

Abstract
The inhibitory leukocyte Ig-like receptor 1 (LIR-1, also known as ILT2, CD85j, or LILRB1) was identified by its high affinity for the human CMV (HCMV) MHC class I homolog gpUL18. The role of this LIR-1-gpUL18 interaction in modulating NK recognition during HCMV infection has previously not been clearly defined. In this study, LIR-1+ NKL cell-mediated cytotoxicity was shown to be inhibited by transduction of targets with a replication-deficient adenovirus vector encoding UL18 (RAd-UL18). Fibroblasts infected with an HCMV UL18 mutant (ΔUL18) also exhibited enhanced susceptibility to NKL killing relative to cells infected with the parental virus. In additional cytolysis assays, UL18-mediated protection was also evident in the context of adenovirus vector transduction and HCMV infection of autologous fibroblast targets using IFN-α-activated NK bulk cultures derived from a donor with a high frequency of LIR-1+ NK cells. A single LIR-1high NK clone derived from this donor was inhibited by UL18, while 3 of 24 clones were activated. CD107 mobilization assays revealed that LIR-1+ NK cells were consistently inhibited by UL18 in all tested donors, but this effect was often masked in the global response by UL18-mediated activation of a subset of LIR-1− NK cells. Although Ab-blocking experiments support UL18 inhibition being induced by a direct interaction with LIR-1, the UL18-mediated activation is LIR-1 independent.
Human CMV (HCMV)5 is a ubiquitous human herpesvirus associated with subclinical primary infections followed by lifelong asymptomatic carriage, during which the innate and adaptive immune responses act in concert to control the virus. Nevertheless, HCMV remains an important pathogen, particularly in individuals with immature (fetus, newborns) or severely compromised (most notably AIDS patients or transplant recipients) immune systems. The NK cell response is crucial in controlling CMV infections. In humans, their role is best demonstrated by the fact that individuals with defects affecting NK cell function are particularly susceptible to HCMV disease (1, 2). In the murine model, impairment of the NK cell function in the host, or associated defense mechanisms in the virus, can have a dramatic effect on murine CMV (MCMV) virulence (3-5).
NK cell function is regulated by integration of signals received from activating and inhibitory receptors on their surface. Although individual NK clones express different combinations of receptors, they are normally maintained in a resting state in vivo by inhibitory signals received mainly from autologous MHC class I molecules. During HCMV infection, MHC class I molecules are efficiently down-regulated from the cell surface by the products of four HCMV-encoded genes (US2, US3, US6, and US11) (reviewed in Ref. 6). In parallel, the synthesis of certain stress-inducible NK cell-activating ligands is increased, including MHC class I-related chains (MIC) A and B, human UL16-binding protein (ULBP) 1, ULBP2, ULBP3, and retinoic acid early transcript (RAET) 1G (7-9). MICA, MICB, ULBP1, ULBP2, ULBP3, ULBP4/RAET1E, and RAET1G proteins are ligands for the NK-activating receptor NKG2D (10-12). Down-regulation of endogenous MHC class I combined with the up-regulation of NK-activating ligands would be expected to promote a NK cell attack. However, HCMV encodes an impressive array of genes that act in concert to suppress NK cell recognition. The UL16 gene product sequesters MICB, ULBP1, and ULBP2 in the endoplasmic reticulum (8, 11). A peptide derived from the leader sequence of HCMV gpUL40 stimulates cell surface expression of the nonclassical MHC class I molecule HLA-E, which binds the NK cell inhibitory receptor CD94/NKG2A (13, 14). The virion tegument protein pp65 (ppUL83) inhibits NK cell cytotoxicity by dissociating the linked CD3ζ chain from the NK-activating receptor NKp30 (15). Infection with HCMV clinical isolates induces much more robust protection against NK cell-mediated cytolysis than observed with the laboratory strain AD169 (14, 16, 17). During extensive passage in vitro, strain AD169 suffered a spontaneous 15-kb deletion in one end of the long unique (UL) region commonly referred to as the UL/b’ sequence. This region is predicted to encode 23 open reading frames (ORFs) designated as UL128–UL150 (18). Consequently, both UL141 and UL142 are missing from strain AD169. UL141 encodes an endoplasmic reticulum-resident glycoprotein that acts to prevent surface expression of CD155 (also called poliovirus receptor), a ligand for NK cell-activating receptors CD226 (DNAM-1) and CD96 (TACTILE) (17, 19, 20). Finally, UL142 helps to protect HCMV-infected fibroblasts from NK cell attack by decreasing surface expression of most MICA alleles (21, 22).
The UL18 ORF was first identified as a MHC class I homolog during analysis of the strain AD169 genome (23) and was shown to encode for a 67-kDa endoglycosidase H-sensitive glycoprotein capable of forming a trimeric complex with β2-microglobulin and endogenous peptides (24, 25). A highly glycosylated 160-kDa endoglycosidase H-resistant species is also expressed in HCMV-infected cells that corresponds to the fully processed, mature form of the UL18 glycoprotein (gpUL18) (26). gpUL18 is expressed in increasing abundance throughout the late phase of infection, when endogenous MHC class I is down-regulated (26, 27). MCMV also encodes a MHC class I homolog, m144, along with 11 additional predicted proteins containing class I-like folds (28, 29). Although the ligand for m144 remains unknown, it is a recognized NK cell evasion protein and deletion of the m144 gene results in impaired virulence in vivo (4). Although a ligand has been defined for gpUL18, its function has been less clear with data supporting UL18-mediated NK cell inhibition (30, 31), UL18-independent NK cell recognition (32), and UL18-mediated NK cell activation (33).
