO), and SNO protein function is exemplified by reversible modification of over 100 proteins [13]. However, loss of regulation leads to biological dysfunction via abnormal spatial or temporal interactions of NO with various target proteins [14].
In mitochondria, NO produces various effects on electron transport, including inhibition of Complex I by SNO protein formation and inhibition of Complex IV by the formation of NO-transition-metal complexes [15-17]. Respiration is inhibited by peroxynitrite (ONOO-) [18] and by endogenous S-nitrosothiols, which S-nitros(yl)ate mitochondrial proteins [19]. Moreover, membrane SNO proteins, ordinarily reduced by glutathione, may persist when the mitochondrial glutathione pool becomes oxidized. Although excessive or prolonged NO exposure is pro-apoptotic [20], physiological NO is anti-apoptotic, in part through cGMP activity and caspase-3 inhibition [21]. NO also promotes anti-apoptotic gene expression, e.g. mtHsp70 (mortalin) opposes oxidant-induced apoptosis [22], and during mitochondrial biogenesis [23-25].
The ambiguity of NOS2 as an immune defense factor led us to investigate its impact on hepatic mitochondria in gram negative peritonitis in mice, where hepatocellular injury is reflected by increases in serum transaminases, alkaline phosphatase, and total bilirubin, and the histology shows prominent inflammatory cell infiltration, micro-abscess formation, cholestasis, hepatocyte vacuolization, and autophagy [26, 27]. In this setting, hepatic NOS2 induction is a protective factor [28], and we expected NOS2-/- mice to demonstrate contrasting effects on mitochondrial equipoise because functional damage from NO would be opposed by activation of mitochondrial biogenesis. NOS2-/- mice did develop multiple deficits, including limited up-regulation of several transcriptional regulatory elements for mitochondrial biogenesis, but most uniquely, showed an impaired S-nitros(yl)ation of Hsp60 that governed its interactions with mitochondrial Tfam, the major mitochondrial DNA transcription factor, and Polγ (A), and the catalytic core of the mtDNA polymerase, respectively. Thus, the novel mechanistic principle is that NOS2 facilitates Hsp60 chaperone interactions with the proteins required to maintain mtDNA transcription and replication after activation of the host antibacterial defenses.
Materials and Methods
Mouse studies
The studies were conducted in 20-25 g male mice on a protocol pre-approved by the Duke University Institutional Animal Care and Use Committee. Wild-type (Wt) C57BL/6J and inbred genetically-engineered NOS2-/- mice on a C57BL/6J background were purchased from Jackson Laboratories (Bar Harbor, ME) and maintained in a pathogen free-barrier facility. Live bacteria were grown from lyophilized E. coli (serotype 086a:K61, American Type Tissue Culture Collection, Rockville, MD), quantified as described [29], and either implanted in a fibrin clot into the peritoneum [30] or heat-inactivated at 65°C and frozen at -80°C. Heat-killed E. coli were thawed once and diluted with sterile, endotoxin-free NaCl to a concentration of 1 × 108/ml and 0.5 ml was injected into the peritoneum. The mice also received 1.0 ml of 0.9% NaCl subcutaneously when the bacteria were administered.
Mitochondrial isolation
Liver mitochondria were prepared by two methods. For respiration studies, liver homogenates prepared by hand-Dounce were centrifuged at 1,000 × g for 10 min and the supernatants placed on a 0.25% sucrose cushion and centrifuged at 15,000 × g for 15 min. State 3 and State 4 respiratory capacity and respiratory control ratios (RCR) were measured at 35°C using calibrated Clark electrodes [31, 32]. Purified mitochondria were obtained from the liver homogenate by discontinuous Percoll gradient centrifugation using a method for highly coupled organelles [33]. The mitochondria-rich band at the interface between Percoll layers was harvested, collecting organelles of the same density. Mitochondria were washed once, re-suspended and placed on ice. Mitoplast and outer membrane fractions were prepared using the method of Greenawalt [34]. Nuclei were obtained from liver homogenates by centrifugation at 1,000 × g for 20 min and then through 1.75 M sucrose at 40,000 × g for 1 h. Protein content was determined using bicinchoninic acid with BSA as a standard (Pierce, Rockford, IL).
Nuclear Expression of Mitochondrial Genes
Cytoplasmic RNA was extracted with TRIzol RNA isolation kits (Invitrogen) [31]. Total RNA (1μg) was reverse-transcribed using oligo(dT) or with gene-specific primers for mouse NADH dehydrogenase subunits 1 (ND1) or cytochrome b (Cytb). Real-time RT-PCR was performed on the ABI Prism 7000 using SYBR Green master mix (Applied Biosystems, Foster City, CA). After PCR, the samples were subjected to melting curve analysis. The threshold cycle (Ct) was determined in the exponential phase and a ΔCT method used to quantify mRNA levels for Cyt b, ND1, NRF-1, NRF-2α, PGC-1α, Polγ, Tfam, and NOS2. Amplification efficiency was checked with an internal 18S rRNA reference over a range of 0.9–90 ng of RNA and gene expression was quantified using ABI Prism 7000 SDS Software. Each sample was assayed in triplicate and the mean values reported.
Mitochondrial DNA copy number
MtDNA was isolated from purified mitochondria and copy number quantified by real-time PCR on a 7700 Sequence Detector System (AB Applied Biosystems). Amplifications were performed on 10 ng total mtDNA using primers designed for cytochrome b (cyt b-s and cyt b-as) with ABI Probe Design software (Applied Biosystem). One copy of linearized pGEMT-cyt b vector was used as a standard for simultaneous mtDNA quantification [35]. The cyt b probe, 5′ FAM-ttcctccacgaaacaggatcaaa-TAMRA 3′, contained at the 5′ end the FAM (6-carboxy-fluorescein) as a fluorescent reporter dye and the 3′ end, the TAMRA (6-carboxy-tetramethyl-rhodamine) as a quencher dye selected from a highly conserved region of mouse cytochrome b gene. Serial dilutions of 105–10 copies of cyt b plasmid were prepared to establish the standard curve. Samples were tested in triplicate for mtDNA at 1:100 and 1:1,000 and the number of mtDNA copies determined relative to known standards and expressed on a log scale.
Western analysis
Total, nuclear, and mitochondrial proteins were separated by SDS-PAGE and transferred to PVDF membranes [35]. Membranes were incubated with validated polyclonal rabbit antibody against mouse PGC-1α, NRF-1, Tfam, and Polγ [31] or with antibodies to caspase-3 (Cell Signaling; Beverly, MA), SOD2, or NOS2 (Santa Cruz Biotechnology). β-actin was used as a loading control for total or nuclear protein and anti-porin for mitochondrial protein (Sigma). After five washes in TBST, the membranes were incubated in HRP-conjugated goat anti-rabbit or anti-mouse IgG (Amersham). Blots were developed with ECL and protein quantification performed on digitized images in the mid-dynamic range and expressed relative to the loading control.
Apoptosis assay
For in situ apoptosis studies, the anti-caspase-3 antibody (Cell Signaling) was used. After perfusion-fixation with 10% formalin, processing, and sectioning, apoptosis was assessed using the TUNEL assay kit and the manufacturer's instructions (Promega). Alexa Fluor 594-labeled streptavidin was used to develop the signal for labeled nuclei. Photomicrographs were taken on a Nikon Eclipse 50i Fluorescence Microscope at 400× and number of apoptotic nuclei counted per 100 hepatocytes.
Immunoblotting/Immunoprecipitation
Mitochondrial proteins (100μg) were incubated overnight at 4°C with 1 μg of Hsp60 antibody (Santa Cruz) or pre-immune serum (negative control) followed by 2 h with 20μl of protein A-Sepharose beads (Santa Cruz; pre-washed and suspended 1:1 in phosphate-buffered saline). After incubation, the beads were washed four times with lysis buffer, the proteins separated by SDS-PAGE, transferred to Immobilon-P (Millipore), and analyzed by immunoblotting with anti-Polγ, anti-Tfam, or anti-NOS2 antibodies (Santa Cruz). Horseradish peroxidase-conjugated secondary antibodies were applied and the signals developed with ECL. Hsp60 and Hsp70 co-immunoprecipitated proteins were checked by Western blotting with validated antibodies.