The leukocyte Ig-like receptor 1 (LIR-1, also known as CD85j, ILT2, or LILRB1) recognizes a broad range of MHC class I molecules (HLA-A, -B, -C, -E, -F, -G) (34-39) and is also able to interact with gpUL18 (34, 39). However, LIR-1 has a >1000-fold higher affinity for gpUL18 than for MHC class I molecules, thus even low levels of gpUL18 could be expected to compete efficiently for binding (35). LIR-1 contains four ITIM in its cytoplasmic domain and is expressed on B lymphocytes, monocytes, dendritic cells, and subsets of T and NK cells (34, 39). The frequency of peripheral blood T cells and NK cells that express LIR-1 is variable among individuals (36, 39). Interestingly, the frequency of T cells and NK cells expressing LIR-1 increases in lung transplant recipients with HCMV disease (40). Furthermore, during persistent HCMV infection, LIR-1 is preferentially expressed on HCMV-specific CD8+ T cells with a differentiated effector memory phenotype, compared with expression on T cells specific for others viruses (41).
As part of a systematic investigation into HCMV NK evasion (13, 14, 17), we sought to gain insight into the potential contribution played by UL18. Functional studies have been constrained by gpUL18 expression being incompatible with the generation of stable cell lines. To overcome this obstacle, UL18 was cloned into a replication-deficient adenovirus (RAd) vector, and expression from the resultant recombinant virus was characterized (26). RAd vectors have proved invaluable in studies of HCMV gene function because they provide for efficient gene delivery (up to 100%) in permissive primary human fibroblasts with minimal perturbation of the target cell. Furthermore, in studies of NK cell modulator functions, this technology enables assays to be performed in an autologous setting. We thereby set out to examine the consequences of gpUL18 expression by target cells on NK cell function. We observed that UL18 was capable of inducing protection against NK recognition both in the context of HCMV infection and when expressed independently from the RAd vector. Importantly, UL18 exclusively inhibited NK cells expressing the LIR-1 receptor, while simultaneously activating a NK subset independent of LIR-1.
Materials and Methods
Cells
Human fetal foreskin fibroblasts (HFFF) and primary skin fibroblasts from volunteer donors were cultured in DMEM supplemented with 10% FCS (Invitrogen Life Technologies). Skin fibroblast cultures from biopsies were immortalized with human telomerase reverse transcriptase (42). No difference was observed when comparing NK cell recognition of nonimmortalized and immortalized skin fibroblasts (data not shown). Human fibroblasts were preferred targets in NK cell assays because they support productive HCMV infection. The established NK cell clone NKL (43) was grown in RPMI 1640 supplemented with 5% human AB serum and 200 IU/ml IL-2 (Proleukin; Chiron). PBMC were prepared from fresh blood from HCMV-seropositive donors or <24-h-old buffy coats (Welsh Blood Service, HCMV serostatus not determined) by Histopaque-1077 (Sigma-Aldrich) density gradient centrifugation. They were incubated for 16 h in SCGM medium (Cellgro) supplemented with 5% human AB serum and 103 IU/ml IFN-α (Roferon-A; Roche) and were washed in PBS before being used in any functional assay. For NK cell cytolysis assays, T cells were depleted from PBMC with anti-CD3-FITC mAb (Serotec) and Dynabeads (Dynal). NK cell cloning was accomplished by single-cell sorting with clones stimulated weekly with irradiated allogeneic PBMC, OKT3 mAb to CD3 (American Type Culture Collection) and IL-2 (103 IU/ml) (44). NK cell clones were maintained in SCGM medium supplemented with 5% human AB serum and were rested from feeder stimulation for at least 1 wk before use in assays. This project was approved by the Bro Taf Local Research Ethics Committee. Prior consent was obtained from all blood and skin biopsy donors.
Viruses
Fibroblasts were infected for 96 h with HCMV (10 PFU/cell) strain AD169 or the previously described AD169 UL18 insertional mutant (ΔUL18; provided by H. Browne, Cambridge University) (45). The UL18 ORF was amplified by PCR from the AD169 genome and inserted into a replication-deficient human adenovirus 5 vector (RAd-UL18) under the control of the HCMV major IE promoter as described previously (26). RAd-UL141 and RAd-UL142 viruses were generated by PCR from the HCMV Toledo strain (17, 21). HFFF (100 PFU/cell) and primary skin fibroblasts (500 PFU/cell) were infected 72 h before NK cell assays with RAd-UL18, RAd-UL141, RAd-UL142, or the empty RAd control (Ctrl) vector (RAd-Ctrl). Different multiplicities of infection were used to compensate for primary skin fibroblasts being less susceptible to RAd vector infection than HFFF.
NK cell cytolysis assays
NK cytotoxicity was measured in standard 4-h 51Cr release assays against mock-infected, HCMV-infected, or RAd-infected fibroblasts. The E:T ratio was adjusted to the number of NK cells present in the CD3-depleted IFN-α-activated PBMC used as effectors. The percentage of specific lysis was calculated as [(experimental mean release − spontaneous mean release)/(maximum mean release − spontaneous mean release)] × 100. Final means and SEMs were determined from triplicate or quadruplicate samples. For receptor-blocking studies, a saturating amount (10 μg/ml) of anti-LIR-1 mAb (clone VMP55; Santa Cruz Biotechnology) or control anti-CD56 mAb (clone B159; BD Biosciences) was added to NKL for 15 min at 37°C before contact with targets and throughout the assays.
NK cell CD107a mobilization assays and flow cytometry
IFN-α-activated PBMC were incubated in duplicate with target cells and anti-CD107a-FITC mAb (4 μl for 106 PBMC; BD Biosciences) for 6 h in the presence of 4 μl/ml BD GolgiStop (BD Biosciences) for the last 5 h. For receptor-blocking studies, a saturating amount (20 μg/ml) of anti-LIR-1 F(ab’)2 (clone HP-F1) (39) or control anti-CD56 mAb was added to effector cells for 15 min at 37°C before contact with targets and throughout the assays. PBMC were then harvested, washed in cold PBS, and stained for 30 min at 4°C with anti-CD3-PerCP (clone SK7; BD Biosciences), anti-LIR-1-PE (clone GHI/75; BD Biosciences, or clone VMP55; Santa Cruz Biotechnology for LIR-1-blocking studies), and anti-CD56-allophycocyanin (clone N901; Beckman Coulter) mAbs. Cells were washed twice in cold PBS and a minimum of 5 × 103 CD3−CD56+ NK cells were acquired from each sample. Surface gpUL18 expression was monitored on infected fibroblasts using anti-gpUL18 (clone 10C7/CRL-2430; American Type Culture Collection) and anti-mouse Ig-PE (BD Biosciences) mAbs. Infected fibroblasts were incubated for 20 min before gpUL18 staining in cold PBS containing 10% human AB serum and mouse IgG1 (10 μg/ml) to block the Ig FcRs encoded by HCMV (46, 47). LIR-1 expression on NK cell surface was analyzed with anti-LIR-1 (clone M405, provided by Amgen) and anti-mouse IgG1-PE (clone A85-1; BD Biosciences) mAbs. A FACSCalibur with CellQuest PRO software (BD Biosciences) was used for flow cytometry and the Cardiff University Central Biotechnology Service with a DakoCytomation MoFlo Sorter provided cell sorting.
Statistical analysis
Probabilities were calculated with two-way ANOVA analysis assuming not-repeated measures followed by Bonferroni post tests. Results with p < 0.05 were considered as significant.
Results
UL18 elicits protection against NKL-mediated cytolysis
Because LIR-1 is the only receptor known to interact with gpUL18 (34), it was natural to focus on this high-affinity interaction as a potential mechanism by which UL18 may exert a direct effect on NK cell function. The transformed NK cell clone NKL was considered to be a suitable effector cell in initial studies because it is associated with high levels of LIR-1 surface expression (Fig. 1a). To directly address the function of UL18 in the context of lytic HCMV infection, HFFF were infected in parallel with strain AD169 and a ΔUL18 mutant generated on the same virus background. In allogeneic cytolysis assays, strain AD169-infected targets were reproducibly less susceptible to NKL cell-mediated cytolysis than either mock-infected HFFF ( p = 0.0116) or cells infected with the ΔUL18 virus ( p = 0.0005) (Fig. 1b). The ablation of gpUL18 expression in cells infected with the mutant virus rendered them more vulnerable to NKL attack, a result consistent with UL18 acting as an inhibitor of NK cell recognition. To function as a MHC class I decoy inhibitory ligand, gpUL18 needs to be expressed on the surface of HCMV-infected cells. HFFF infected in parallel with strain AD169 and the ΔUL18 mutant were analyzed for gpUL18 surface expression by flow cytometry (96 h postinfection) after incubating cells in human AB serum and mouse IgG1 mAb to block interactions with FcRs expressed in HCMV-infected cells. A small, yet reproducible increase in staining was detected with the gpUL18-specific mAb 10C7 following infection with strain AD169 (median fluorescence intensity (MFI), 13.2) relative to ΔUL18-infected HFFF (MFI, 8.7) or following staining with a control IgG1 isotype (MFI, 9.0) (Fig. 1c). Compatible with its observed function in inhibiting NKL cytolysis, gpUL18 was detectable on the surface of HCMV-infected cells and consequently available to interact with receptors on immune cells.
FIGURE 1.
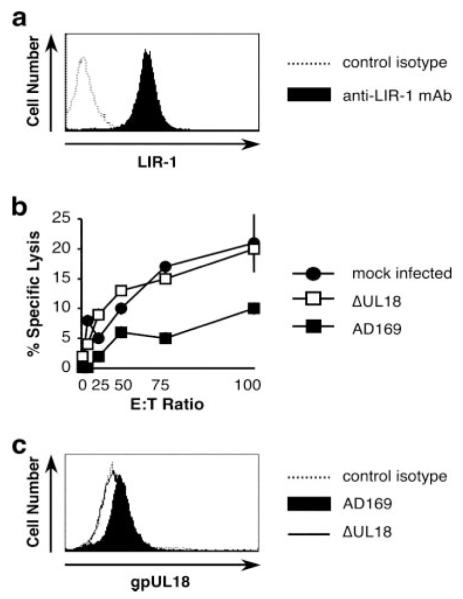
gpUL18 expression inhibits NKL cytotoxicity. HFFF were mock infected or were infected for 96 h with HCMV strains AD169 or AD169 UL18 mutant (ΔUL18) (10 PFU/cell). a, LIR-1 surface expression on NKL was analyzed by flow cytometry with anti-LIR-1 and control isotype mAbs. b, NKL cells were used as effectors in allogeneic chromium release assays. Results are means ± SEM of triplicate samples and are representative of three independent experiments (two-way ANOVA analysis: ΔUL18 vs AD169, p = 0.0005). c, gpUL18 surface expression on HFFF was detected by flow cytometry with anti-gpUL18 and control isotype mAbs.