Biotin-switch labeling of SNO proteins
Liver mitochondria were prepared and processed for the biotin switch assay in dark conditions by established methods [36-38]. The mitochondria (∼3mg) were suspended in 1 ml HEN (250mM HEPES, 1mM EDTA, and 0.1mM neocuproine at pH 7.7), and fresh MMTS (methyl methanethiosulfonate) in N,N-dimethylformamide and SDS added at final concentrations of 0.2 and 2.25% respectively. The samples were incubated for 20 min at 50°C with frequent mixing before precipitation in cold acetone [36, 37]. For CSNO studies, 0 to 50 μM final CSNO was added to isolated mitochondria in 1.5 ml HEN buffer for 15 min at room temperature followed by MMTS/SDS and acetone precipitation. The precipitated protein was re-suspended in 850 μl HENS and fresh ascorbate (20 mM) and 0.4 mM biotin-HPDP (Pierce) were added for 1h followed by precipitation, washing, and suspension in Laemmli buffer for SDS–PAGE. Biotinylation was detected with streptavidin-HRP (Amersham). Alternatively, the biotinylated proteins were suspended in 250 μl HEN plus 750 μl of neutralization buffer (20 mM HEPES, 100 mM NaCl, 1 mM EDTA, 0.5% Triton X-100) and precipitated overnight in 50 μl of pre-washed streptavidin–agarose beads. The beads were washed five times in 600 μM NaCl neutralization buffer at 4°C and biotinylated proteins eluted in 50μl elution buffer (25 mM HEPES, 100 mM NaCl, 1 mM EDTA, 100 mM mercaptoethanol) and heated in reducing buffer for 5 min at 95°C before SDS–PAGE. Western blotting was done with anti-Hsp60 or anti-Hsp70 (Santa Cruz). Gel loading was checked by Coomassie blue or silver staining.
Hsp60-expressing plasmids and mutagenesis
A Myc-DDK-tagged clone of the open-reading frame (ORF) of the human Hsp60 expression vector was obtained from Origen (RC201281). The Hsp60 ORF cloned in pCMV6-Entry Vector was expressed in mammalian cells as a tagged protein with C-terminal Myc-DDK tags (the same as FLAG). Mutant Hsp60 (mut-Hsp60) was created in which 237Cys in a nitros(yl)ation motif was replaced with alanine by mutagenesis using the QuikChange II kit (Stratagene) and mut-Hsp60 primers: sense (5′-CATCAAAAGGTCAGAAAGCTGA ATTCCAGGATGCCTATG -3′) and antisense (5′-CATAGGCATCCTGGAATTCAGCTTTC TGACCTTTTGATG -3′). Rat H4IIE hepatoma cells were transfected at 50–70% confluence with Wt or mutant Hsp60 or empty vector using the FuGene system (Roche) and then grown to 80–90% confluence. Wt and mut-Hsp60 cell and mitochondrial lysates were tested for protein expression using anti-tag antibody (Origene).
Control and mut-Hsp60 transfected H4IIE cells were treated with LPS + TNF-α (10 μM each) for 24 h and then washed with cold phosphate-buffered saline (PBS). The mitochondria were isolated and placed in lysis buffer (20 mM Tris, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton ×100, 1 μg/ml leupeptin, 1 mM PMSF) at pH 7.4 for 20 min. The lysates (40 μg) were incubated overnight at 4°C with anti-flag antibody and then with protein A-agarose beads (Santa Cruz) for 2h. The beads were washed twice in lysis buffer and twice in PBS, and eluted in SDS sample buffer for Western blotting with anti- Hsp-60 or Tfam.
Statistics
Grouped data are reported as means ± standard error. Statistical analysis was by computer software with Student's t-tests or ANOVA followed by Tukey's post-hoc test as appropriate. The n values indicated in the figure legends are for independent replicates. Significance was P < 0.05 for all experiments.
Results
The fibrin-clot model
Implantation of infected fibrin clots into the mouse peritoneum produces dose-dependent hepatic damage with focal necrosis and apoptosis by 24-48h [30]. More pronounced and diffuse hepatic apoptosis was encountered in NOS2-/- than in Wt mice, where it was sporadic and closely associated with inflammatory foci (Fig. 1A). By day 3, non-neutrophilic TUNEL-positive cell counts in control and infected Wt mice were 2.1 and 5.5%, respectively, while the counts in NOS2-/- mice were 3.4 and 11.2%, respectively (P<0.05 between strains). In NOS2-/- mice, apoptosis involved caspase-3 activation (Fig. 1B), and lethality was higher in NOS2-/- mice than in Wt mice (Figure 1C). Since NOS2-/- mice demonstrated early cell death and increased mortality, and differences in bacterial clearance in the mouse strains can impact on survival, we performed mechanistic studies with non-lethal heat-inactivated E. coli to eliminate this bias and improve specificity for NOS2 effects by delta-function activation of Toll-like receptor-4 (TLR4) [29, 39].
Figure 1. Hepatic apoptosis and survival after live E. coli peritonitis in mice.
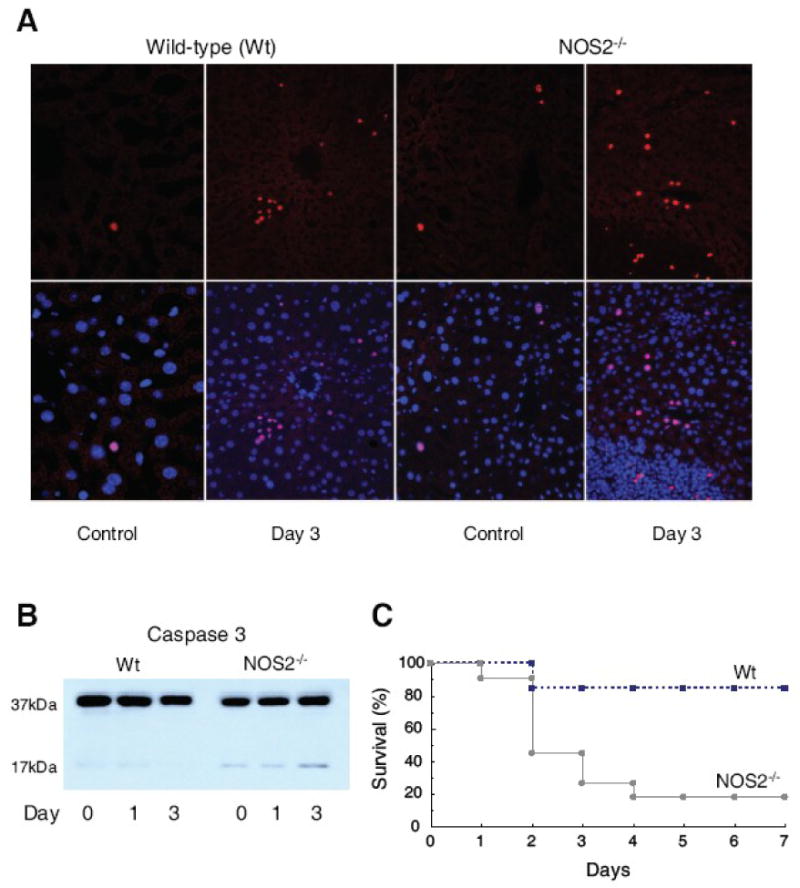
E. coli inoculation is 1× 107 CFU. Fig. 1A shows TUNEL staining of liver sections of Wt and NOS2-/- mice pre and 3 days post fibrin clot implantation. Top row, Wt mice showed occasional scattered small clusters and independent TUNEL-positive cell nuclei (red), while NOS2-/- mice show many stained nuclei throughout. Bottom images show TUNEL merger with DAPI nuclear staining. Fig. 1B: NOS2-/- mice showed greater hepatic cleavage of caspase-3 than Wt mice. Fig. 1C: NOS2-/- mice have a higher 7-day mortality than Wt mice (P<0.05).
Transcriptional program of mitochondrial biogenesis in Wt and NOS2-/- mice
Transcriptional activation of nuclear-encoded activators of mitochondrial biogenesis was evaluated by real time RT-PCR. An mRNA analysis of the NRF-1 and NRF-2 transcription factors and the PGC-1α nuclear co-activator of mitochondrial biogenesis indicated that all three responded in Wt and NOS2-/- mice (Fig. 2A-C) followed by transient mRNA elevations for two genes, Tfam and Polγ, required for mtDNA transcription and replication (Fig. 2D, E). The mRNA profiles for NRF-1 and PGC-1α were attenuated in NOS2-/- compared with Wt mice, and the responses were time-shifted by 1 day. NOS2-/- mice showed lower mRNA levels than Wt mice for the mitochondrial transcriptosome proteins in the first three days after challenge.