During lytic infection, HCMV-infected cells express multiple proteins capable of both promoting and suppressing NK cell recognition. To investigate the effects of UL18 in isolation, a RAd vector was exploited to deliver and express the strain AD169 UL18 gene in HFFF targets. Potential vector effects were controlled by comparing the RAd-UL18 to the RAd vector without transgene insert (RAd-Ctrl). Surface gpUL18 expression on RAd-UL18-infected fibroblasts was analyzed by flow cytometry at 72 h postinfection. The fluorescence observed with RAd-UL18-infected cells using the anti-gpUL18 mAb 10C7 (MFI, 13.8) was much higher than for cells infected with RAd-Ctrl (MFI, 3.1) or when a control isotype IgG1 was used to stain RAd-UL18-infected cells (MFI, 3.7) (Fig. 2a). RAd-UL18- and RAd-Ctrl-infected HFFF were then compared as targets in a NK cell cytolysis assay. When expressed in isolation using the RAd vector, UL18 elicited clear protection from NKL killing ( p = 0.0014), with the specific cytotoxicity dropping, respectively, from 44 ± 1% to 20 ± 2% at an E:T ratio of 100:1 ( p < 0.001; Fig. 2b). When the same chromium release assay was repeated with saturating amounts of mAbs (10 μg/ml), the NKL killing of RAd-UL18-infected targets was entirely abrogated in the presence of the blocking anti-LIR-1 mAb compared with the control anti-CD56 mAb ( p < 0.0001; Fig. 2c). This experiment is consistent with the UL18-mediated NKL inhibition being due to the gpUL18-LIR-1 interaction.
FIGURE 2.

gpUL18 expression inhibits NKL cytotoxicity. HFFF were infected for 72 h with a RAd vector containing the complete AD169 UL18 ORF (RAd-UL18) or with the empty control adenovirus (RAd-Ctrl) (100 PFU/cell). a, gpUL18 surface expression on HFFF was detected by flow cytometry with anti-gpUL18 and control isotype mAbs. b, NKL cells were used as effectors in allogeneic chromium release assays (two-way ANOVA analysis: RAd-UL18 vs RAd-Ctrl, p = 0.0014). c, A saturating amount (10 μg/ml) of anti-LIR-1 or control anti-CD56 mAbs was added on NKL for 15 min at 37°C before contact with targets and throughout the allogeneic chromium release assays (two-way ANOVA analysis: RAd-UL18 + anti-CD56 vs RAd-UL18 + anti-LIR-1, p < 0.0001). Results are means ± SEM of triplicate samples and are representative of three independent experiments.
UL18-mediated protection against polyclonal NK cells
Having established UL18 was capable of exerting an inhibitory effect on NKL, it was important to investigate its function using a more natural effector cell population. Certain NK cell receptors are selectively sensitive to allogeneic MHC class I, and thus an autologous assay system was set up using polyclonal NK cells and skin fibroblasts derived from volunteer donors. We analyzed the broad NK cell response from three HCMV-seropositive healthy individuals using PBMC that were depleted of T cells and activated over-night with IFN-α. The proportion of NK cells expressing the LIR-1 receptor 1) on fresh PBMC and 2) after T cell depletion and over-night incubation with IFN-α were similar, indicating that the treatment was not exerting an obvious bias (Table I). However, differences were observed in the proportion of LIR-1+ NK cells present in the donors, specifically: LIR-1+ NK cells after T cell depletion were on the order of 2-fold more frequent in donor 3 (64.8%) than in the two other individuals (25.3 and 33.2%, respectively, for donors 1 and 2; Table I).
Table I.
LIR-1 expression on polyclonal NK cells from three individuals
| Fresh PBMC |
PBMC after T Cell Depletiona |
|||
|---|---|---|---|---|
| Donor | % NKb | % LIR-1+ NKc | % NKb | % LIR-1+ NKc |
| 1 | 22.5 | 28.0 | 67.0 | 25.3 |
| 2 | 7.4 | 35.1 | 19.3 | 33.2 |
| 3 | 38.2 | 66.0 | 70.1 | 64.8 |
PBMC were depleted of CD3+ T cells and incubated for 16 h with IFN-α.
Percentages of CD3−CD56+ NK cells among lymphocytes based on side scatter/forward scatter parameters.
Percentages of LIR-1+ cells among CD3−CD56+ NK cells.
The effect of UL18 on cytolysis was examined using IFN-α-activated polyclonal NK cells from the three donors. Using targets infected with either strain AD169 or the ΔUL18 mutant, no clear difference in cytolysis was apparent for donors 1 and 2 (Fig. 3a). However, for donor 3, strain AD169-infected fibroblasts were significantly more resistant to killing than ΔUL18-infected cells ( p < 0.0001; Fig. 3a). Chromium release assays were also performed in parallel using RAd-Ctrl and RAd-UL18-infected autologous skin fibroblast targets (Fig. 3b). No statistically significant difference in killing was observed using polyclonal NK cells from donors 1 and 2. However, UL18 expression from the RAd vector afforded a significant level of protection against polyclonal NK cells from donor 3 ( p < 0.0001; Fig. 3b). The results obtained comparing HCMV-infected targets with those infected with RAd vectors were compatible and both supportive of UL18 being able to promote protection against NK recognition.
FIGURE 3.
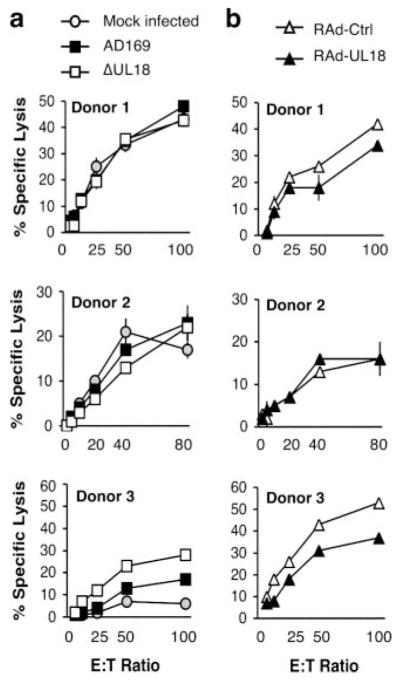
gpUL18 protection from polyclonal NK cell killing differs between donors. Autologous NK cytolysis assays were set up with T cell-depleted IFN-α-activated PBMC from three HCMV+ healthy donors and primary skin fibroblasts from the same volunteers. a, Fibroblasts were mock infected or infected for 96 h with HCMV strains AD169 and AD169 UL18 mutant (ΔUL18) (10 PFU/cell). ΔUL18 vs AD169 recognition was analyzed with a two-way ANOVA test assuming not-repeated measures for donor 1 (NS), donor 2 (NS), and donor 3 (p < 0.0001). b, Fibroblasts were infected for 72 h with a RAd vector containing the complete AD169 UL18 ORF (RAd-UL18) or with the empty control adenovirus (RAd-Ctrl) (500 PFU/cell). RAd-UL18 vs RAd-Ctrl recognition was analyzed with a two-way ANOVA test assuming not-repeated measures for donor 1 (NS), donor 2 (NS), and donor 3 (p < 0.0001). Results are means ± SEM of triplicate or quadruplicate samples and are representative of three independent experiments.
Inhibition of NK clones
NK clones can prove invaluable in analyzing virus-encoded NK cell evasion functions. NK clone activation is directly correlated with the expression of specific activating and inhibitory receptors. A panel of 24 NK cell clones was therefore selected from donor 3. The clones were deemed functional based on their capacity to kill the MHC class I-deficient cell line K562 by a specific lysis of ≥20%. Flow cytometric analysis using an anti-LIR-1 mAb (M405 clone) and the isotype control mouse IgG1 mAb showed that the clones exhibited a broad range of LIR-1 surface expression (MFI, 1–145; Table II). Remarkably, only 2 NK clones (clone B MFI, 145, and clone H MFI, 82) of the 24 selected from donor 3 PBMC expressed LIR-1 to a higher MFI than the polyclonal NK population from this individual (MFI, 75). This data implied that NK clones exhibiting elevated expression of the LIR-1 inhibitory receptor proliferate less efficiently in vitro than other NK cells.
Table II.
Effect of gpUL18, gpUL142, and gpUL141 expression on cytotoxicity of NK clones from donor 3
| Targetsb |
||||||
|---|---|---|---|---|---|---|
| NK Clone | E:T Ratio | LIR-1a | RAd-Ctrl | RAd-UL18 | RAd-UL142 | RAd-UL141 |
| A | 30:1 | 6 | 37 ±2 | 31± 4 | 23 ± 0 c | 9 ± 1 c |
| B | 50:1 | 145 | 48 ± 3 | 28 ± 2 c | 17 ± 1 c | 13 ± 2 c |
| C | 30:1 | 17 | 45 ±3 | 36± 1c | 19 ± 1 c | 11 ± 2 c |
| D | 30:1 | 54 | 35 ±3 | 35± 1 | 10 ± 1 c | 0 ± 0 c |
| E | 30:1 | 38 | 70 ±1 | 62± 3c | 73 ± 4 | 52 ± 6 c |
| F | 40:1 | 9 | 62 ±3 | 56± 3 | 49 ± 4 c | 28 ± 3 c |
| G | 30:1 | 13 | 49 ±3 | 50± 3 | 29 ± 1 c | 23 ± 2 c |
| H | 30:1 | 82 | 31 ±2 | 34± 4 | 13 ± 3 c | 12 ± 2 c |
| I | 40:1 | 11 | 15 ±3 | 14±2 | 17 ± 4 | 3 ± 1 c |
| J | 80:1 | 12 | 59 ±5 | 71± 6c | 40 ± 2 c | 26 ± 3 c |
| K | 30:1 | 7 | 41 ±4 | 38± 4 | 22 ± 1 c | 11 ± 1 c |
| L | 60:1 | 26 | 10 ±3 | 7±2 | 6±1 | 1± 1c |
| M | 90:1 | 11 | 60 ±3 | 57± 4 | 38 ± 1 c | 15 ± 3 c |
| N | 40:1 | 12 | 50 ±4 | 49± 4 | 30 ± 3 c | 14 ± 2 c |
| O | 30:1 | 1 | 24 ±1 | 18±1 | 16 ± 0c | 7 ± 1 c |
| P | 60:1 | 3 | 46 ±3 | 48±7 | 46 ± 3 | 31 ± 4 c |
| Q | 90:1 | 18 | 41 ±8 | 52± 3c | 27 ± 1 c | 13 ± 1 c |
| R | 100:1 | 14 | 49 ±5 | 58± 5c | 50 ± 5 | 38 ± 5 c |
| S | 30:1 | 3 | 26 ±3 | 22±2 | 16 ± 3c | 6 ± 1 c |
| T | 40:1 | 4 | 34 ±2 | 30±1 | 28 ± 2 | 12 ± 3 c |
| U | 30:1 | 5 | 4 ±2 | 6±1 | 4±1 | 3± 1 |
| V | 40:1 | 2 | 52 ±3 | 57±4 | 42 ± 4c | 25 ± 2 c |
| W | 30:1 | 4 | 6 ±3 | 5±1 | 4±1 | 2± 0 |
| X | 50:1 | 3 | 40 ±4 | 53± 5c | 31 ± 1c | 18 ± 1 c |
LIR-1 MFI of NK clones.