Figure 2. NOS2 effect on transcriptional control of mitochondrial biogenesis.
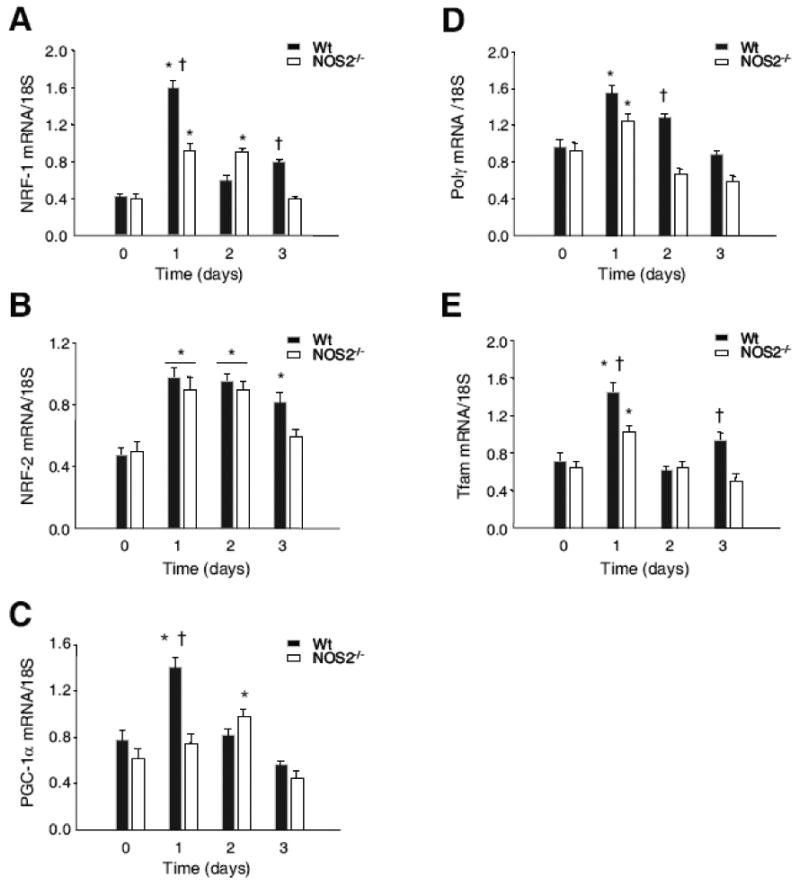
Fig. 2A-C: Real time RT-PCR analysis of nuclear-encoded transcriptional activators of mitochondrial biogenesis, NRF-1 and NRF-2, and the PGC-1α co-activator, indicated increases in all three mRNA levels after E. coli challenge in Wt and NOS2-/- mice. NRF-1 and PGC-1α transcript responses were attenuated and PGC-1α response was late-shifted in NOS2-/- mice. Fig. 2D&E: Transient elevations of nuclear-encoded transcript levels for Polγ and Tfam, downstream genes involved in mtDNA transcription and replication. In NOS2-/- mice, transcript levels for these mitochondrial proteins were attenuated compared with the Wt responses (n=4 mice for all time points; * P<0.05 vs. baseline; † P<0.05 Wt vs. NOS2-/-).
The differences in NRF-1 and PGC-1α, which together regulate downstream Tfam and Polγ genes, were evaluated by nuclear Western analysis (Fig. 3A). By gel densitometry, the nuclear accumulation of NRF-1 and PGC-1α was significantly delayed in NOS2-/- mice by at least one day (Fig. 3B).
Figure 3. Nuclear Western analysis of NRF-1 and PGC-1α proteins.
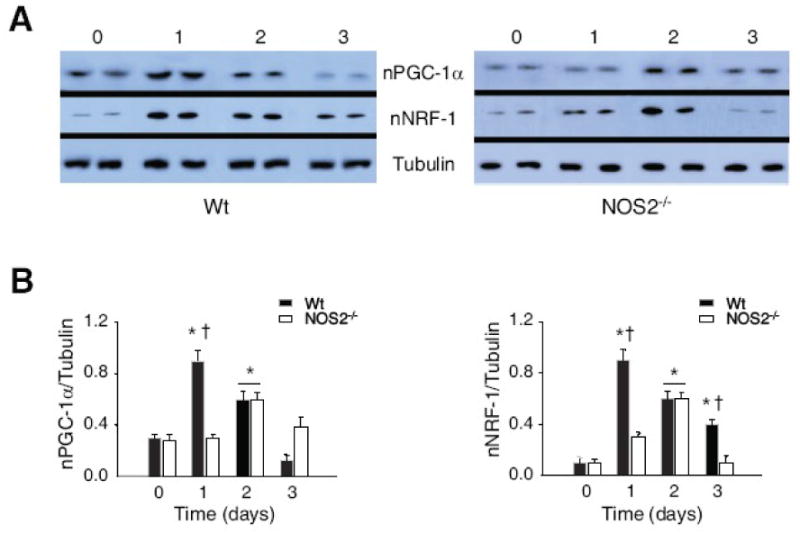
Nuclear protein levels for upstream regulators of Tfam and Polγ gene expression by Western analysis at 0, 1, 2, and 3 days after E. coli administration in Wt and NOS2-/- mice (Fig. 3A). Densitometry confirms delayed nuclear enrichment of NRF-1 and PGC-1α in NOS2-/- compared with Wt mice (Fig. 3B; n= 4 samples at each time point; * P<0.05 vs. baseline; † P<0.05 Wt vs. NOS2-/- mice).
Mitochondrial function
The proliferation and function of liver mitochondria in Wt and NOS2-/- mice were compared before and after E. coli administration in several ways. Because mtDNA damage can impair mitochondrial gene transcription, mtDNA content was measured by real time PCR. In both lines of mice, hepatic mtDNA copy number decreased at day 1 after E. coli, and Wt but not NOS2-/- mice recovered by day 2 (Fig. 4A). At day 3, the mtDNA deficit in NOS2-/- mice remained significant, indicating clear differences between NOS2-/- and Wt mice in the regulation of mtDNA copy number. To assess this difference, mRNA levels were assayed for two mtDNA-encoded genes, cytochrome b (Cyt b) and NADH dehydrogenase subunit 1 (ND1). Cyt b and ND1 mRNA levels increased significantly in Wt but not NOS2-/- mice, which is compatible with the differences in mtDNA content (Fig. 4B&C ). In the fibrin-clot live bacteria model, hepatic State 3 respiration improved significantly over 3 days for both NADH- and FADH2-linked substrates in Wt but not in NOS2-/- mice (Fig. 4D). RCR was in the range of 4-8, and although NOS2-/- mice showed a trend toward looser coupling at day 3, overall State 4 rates and respiratory coupling were not statistically different over the 3 days.
Figure 4. Molecular and functional responses in liver mitochondria of Wt and NOS2-/- mice.
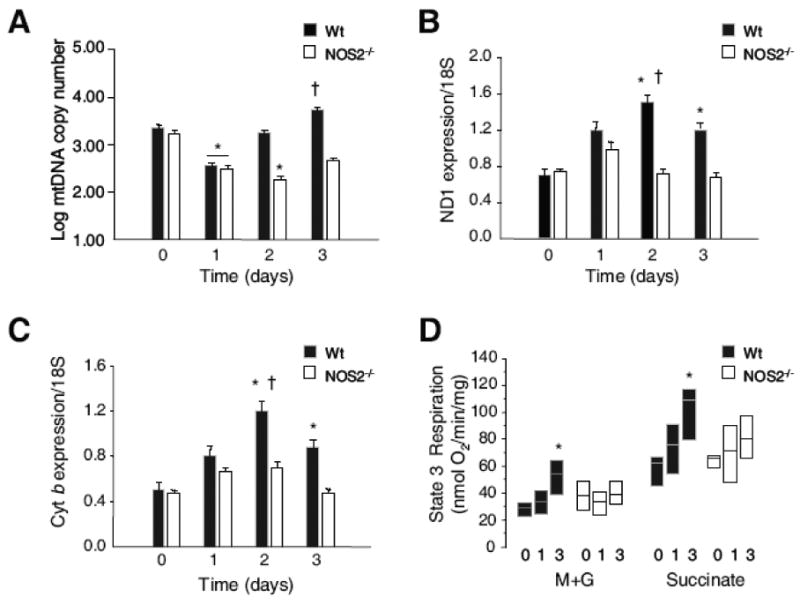
Fig. 4A: Administration of E. coli decreased hepatic mtDNA copy number at day 1 in both lines with recovery by day 2 in Wt but not in NOS2-/- mice. MtDNA copy number remained significantly reduced in NOS2-/- mice at day 3 (P<0.05). Fig. 4B & C: Inter-genotype differences in mtDNA transcription measured by mRNA levels for cytochrome b (Cyt b) and NADH dehydrogenase subunit 1 (ND1) showed significant increases by day 2 for Cyt band ND1 in Wt mice but not in NOS2-/- mice. Fig. 4D: Box plots of State 3 (peak) respiration rates for succinate (S) and malate + glutamate (M+G) in mitochondria isolated from mice with E. coli peritonitis showed significant expansion of State 3 respiration by day 3 in Wt but not NOS-/- mice (n=3-4 mice for all time points; * P<0.05 vs. baseline; † P<0.05 Wt vs. NOS2-/-).
Mitochondrial proteins
The late recovery of mtDNA copy number in NOS2-/- mice may have reflected different rates of nitrosative damage and/or capacity to replicate mtDNA; e.g. due to compromised mitochondrial protein importation. First, we evaluated NOS2 localization after the infection by Western analysis of whole homogenate (Fig. 5A) and liver mitochondria (Fig. 5B) and identified a significant fraction of the enzyme associated with mitochondria. As NOS2 lacks a known mitochondrial leader sequence, gentle digitonin-stripping of the outer mitochondrial membrane was performed, and most of the enzyme was removed (Fig. 5C).
Figure 5. NOS2-dependent association of Hsp60 with mitochondrial Tfam and Polγ.
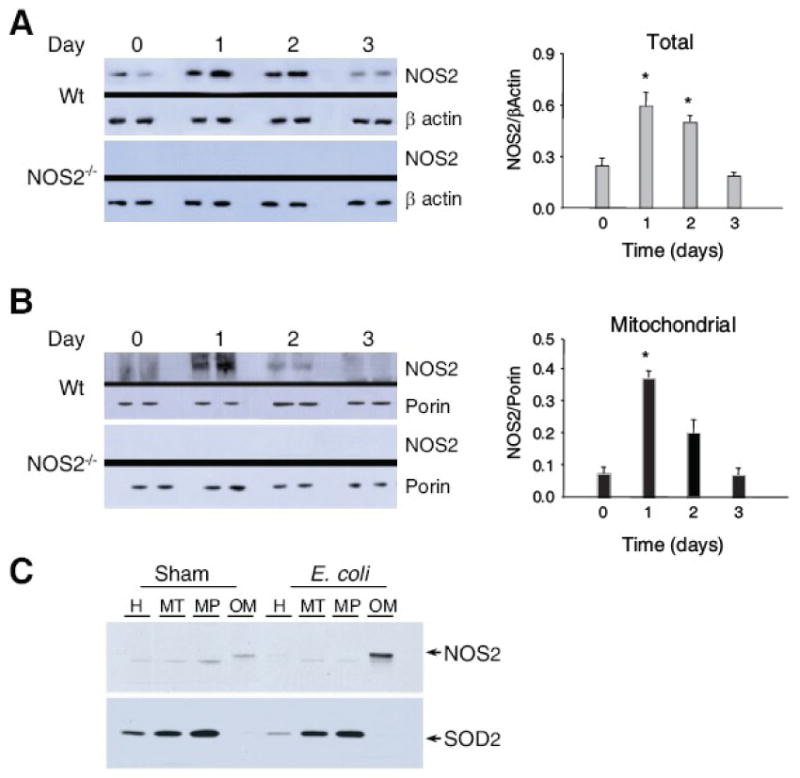
Fig. 5A: Western blots of NOS2 in liver at days 0, 1, 2, and 3 after E. coli challenge. NOS2 increased similarly in total homogenate and in the mitochondrial fraction with a peak response at day 1 (Fig. 5B). Comparable samples from NOS2-/- mice are provided as negative controls (*P<0.05 vs. baseline).
Next, the quantities of the mtDNA binding proteins Tfam and Polγ protein were examined by Western blot relative to porin (VDAC), a stable outer membrane protein (Fig 6A). Unlike the mRNA levels, where NOS2-/- mice were only modestly below Wt levels, mitochondrial Tfam and Polγ increased in Wt but remained unchanged in NOS2-/- mice. To assess mitochondrial oxidative stress, we checked hepatic SOD2 mRNA and mitochondrial protein levels and found that SOD2 mRNA levels increased by only ∼50% in both strains after E. coli and then recovered (Fig. 6B left panel). The mitochondrial SOD2 protein increased slightly more in Wt than in NOS2-/- mice, but overall, the changes were less than twofold (Fig. 6B right).
Figure 6. Mitochondrial Hsp Tfam and Polγ proteins.
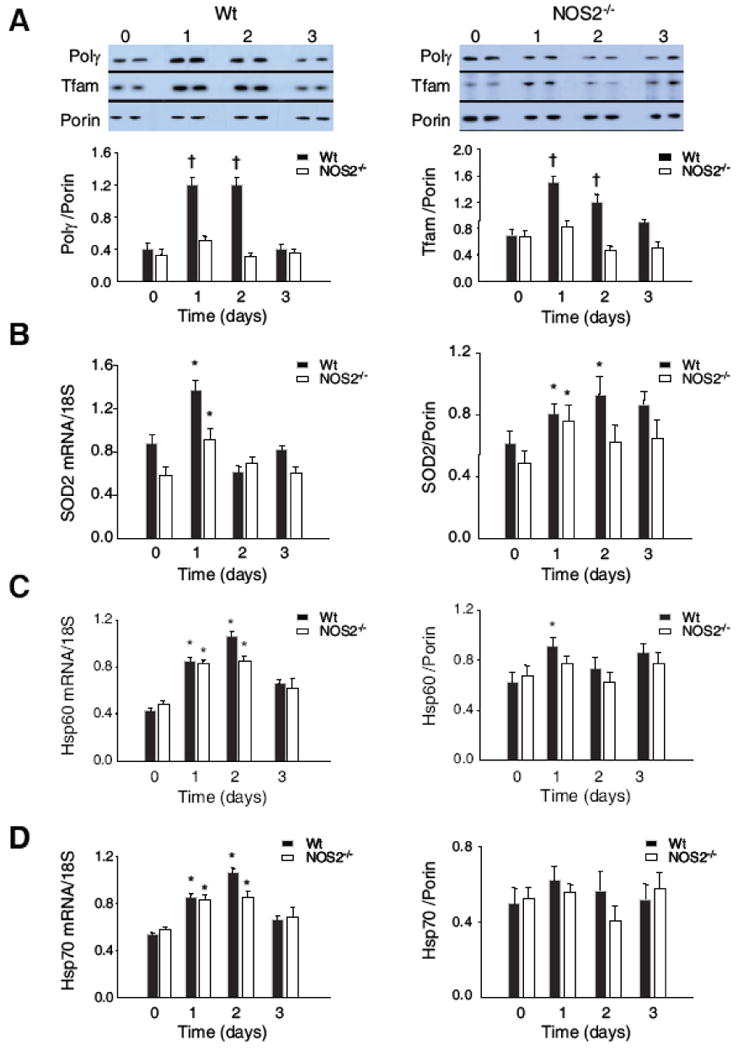
Tfam and Polγ protein in Wt and NOS2-/- mouse liver mitochondrial extract by Western blot relative to porin (Fig. 6A). These mitochondrial proteins increased significantly in Wt, but not in NOS2-/- mice. Mitochondrial Hsp60 and Hsp70 content showed a small increase in Hsp60 relative to porin after E. coli in Wt, but a minimal difference compared with NOS2-/- mice. Hsp70 levels were stable in both types of mice. Fig. 6B: SOD2 mRNA increased on day 1 after E. coli by 50%, but then returned to baseline in both lines. Mitochondrial SOD2 protein responded with less than a twofold increase in NOS2-/- and Wt mice. Fig. 6C: Total hepatic mRNA and mitochondrial protein levels for Hsp60 increased comparably experiment-wide in both types of mice. Fig. 6D: Total hepatic mRNA and mitochondrial protein levels for Hsp 70 as in 6C (Densitometry for 6A-D is n= 4 samples per time point; * P<0.05 vs. baseline; † P<0.05 vs. baseline and vs. NOS2-/-).