Autologous fibroblasts infected with the indicated RAd, numbers show percentages of specific lysis ± SEM. Bold numbers indicate an inhibition >10% compared to RAd-Ctrl-infected target lysis.
Significant, p < 0.05, where p values compare RAd-Ctrl and RAd-UL18-, RAd-UL142-, or RAd-UL141-infected target killing (two-way ANOVA analysis and Bonferroni post test). RAd-UL18 vs RAd-Ctrl for clone B, p < 0.001.
All NK clones were tested in cytotoxicity assays against RAd-UL18-, RAd-UL141-, RAd-UL142-, and RAd-Ctrl-infected autologous skin fibroblasts. RAd-UL141 and RAd-UL142 were used as controls for NK inhibition. UL141 was selected as the most robust NK evasion molecule in our assays: RAd-UL141 infection previously protected target cells against 66% of NK clones tested (17). UL142 was selected because it is the only other member of the HCMV UL18 gene family (48) and also a recognized NK evasion function (21, 22). In cytotoxicity assays, a difference in percentage of lysis of >10 relative to RAd-Ctrl-infected target killing was required to be scored as a positive result (Table II). Of the 24 NK clones analyzed, 21 were inhibited by UL141 and 11 inhibited by UL142 expression. Consistent with their characterized functions, neither UL141- nor UL142-mediated NK inhibitions were correlated with LIR-1 expression on NK clones. For example, clones O and V (LIR-1 MFI, 1 and 2, respectively) were inhibited by UL141 expression and clone A (LIR-1 MFI, 6) was inhibited by UL142,whereas clone E was not (LIR-1 MFI, 38). Three NK clones (L, W, and U) exhibited low levels of cytotoxicity against RAd-Ctrl-infected targets (≤10%) and, thus, inhibition of these clones could not be scored.
Although the broad NK cell population from donor 3 was clearly inhibited by gpUL18 expression (Fig. 3), only the NK clone with the highest LIR-1 MFI (clone B MFI, 145) showed a decrease in cytotoxicity against RAd-UL18-infected fibroblasts >10%. A high density of LIR-1 receptors may thus be required on the NK cell surface to counteract signals given by NK activatory receptors. Interestingly, 3 of the 24 NK clones (J, Q, and X) were activated by gpUL18 expression. These clones exhibiting low LIR-1 expression (MFI, 12, 18, and 3, respectively) killed RAd-UL18-infected targets by >10% compared to RAd-Ctrl.
LIR-1+ NK cell activation is inhibited by gpUL18 expression on target cells
Because LIR-1high NK cells were not efficiently amplified during in vitro culture, even a large panel of NK clones would not constitute a truly accurate representation of the broad NK cell response. A CD107a mobilization assay was thus performed to directly investigate the effect of UL18 expression on the function of NK cells, depending on their level of LIR-1 expression. The binding of the CD107a mAb detects degranulation of cytolytic cells and thereby allows the analysis of the activation of specific PBMC subsets directly in mixed population, without prolonged in vitro culture required to propagate clones. The CD107a mobilization assay was set up by incubating IFN-α-activated PBMC from donor 3 with RAd-UL18-, RAd-UL142-, and RAd-Ctrl-infected autologous skin fibroblasts. PBMC incubated without any target were used as a negative control, while PBMC incubated with the K562 cell line were used as a sensitive test for NK cell activation (Fig. 4a). The proportion of activated (CD107a+) LIR-1− NK cells was similar with RAd-UL18- and RAd-Ctrl-infected fibroblasts (10 and 10.7%, respectively), but 43.8% of LIR-1+ NK cells were inhibited comparing percentages of LIR-1+ NK cells recognizing RAd-UL18-(6.8%) vs RAd-Ctrl-infected targets (12.1%) (Fig. 4b). This reduction of LIR-1+ NK cell degranulation was significant ( p < 0.01) and specific for gpUL18 expression. UL142-mediated NK inhibition was independent of the LIR-1 NK cell phenotype: both LIR-1+ and LIR-1− NK cells were less activated by RAd-UL142-(12.8% for both NK subsets) than by RAd-Ctrl-infected targets (13.7% for LIR-1+ NK cells and 13.4% for LIR-1−) (Fig. 4c). Furthermore, the global NK cell inhibition of 26.3% induced by gpUL18 expression in this assay was comparable to the polyclonal NK inhibition obtained in cytolysis assays with T cell-depleted PBMC from donor 3 (Fig. 3b).
FIGURE 4.

gpUL18 expression inhibits LIR-1+ NK cell activation from donor 3. Autologous CD107a mobilization assays were set up with IFN-α-activated PBMC against primary skin fibroblasts from donor 3. Dot plots were gated on CD3−CD56+ NK cells. a, PBMC incubated without any target or with K562 cells are shown as controls. b, Fibroblasts were infected for 72 h with RAd vectors containing the UL18 ORF (RAd-UL18) or the empty control adenovirus (RAd-Ctrl) (500 PFU/cell). c, Fibroblasts were infected for 72 h with RAd vectors containing the UL142 ORF (RAd-UL142) or with the empty control adenovirus (RAd-Ctrl) (500 PFU/cell). d, The dot plot quadrants were adjusted on the LIR-1+ NK cell MFI to allow a comparison of the LIR-1high NK cell response on the upper quadrants with the LIR-1low NK cell degranulation on the lower quadrants. Results are representative of duplicate samples in three independent experiments.