This discrepancy suggested that NOS2 influences mitochondrial protein importation, which was examined using mRNA and protein for two critical chaperones, Hsp60 (Fig. 6C) and mtHsp70 (mortalin; Fig. 6D). The mRNA levels for these chaperones were similar in Wt and NOS2-/- mice after E. coli challenge, and protein expression was marginally increased relative to porin. Similar mitochondrial levels of both chaperones were detected in NOS2-/- and Wt mice (Fig. 6C and 6D); thus, differences in mitochondrial Hsp60 and Hsp70 protein regulation did not explain the failure of mitochondrial Tfam and Polγ to increase in NOS2-/- mice.
Fluorescence microscopy
Mitochondrial Hsp60 was localized by fluorescence microscopy of fixed liver sections in mitochondrial reporter mice that express GFP constitutively in the mitochondria. Control sections revealed a typical monotonic distribution of mitochondria and modest Hsp60 staining. At 24h after E. coli administration, strong Hsp60 localization to mitochondria was present, as shown in supplemental Figure 1 (Fig. S1A, B, yellow color). However, when 1400W was administered to inhibit NOS2 before E. coli challenge, the induction of Hsp60 was heterogeneous and the protein distribution did not effectively match that of mitochondria (Fig. S1C, D, orange color). These co-localization data provided evidence that NOS2 facilitates functional integration of Hsp60 into the hepatic mitochondrial system.
NOS2-dependent Hsp60 association with Tfam and Polγ
Levels of several other antioxidant anti-apoptotic proteins in mitochondria were checked, i.e. thioredoxin-2 (Trx2), thioredoxin reductase-2 (TrxR2), and peroxiredoxin-3 (Prxd3), which are also involved in mitochondrial NO transfer (Fig. 7A). Mitochondrial protein levels for all three proteins increased on day 1 in Wt mice, but similar to Tfam and Polγ, did not increase in NOS2-/- mice.
Figure 7. Mitochondrial Hsp60 interactions with Tfam and Polγ protein.
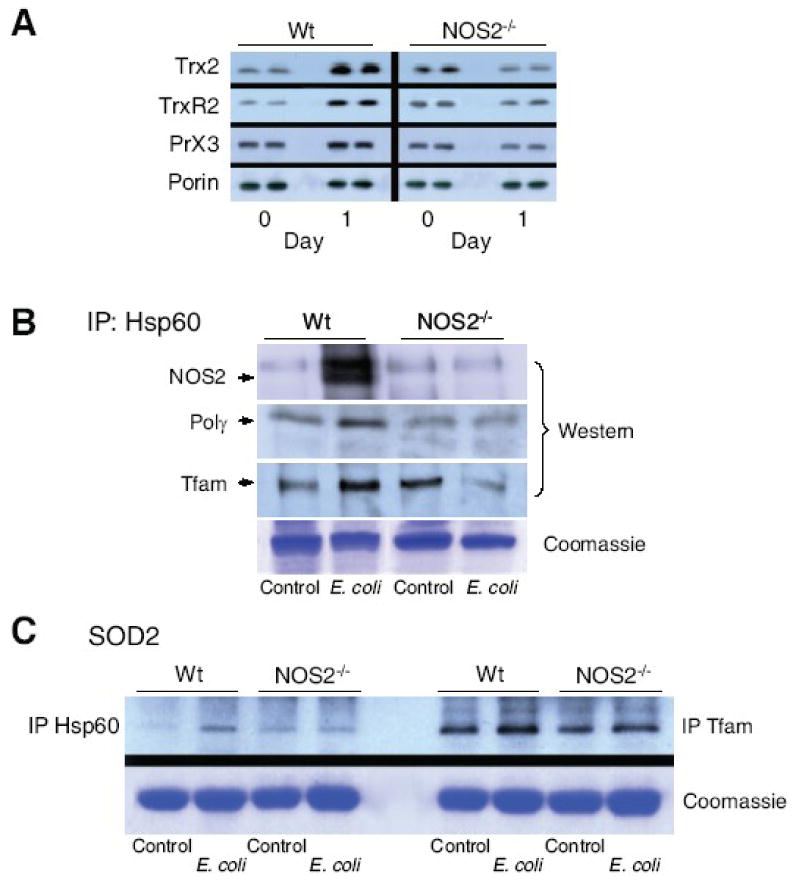
Fig. 7A: Hepatic Hsp60 total/mitochondrial ratio at day 0, and day 1 and 3 after E. coli peritonitis for both lines of mice. Hsp60 was stable in mitochondria relative to porin and total Hsp60 in the extracts. Fig. 7B: Mitochondrial antioxidant, anti-apoptotic proteins, thioredoxin-2 (Trx2), thioredoxin reductase-2 (TrxR2), and peroxiredoxin-3 (Prx3) increased by Western analysis in Wt mice but failed to respond in NOS2-/- mice (* P<0.05 vs. baseline; † P<0.05 vs. baseline and vs. NOS2-/-). In Fig. 7C, NOS2 protein was not detectable in mitochondria pre-challenge, but co-precipitated with mitochondrial Hsp60 at day 1 post E.coli (top gel). Hsp60 co-precipitated with Tfam and Polγ before, but more strongly at 1 day post challenge in Wt mice. In NOS2-/- mice, the Hsp60 association was unchanged or declined after challenge (second and third gels). Fig. 7D: In contrast, in both lines of mice, mitochondrial Hsp60 co-precipitated weakly with SOD2 (left 4 lanes), while SOD2 co-precipitated strongly with Tfam before and after challenge (right 4 lanes).
Mitochondrial protein-protein interactions among NOS2, Hsp60 (or mtHsp70), Tfam, and Polγ were evaluated by protein immunoprecipitation and Western analysis before and at 1 day after E. coli challenge. Anti-Hsp60 was used to precipitate mitochondrial proteins and strong NOS2 co-precipitation was identified post-challenge (Fig. 7B; top). Hsp60 also associated with Tfam and Polγ in Wt mice, but this association was diminished in NOS2-/- mice (Fig 7B; middle, lower panels). Analogously, Tfam and Polγ interacted similarly with Hsp70 protein (data not shown). By contrast, Hsp60 associated only weakly with SOD2, but SOD2 and Tfam strongly co-precipitated before and 1 day after E. coli in the mitochondria of both strains (Fig. 7C).
Biotin switch assays
The biotin switch demonstrated constitutive hepatic Hsp60 and mtHsp70 SNO protein formation, but after E. coli administration, SNO levels for both proteins increased only in Wt mice (Fig. 8A; left panel). In contrast, SNO levels were stable or actually declined in NOS2-/- mice after E. coli (Fig. 8A). In isolated whole liver mitochondria, the low molecular weight NO donor, CSNO, fully S-nitros(yl)ated Hsp60 at 15μM and Hsp70 at 50μM CSNO (Fig. 8A; middle panel). Fig. 8A (right panel) also demonstrated that mitochondria of Wt control and NOS2-/- mice generate ascorbate-dependent Hsp60 and Hsp70 SNO formation in vitro.
Figure 8. Biotin switch assays of mitochondrial Hsp60 and Hsp70 and site-directed mutagenesis of Hsp60.
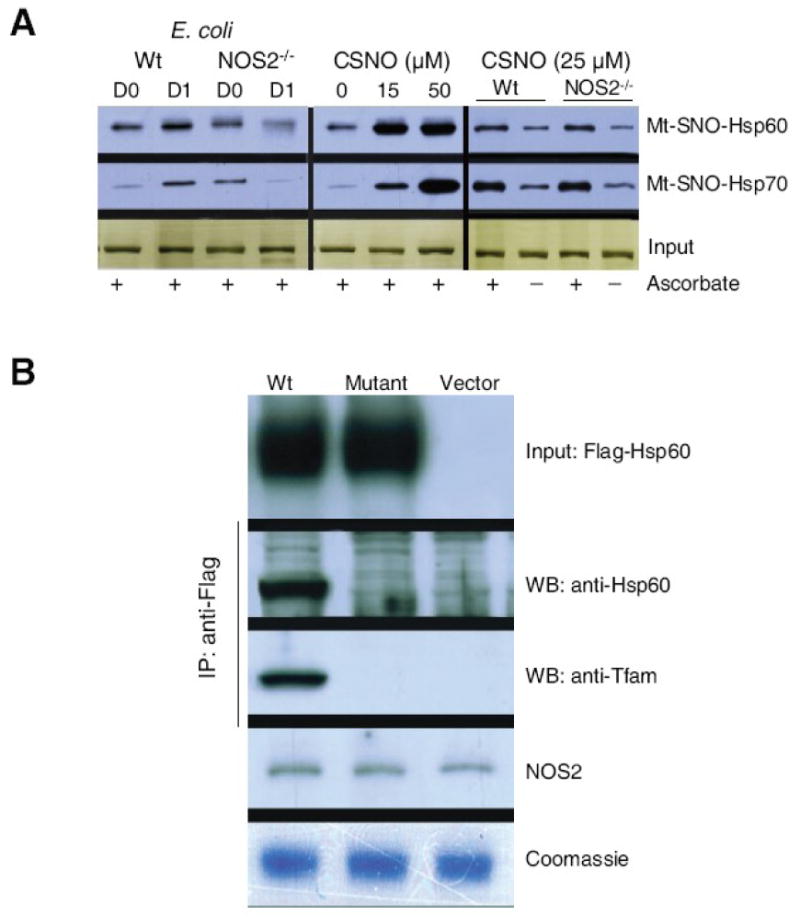
Constitutive S-nitros(yl)ation of both chaperones in Wt and NOS2-/- mice. SNO protein levels increased in Wt mice after E. coli challenge (Fig. 8A; left panel) but fell in NOS2-/- mice. In liver mitochondria, the low molecular weight NO donor, CSNO, fully nitros(yl)ated Hsp60 at 15μM and Hsp70 at 50μM CSNO (Fig. 8A middle panel). Equivalent ascorbate-dependent Hsp60- and Hsp70-SNO formation was demonstrated in control Wt and NOS2-/- hepatic mitochondria (Fig. 8A right panel). Fig. 8B shows studies of flagged wild-type and mutant Hsp60 protein in H4IIE rat hepatocytes. Hsp60 in mitochondria and association with Tfam was demonstrated in cells transfected with flagged native Hsp60 and treated for 24h with LPS+TNF-α to induce NOS2, but neither occurs when 237Cys is replaced by Ala in mut-Hsp60. Mitochondrial NOS2 and protein loading are shown for comparison.
Site-directed mutagenesis and mitochondrial localization of Hsp60
After the conversion of 237Cys to Ala in the Hsp60 KCE motif by site-directed mutagenesis, rat H4IIE hepatocytes were transfected with control or mutant Hsp60 (Fig. 8B). Mitochondrial immunoprecipitation studies demonstrated that native Hsp60 associates with mitochondria and binds Tfam after NOS2 induction by LPS/TNFα administration. However, mut-Hsp60 expressing cells demonstrated neither tagged mitochondrial Hsp60 nor Tfam binding despite the presence of endogenous NOS2 (Fig. 8B). The inability of mut-Hsp60 to attach to mitochondria or bind to Tfam after NOS2 induction provided further evidence that the enzyme S-nitros(yl)ates Hsp60 and targets it to mitochondria.
Discussion
The role of NOS2 induction in the mammalian antibacterial defenses has been difficult to define since it became apparent that NO over-production adversely affects outcome [7, 10]. The systemic release of enteric bacteria activates the innate immune response primarily through TLR4 and classical NF-kB signaling, which generates early-phase inflammatory cytokines and induces NOS2 [29, 39, 40]. Although NOS2 causes deleterious vasodilation and hypotension, in the liver, a sentinel organ of host defense, and particularly in the Kupffer cell, NOS2 is protective. Here, NOS2-/- mice demonstrated higher mortality in infective E. coli peritonitis compared with Wt mice as well as greater hepatic caspase-3 activation and apoptosis. Whether the mortality difference is related to mitochondrial damage is not clear, but the involvement of NOS2 in the pro-survival program of mitochondrial biogenesis is important because mtDNA depletion is associated with higher apoptosis rates in accordance with the known susceptibility to intrinsic apoptosis produced by a lack of NO [21].
NOS3 and NOS1 are known to participate in mitochondrial biogenesis e.g. via cGMP-linked expression of the PGC-1α co-activator and its transcription factor partners; however, such a role for NOS2, apart from late transcriptional activation of the program in the heart in NOS2-/- mice, has not been established [41]. Here NOS2-/- mice displayed slower recovery of hepatic mtDNA copy number compared with Wt mice despite comparable early declines in copy number after E. coli challenge. Post-challenge mRNA levels for mitochondrial Cyt b and ND1 in NOS2-/- mice also did not respond, implying that the low mtDNA copy number interfered with mtDNA transcription. This could explain the restricted capacity of NOS2-/- mice to expand State 3 respiration rates compared with Wt mice, which has important implications for cell survival and organ failure, especially during continuing inflammatory stress.
The initial phase of this study described the effects of NOS2 deficiency on the major nuclear transcriptional regulatory factors for hepatic mitochondrial biogenesis. NOS2-/- mice did show modest induction of NRF-1, NRF-2 (GABPα), and PGC-1α, but the nuclear accumulation of NRF-1 and PGC-1α protein was impaired. Any resulting NOS2-dependent differences in gene expression should contribute to a delay in recovery from organ damage; for instance, the delayed expression of Tfam and Polγ mRNA probably contributed to the lack of accumulation of these proteins in the mitochondria. In combination, these events would have a braking effect on the mtDNA transcription and replication necessary for mitochondrial biogenesis.
The disparity between Wt and NOS2-/- mice in the mitochondrial content of the Tfam and Polγ mtDNA-binding proteins, despite the comparable early mtDNA depletion, implicated NOS2 primarily in mtDNA recovery rather than in mtDNA damage in this model. Moreover, mitochondrial thioredoxin-2 and peroxiredoxin-3 increased relative to porin in Wt but not NOS2-/- mice, reflecting selective NO influences over mitochondrial protein composition [42, 43]. Mitochondrial SOD2 levels however were not appreciably affected, presumably indicating that superoxide leak rates and retrograde signaling for angiogenesis [44] and mitochondrial biogenesis were comparable in the two strains [45].
Functional linkage of NOS2 to the mitochondrial transcriptome is also implicit in the different ratios of the transcriptome proteins to porin, a reasonably stable outer membrane protein [46]. The strong imbalance in mitochondrial Tfam/Polγ protein levels between strains after E. coli is telling, as these proteins tripled in Wt mice but failed to increase in NOS2-/- mice. A general interpretation of this observation is that mitochondrial protein importation for maintenance of mtDNA copy number is governed by NO availability. Mitochondrial protein importation depends on transit peptides that target pre-proteins to a translocase system spanning the outer and inner mitochondrial membranes [47]. This highly-ordered system also incorporates the outer membrane sorting and assembly machinery (SAM) and uses energy from ATP or from the transmembrane proton gradient. The translocase guides unfolded cytosolic pre-proteins inward via matrix chaperones, most importantly Hsp60 and mtHsp70, which are part of a poorly understood molecular motor [48]. Nonetheless, these chaperones clearly facilitate macromolecular assembly, including the initial folding as well as the refolding of polypeptides denatured by matrix stress.
Mitochondrial chaperones are also distinctly pro-biogenesis and anti-apoptotic, but if Hsp60 accumulates in the cytosol, it can promote apoptosis by interacting with caspase-3 [49]. Here, Hsp60 and Hsp70 mRNA levels increased after E. coli, but the protein levels remained stable or increased slightly in both strains, implying that lack of translation of these chaperones did not explain why NOS2-/- mice failed to maintain mitochondrial transcriptome protein levels.
NOS2 lacks a canonical mitochondrial leader sequence, but still associates periodically with mitochondria [50], and here after E. coli exposure was found loosely attached to mitochondria in amounts commensurate with that in total liver homogenate. NOS2 is active at the mitochondrion [50], and we found it associated with differential enrichment of imported mitochondrial proteins. Immunoprecipitation studies indicated that mitochondria-associated NOS2 binds to Hsp60, and Hsp60 to Tfam and to Polγ, linking NOS2 to a functional mitochondrial transcriptome independently of any intrinsic mitochondrial NOS. In checking for Hsp60 binding to mitochondrial Tfam and Polγ, increased binding of both was found after E. coli in Wt, but not in NOS2-/- mice. This finding, together with an inability of NOS2-/- mice to increase mitochondrial Tfam and Polγ protein content, implicated NOS2 in the Hsp60 chaperone function for these transcriptome proteins; however, a detailed spatial understanding of NOS2 at the mitochondrial-cytosolic interface will require better resolution. It is reiterated that this does not require a matrix NOS [51, 52], the presence of which is controversial [53], but simply an association of NOS2 with the outer mitochondrial membrane.