Focusing on the LIR-1+ NK cells, a further analysis of CD107a expression was conducted defined by their level of LIR-1 expression; those with a LIR-1 fluorescence greater than the LIR-1+ NK cell MFI were considered as LIR-1high and cells with LIR-1 expression under the MFI as LIR-1low. In Fig. 4d, the dot plot quadrants were adjusted on the LIR-1+ NK cell MFI to allow a comparison of the LIR-1high NK cell response on the upper quadrants with the LIR-1low NK cell degranulation on the lower quadrants. In this analysis, the gpUL18-mediated NK inhibition was much more potent for LIR-1high NK cells, with the level of degranulation falling by 55% ( p < 0.01) against gpUL18-expressing targets, whereas LIR-1low NK cells were only inhibited by 28.8% ( p < 0.01) when comparing RAd-UL18- to RAd-Ctrl-infected fibroblasts. These data indicate that the efficiency of gpUL18-mediated NK inhibition was related to the level of LIR-1 receptors on the NK cell surface.
Interestingly, a difference was also observed in the proportion of LIR-1+ NK cells (CD107a+ and CD107a−) detected in assays comparing RAd-UL18- and RAd-Ctrl-infected targets. As the proportion of CD107a+LIR-1+ NK cells recognizing gpUL18-expressing targets decreased, a compensating increase in CD107a−LIR-1+ NK cells was not detected. Rather, there was an increased proportion of CD107a−LIR-1− NK cells (Fig. 4b). These data suggest that surface expression of LIR-1 may be down-regulated after interaction with UL18. This outcome would not be unique, NKG2D, for example, has been observed to be internalized by NK cells following receptor-ligand interactions (49).
UL18 protection is conferred by the LIR-1 receptor on NK cells
Because UL18-mediated inhibition of NK recognition correlated with LIR-1 expression, we tested directly whether this function was mediated by direct binding of gpUL18 to the LIR-1 inhibitory receptor. The CD107a mobilization assay was therefore repeated using IFN-α-activated PBMC from donor 3 against RAd-UL18- and RAd-Ctrl-infected autologous skin fibroblasts in the presence of saturating amounts of a blocking anti LIR-1 F(ab’) 2 or a control anti-CD56 mAb (Fig. 5). In this experiment, LIR-1+ NK cell degranulation was inhibited by 54.3% ( p < 0.001) when comparing proportions of CD107a+ cells obtained after interaction with RAd-UL18- vs RAd-Ctrl-infected targets in the presence of the anti-CD56 control mAb. When the LIR-1 interaction with target ligands was blocked by the anti-LIR-1 F(ab’) 2, LIR-1+ NK cell degranulation was only inhibited by 6.4%. This substantial reversal of the gpUL18-mediated NK inhibition ( p < 0.001) supports a role for a direct interaction between LIR-1 and gpUL18 as responsible for UL18-mediated suppression of NK cell function. A slight increase in LIR-1+ NK cell activation was observed with RAd-Ctrl targets in the presence of the anti-LIR-1 F(ab’) 2. This effect may be attributed to abrogation of the low-affinity interaction between LIR-1 and MHC class I.
FIGURE 5.
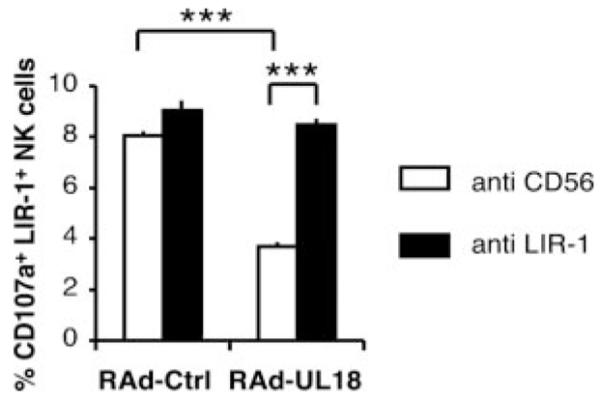
gpUL18 NK inhibition is mediated via the LIR-1 receptor. Autologous CD107a mobilization assays were set up with IFN-α-activated PBMC against primary skin fibroblasts from donor 3. Fibroblasts were infected for 72 h with RAd vectors containing the UL18 ORF (RAd-UL18) or with the empty control adenovirus (RAd-Ctrl) (500 PFU/cell). A saturating amount (20 μg/ml) of anti-LIR-1 F(ab’)2 or control anti-CD56 mAb was added on PBMC for 15 min at 37°C before contact with targets and throughout the assays (***, p < 0.001 with two-way ANOVA analysis assuming not-repeated measures followed by Bonferroni post tests). Results are means ± SEM of duplicate samples and are representative of three independent experiments.