The comparative stability of Hsp60 and 70 mitochondrial protein levels suggested that NOS2 has a distributive role in the chaperone function. Like the other NOS isoforms, NOS2 is capable of S-nitros(yl)ation of appropriate proteins, although by biotin switch, constitutive S-nitrosyl-Hsp60 and -Hsp70 was detected that was quantitatively indistinguishable from Wt controls in NOS2-/- mice. After E. coli however, mitochondrial Hsp60 S-nitros(yl)ation increased only in Wt mice. This finding may reflect a basal compensatory mechanism in NOS2-/- mice that does not respond to activation of the LPS receptor, as in Wt mice. The fall-off in Hsp60-Tfam binding in stressed mitochondria in NOS2-/- mice may simply indicate that Tfam protein is lost by degradation or mitophagy at a faster rate than it can be imported.
Although both Hsp60 and Hsp70 showed greater S-nitros(yl)ation and association with mitochondrial Tfam in Wt than in NOS2-/- mice after E. coli, we focused on Hsp60 because the protein in intact mitochondria could be S-nitros(yl)ated at low micromolar concentrations of CSNO. Hsp60 contains three cysteines (UniProt Knowledgebase; Swiss-Prot/TrEMBL), but only 237Cys is found in a KCE motif amenable to SNO formation [13]. MtHsp70 has four cysteines, one in a SNO motif at 317Cys, while all four cysteines in Tfam are in the leader sequence, and there are no clear SNO motifs. Polγ (A) contains 20 cysteines, but no obvious SNO motifs. Therefore, we performed site-directed mutagenesis for Hsp60 237Cys, where this residue, some distance from the 27 amino acid mitochondrial targeting sequence, was shown to facilitate Hsp60 localization to mitochondria following NOS2 induction by LPS plus TNFα. This finding does not preclude SNO formation at other cysteines or provide a definitive site of molecular translocation, but it does demonstrate an unequivocal chaperone function for Hsp60 for mitochondrial Tfam and Polγ as well as a requirement for the Hsp60 SNO-protein. This biochemistry appears operate in vivo as demonstrated by failure of Hsp60 to internally co-localize with mitochondrial GFP in reporter mice treated with the 1400W NOS2 inhibitor before E. coli challenge.
In conclusion, NOS2 factors into at least two aspects of mitochondrial biogenesis during the host response to E. coli infection. The first is the facilitation and appropriate timing of nuclear gene and protein expression for the NRF-1 transcription factor and the PGC1-α co-activator, which among other genes regulate the nuclear-encoded expression of mitochondrial transcriptome proteins. Although the impact of NO regulation of mitochondrial biogenesis on organ function is not yet resolved, NOS2 clearly enhances S-nitros(yl)ation of Hsp60, which improves Hsp60 mitochondrial targeting and Tfam protein binding. Conversely, failure to S-nitros(yl)ate Hsp60 interferes with intra-mitochondrial trafficking of the critical mtDNA binding proteins required for mtDNA maintenance. Therefore, we surmise that NOS2 induction supports mitochondrial function by ensuring mtDNA transcription and replication, which maintains and expands cellular respiratory capacity. Moreover, by implication, interference with NOS2 after innate immune activation would limit the cell's capacity for oxidative phosphorylation and predispose to apoptosis and organ dysfunction during progressive inflammation.
Supplementary Material
Reporter mice that constitutively express mitochondrial GFP show increases in Hsp60 localization to mitochondria at 1 day in the E. coli model (A&B, yellow color). However, NOS2 inhibition with 1400W before E. coli exposure led to heterogeneously distributed Hsp60 as well as failure of the protein to co-localize sharply within the internalized mitochondrial GFP (C&D, orange color—two sets of samples from different regions of the liver are shown in D).
Acknowledgments
The authors thank Craig Marshall, Susan Fields, and Marta Salinas for excellent technical assistance. This work was supported by R01 AI0664789 (CAP).
Abbreviations
- sGC
soluble guanylate cyclase
- Hsp
heat shock protein
- mtDNA
mitochondrial DNA
- ND1
NADH dehydrogenase subunit 1
- NOS
nitric oxide synthase
- NRF
nuclear respiratory factor
- PGC-1α
PPARγ coactivator 1α
- Polγ
DNA polymerase-γ
- SNO
S-nitros(yl)ation/nitrosation
- SOD2
managanese superoxide dismutase
- Tfam
mitochondrial transcription factor-A
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Protti A, Singer M. Bench-to-bedside review: potential strategies to protect or reverse mitochondrial dysfunction in sepsis-induced organ failure. Crit Care. 2006;10:228. doi: 10.1186/cc5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zamora R, Vodovotz Y, Billiar TR. Inducible nitric oxide synthase and inflammatory diseases. Mol Med. 2000;6:347–373. [PMC free article] [PubMed] [Google Scholar]
- 4.Levy RM, Prince JM, Billiar TR. Nitric oxide: a clinical primer. Crit Care Med. 2005;33:S492. doi: 10.1097/01.ccm.0000191726.33105.f2. [DOI] [PubMed] [Google Scholar]
- 5.Liew FY, Wei XQ, Proudfoot L. Cytokines and nitric oxide as effector molecules against parasitic infections. Philos Trans R Soc Lond B Biol Sci. 1997;352:1311–1315. doi: 10.1098/rstb.1997.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stuehr DJ, Santolini J, Wang ZQ, Wei CC, Adak S. Update on mechanism and catalytic regulation in the NO synthases. J Biol Chem. 2004;279:36167–36170. doi: 10.1074/jbc.R400017200. [DOI] [PubMed] [Google Scholar]
- 7.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 8.Nathan C. Inducible nitric oxide synthase: what difference does it make? J Clin Invest. 1997;100:2417–2423. doi: 10.1172/JCI119782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gunnett CA, Chu Y, Heistad DD, Loihl A, Faraci FM. Vascular effects of LPS in mice deficient in expression of the gene for inducible nitric oxide synthase. Am J Physiol. 1998;275:H416–421. doi: 10.1152/ajpheart.1998.275.2.H416. [DOI] [PubMed] [Google Scholar]
- 10.Laubach VE, Shesely EG, Smithies O, Sherman PA. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc Natl Acad Sci U S A. 1995;92:10688–10692. doi: 10.1073/pnas.92.23.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwentker A, Billiar TR. Nitric oxide and wound repair. Surg Clin North Am. 2003;83:521–530. doi: 10.1016/S0039-6109(02)00207-4. [DOI] [PubMed] [Google Scholar]
- 12.Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto A, Comatas KE, Liu L, Stamler JS. Screening for nitric oxide-dependent protein-protein interactions. Science. 2003;301:657–661. doi: 10.1126/science.1079319. [DOI] [PubMed] [Google Scholar]
- 14.Kone BC, Kuncewicz T, Zhang W, Yu ZY. Protein interactions with nitric oxide synthases: controlling the right time, the right place, and the right amount of nitric oxide. Am. J Physiol Renal Physiol. 2003;285:F178. doi: 10.1152/ajprenal.00048.2003. [DOI] [PubMed] [Google Scholar]
- 15.Brown GC. Nitric oxide and mitochondria. Front Biosci. 2007;12:1024–1033. doi: 10.2741/2122. [DOI] [PubMed] [Google Scholar]
- 16.Erusalimsky JD, Moncada S. Nitric oxide and mitochondrial signaling: from physiology to pathophysiology. Arterioscler Thromb Vasc Biol. 2007;27:2524–2531. doi: 10.1161/ATVBAHA.107.151167. [DOI] [PubMed] [Google Scholar]
- 17.Borutaite V, Moncada S, Brown GC. Nitric oxide from inducible nitric oxide synthase sensitizes the inflamed aorta to hypoxic damage via respiratory inhibition. Shock. 2005;23:319–323. doi: 10.1097/01.shk.0000156672.36439.2d. [DOI] [PubMed] [Google Scholar]
- 18.Carreras MC, Poderoso JJ. Mitochondrial nitric oxide in the signaling of cell integrated responses. Am J Physiol Cell Physiol. 2007;292:C1569. doi: 10.1152/ajpcell.00248.2006. [DOI] [PubMed] [Google Scholar]
- 19.Dahm CC, Moore K, Murphy MP. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: implications for the interaction of nitric oxide with mitochondria. J Biol Chem. 2006;281:10056–10065. doi: 10.1074/jbc.M512203200. [DOI] [PubMed] [Google Scholar]
- 20.Kim YM, Bombeck CA, Billiar TR. Nitric oxide as a bifunctional regulator of apoptosis. Circ Res. 1999;84:253–256. doi: 10.1161/01.res.84.3.253. [DOI] [PubMed] [Google Scholar]
- 21.Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 22.Chung HT, Pae HO, Choi BM, Billiar TR, Kim YM. Nitric oxide as a bioregulator of apoptosis. Biochem Biophys Res Commun. 2001;282:1075–1079. doi: 10.1006/bbrc.2001.4670. [DOI] [PubMed] [Google Scholar]
- 23.Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, Pisconti A, Brunelli S, Cardile A, Francolini M, Cantoni O, Carruba MO, Moncada S, Clementi E. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci U S A. 2004;101:16507–16512. doi: 10.1073/pnas.0405432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 25.Nisoli E, Carruba MO. Nitric oxide and mitochondrial biogenesis. J Cell Sci. 2006;119:2855–2862. doi: 10.1242/jcs.03062. [DOI] [PubMed] [Google Scholar]
- 26.Crouser ED, Julian MW, Huff JE, Struck J, Cook CH. Carbamoyl phosphate synthase-1: a marker of mitochondrial damage and depletion in the liver during sepsis. Crit Care Med. 2006;34:2439–2446. doi: 10.1097/01.CCM.0000230240.02216.21. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe E, Muenzer JT, Hawkins WG, Davis CG, Dixon DJ, McDunn JE, Brackett DJ, Lerner MR, Swanson PE, Hotchkiss RS. Sepsis induces extensive autophagic vacuolization in hepatocytes: a clinical and laboratory-based study. Lab Invest. 2009;89:549–561. doi: 10.1038/labinvest.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarady JK, Zuckerbraun BS, Bilban M, Wagner O, Usheva A, Liu F, Ifedigbo E, Zamora R, Choi AM, Otterbein LE. Carbon monoxide protection against endotoxic shock involves reciprocal effects on iNOS in the lung and liver. Faseb J. 2004;18:854–856. doi: 10.1096/fj.03-0643fje. [DOI] [PubMed] [Google Scholar]
- 29.Suliman HB, Welty-Wolf KE, Carraway MS, Schwartz DA, Hollingsworth JW, Piantadosi CA. Toll-like receptor 4 mediates mitochondrial DNA damage and biogenic responses after heat-inactivated E. coli. Faseb J. 2005;19:1531–1533. doi: 10.1096/fj.04-3500fje. [DOI] [PubMed] [Google Scholar]
- 30.Haden DW, Suliman HB, Carraway MS, Welty-Wolf KE, Ali AS, Shitara H, Yonekawa H, Piantadosi CA. Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. Am J Respir Crit Care Med. 2007;176:768–777. doi: 10.1164/rccm.200701-161OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suliman HB, Carraway MS, Piantadosi CA. Postlipopolysaccharide oxidative damage of mitochondrial DNA. Am J Respir Crit Care Med. 2003;167:570–579. doi: 10.1164/rccm.200206-518OC. [DOI] [PubMed] [Google Scholar]
- 32.Suliman HB, Welty-Wolf KE, Carraway M, Tatro L, Piantadosi CA. Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc Res. 2004;64:279–288. doi: 10.1016/j.cardiores.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 33.Kantrow SP, Taylor DE, Carraway MS, Piantadosi CA. Oxidative metabolism in rat hepatocytes and mitochondria during sepsis. Arch Biochem Biophys. 1997;345:278–288. doi: 10.1006/abbi.1997.0264. [DOI] [PubMed] [Google Scholar]
- 34.Greenawalt JW. The isolation of outer and inner mitochondrial membranes. Methods Enzymol. 1974;31:310–323. doi: 10.1016/0076-6879(74)31033-6. [DOI] [PubMed] [Google Scholar]
- 35.Suliman HB, Carraway MS, Welty-Wolf KE, Whorton AR, Piantadosi CA. Lipopolysaccharide stimulates mitochondrial biogenesis via activation of nuclear respiratory factor-1. J Biol Chem. 2003;278:41510–41518. doi: 10.1074/jbc.M304719200. [DOI] [PubMed] [Google Scholar]
- 36.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 37.Burwell LS, Nadtochiy SM, Tompkins AJ, Young S, Brookes PS. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem J. 2006;394:627–634. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Forrester MT, Foster MW, Stamler JS. Assessment and application of the biotin switch technique for examining protein S-nitrosylation under conditions of pharmacologically induced oxidative stress. J Biol Chem. 2007;282:13977–13983. doi: 10.1074/jbc.M609684200. [DOI] [PubMed] [Google Scholar]
- 39.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 40.Frost RA, Nystrom GJ, Lang CH. Lipopolysaccharide stimulates nitric oxide synthase-2 expression in murine skeletal muscle and C(2)C(12) myoblasts via Toll-like receptor-4 and c-Jun NH(2)-terminal kinase pathways. Am J Physiol Cell Physiol. 2004;287:C1605. doi: 10.1152/ajpcell.00010.2004. [DOI] [PubMed] [Google Scholar]
- 41.Reynolds CM, Suliman HB, Hollingsworth JW, Welty-Wolf KE, Carraway MS, Piantadosi CA. Nitric oxide synthase-2 induction optimizes cardiac mitochondrial biogenesis after endotoxemia. Free Radic Biol Med. 2008 doi: 10.1016/j.freeradbiomed.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foster MW, Hess DT, Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Connor KM, Subbaram S, Regan KJ, Nelson KK, Mazurkiewicz JE, Bartholomew PJ, Aplin AE, Tai YT, Aguirre-Ghiso J, Flores SC, Melendez JA. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J Biol Chem. 2005;280:16916–16924. doi: 10.1074/jbc.M410690200. [DOI] [PubMed] [Google Scholar]
- 45.Suliman HB, Carraway MS, Tatro LG, Piantadosi CA. A new activating role for CO in cardiac mitochondrial biogenesis. J Cell Sci. 2007;120:299–308. doi: 10.1242/jcs.03318. [DOI] [PubMed] [Google Scholar]
- 46.Krimmer T, Rapaport D, Ryan MT, Meisinger C, Kassenbrock CK, Blachly-Dyson E, Forte M, Douglas MG, Neupert W, Nargang FE, Pfanner N. Biogenesis of porin of the outer mitochondrial membrane involves an import pathway via receptors and the general import pore of the TOM complex. J Cell Biol. 2001;152:289–300. doi: 10.1083/jcb.152.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wiedemann N, Pfanner N, Chacinska A. Chaperoning through the mitochondrial intermembrane space. Mol Cell. 2006;21:145–148. doi: 10.1016/j.molcel.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 48.Deocaris CC, Kaul SC, Wadhwa R. On the brotherhood of the mitochondrial chaperones mortalin and heat shock protein 60. Cell Stress Chaperones. 2006;11:116–128. doi: 10.1379/CSC-144R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chandra D, Choy G, Tang DG. Cytosolic accumulation of HSP60 during apoptosis with or without apparent mitochondrial release: evidence that its pro-apoptotic or pro-survival functions involve differential interactions with caspase-3. J Biol Chem. 2007;282:31289–31301. doi: 10.1074/jbc.M702777200. [DOI] [PubMed] [Google Scholar]
- 50.Oess S, Icking A, Fulton D, Govers R, Muller-Esterl W. Subcellular targeting and trafficking of nitric oxide synthases. Biochem J. 2006;396:401–409. doi: 10.1042/BJ20060321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elfering SL, Sarkela TM, Giulivi C. Biochemistry of mitochondrial nitric-oxide synthase. J Biol Chem. 2002;277:38079–38086. doi: 10.1074/jbc.M205256200. [DOI] [PubMed] [Google Scholar]
- 52.Lores-Arnaiz S, D'Amico G, Czerniczyniec A, Bustamante J, Boveris A. Brain mitochondrial nitric oxide synthase: in vitro and in vivo inhibition by chlorpromazine. Arch Biochem Biophys. 2004;430:170–177. doi: 10.1016/j.abb.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 53.Venkatakrishnan P, Nakayasu ES, Almeida IC, Miller RT. Absence of nitric-oxide synthase in sequentially purified rat liver mitochondria. J Biol Chem. 2009;284:19843–19855. doi: 10.1074/jbc.M109.003301. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Reporter mice that constitutively express mitochondrial GFP show increases in Hsp60 localization to mitochondria at 1 day in the E. coli model (A&B, yellow color). However, NOS2 inhibition with 1400W before E. coli exposure led to heterogeneously distributed Hsp60 as well as failure of the protein to co-localize sharply within the internalized mitochondrial GFP (C&D, orange color—two sets of samples from different regions of the liver are shown in D).