Differential modulation of LIR-1+ and LIR-1− NK cells
There were clear differences observed in the inhibition of the polyclonal NK cells conferred by UL18 expression in the different donors (Fig. 3). Although donor 3 possessed the highest proportion of LIR-1+ NK cells (64.8%) and the polyclonal NK cells were inhibited by UL18, a substantial proportion of polyclonal NK cells from donors 1 and 2 also expressed LIR-1 (25.3 and 33.2%, respectively; Table I), although there was no clear UL18-mediated inhibition for those donor NK cells. To expand donor numbers, CD107a mobilizations assays were therefore performed in an allogeneic setting using PBMC obtained from buffy coats. IFN-α-activated PBMC for six additional donors were both assessed for LIR-1 expression (Table III) and incubated with RAd-UL18- or RAd-Ctrl-infected HFFF. The percentages of CD107a+ NK cells (CD3−CD56+) were determined for LIR-1+ and LIR-1− NK cell subsets (Fig. 6). The result of this experiment provided significant insight into how gpUL18 was impacting the polyclonal NK population (Table III). First, degranulation of LIR-1+ NK cell subpopulations from all six donors was decreased when exposed to gpUL18-expressing targets compared to RAd-Ctrl-infected HFFF. The mean level of inhibition was 39.2%, with a range from 28.7% for donor 421V to 57.7% for donor 683N. NK cells expressing the LIR-1 receptor were therefore sensitive to UL18-mediated inhibition in all donors tested. Second, remarkably, gpUL18 expression was broadly associated with an increase in CD107a+LIR-1− NK cells. The mean level of activation was 33%, with a range from 18.3% for donor 421V to 53.3% for donor 705G. This result indicated that gpUL18 expression was activating LIR-1− NK cells which is compatible with the UL18-mediated activation of three NK clones with low LIR-1 MFI (Table II). Third, the combination of LIR-1+ NK subset inhibition and LIR-1− NK subpopulation activation produced different patterns of polyclonal NK response between the donors. The broad NK cell response of four of the six individuals remained stable (inhibition or activation <10%) with RAd-UL18- and RAd-Ctrl-infected HFFF, whereas NK cell populations from donors 705G and 683N were activated by targets expressing gpUL18 (24.6 and 29.2%, respectively). Therefore, in most donors, the UL18-mediated LIR-1− NK cell activation was able to mask the LIR-1+ NK cell inhibition; a finding that provides explanation for why UL18 did not elicit overt protection in the cytotoxicity experiments performed using polyclonal NK cells from donors 1 and 2 (Fig. 3).
Table III.
NK cell response from six donors against targets expressing gpUL18
| Donor | % LIR-1+ NKa |
% LIR-1+ NK Inhibitionb |
% LIR-1− NK Activationb |
% Total NK Activationb |
|---|---|---|---|---|
| 705G | 28.6 | 34.6 ± 6 | 53.3 ± 1 | 24.6 ± 1 |
| 683N | 17.5 | 57.7 ± 9 | 52.7 ± 9 | 29.2 ± 10 |
| 420A | 55.5 | 44.5 ± 1 | 32.9 ± 12 | NSc |
| 004W | 17.1 | 40.2 ± 13 | 21.6 ±7 | NS |
| 907M | 30 | 29.6 ± 9 | 19.2 ±1 | NS |
| 421V | 55.4 | 28.7 ± 3 | 18.3 ± 10 | NS |
Percentages of LIR-1+ cells among CD3−CD56+ NK cells after PBMC incubation with IFN-α for 16 h.
Percentages of inhibition or activation comparing NK degranulation ± SEM between RAd-Ctrl- and RAd-UL18-infected targets.
NS, with NK inhibition or activation <10%.
FIGURE 6.
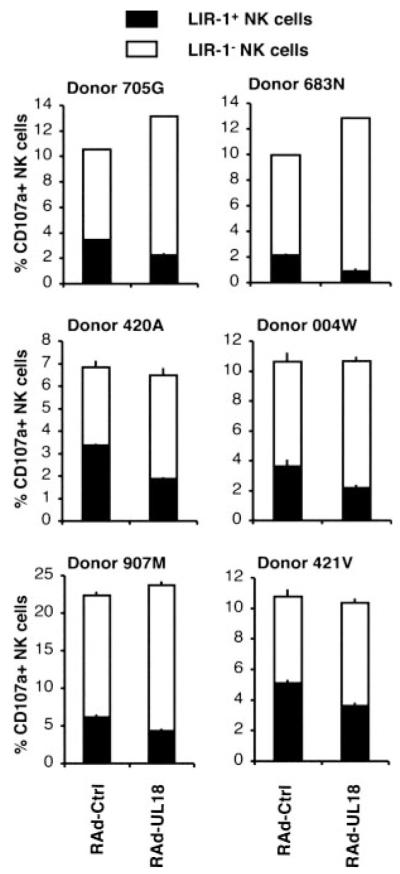
gpUL18 expression inhibits LIR-1+ NK cell activation from six tested donors. Allogeneic CD107a mobilization assays were set up with IFN-α-activated PBMC against HFFF infected for 72 h with a RAd vector containing the UL18 ORF (RAd-UL18) or with the empty control adenovirus (RAd-Ctrl) (100 PFU/cell). Results are means ± SEM of duplicate samples and are representative of three independent experiments.
Discussion
Despite the well-characterized interaction between gpUL18 and LIR-1, UL18 function has long remained an enigma. HCMV is the most complex human virus, encoding on the order of 120 genes that are not essential for virus replication in vitro (50). Many of these ORFs encode proteins implicated in immune modulation including an impressive array of genes that act in concert to suppress NK cell recognition. Studying individual HCMV NK evasion mechanisms in the context of a productive infection is therefore subject to interference by other immunosuppressive and immuno-stimulatory functions. In this study, NK cell assays were performed using the laboratory strain AD169, which lacks NK evasion genes associated with the UL/b’ sequence, specifically UL141 and UL142. Nevertheless, strain AD169 still encodes three characterized NK evasion genes (UL16, UL40, and UL83) plus additional candidates currently being evaluated. When expressed in isolation using mammalian expression vectors, the UL18 gene has proved to be highly problematic: UL18 has been found to be incompatible with the generation of stable cell lines by a number of laboratories. Much of the protein is degraded on synthesis and the extensive posttranslational events required to generate the mature 160-kDa glycoform can take up to 8 h (26). Even using a RAd vector, gpUL18 expression levels were not easily detectable. During productive HCMV infection, the protein could only be detected by using very sensitive detection systems (Fig. 1c and Ref. 26). Nevertheless, in this study, UL18 function was successfully evaluated in autologous NK cell assays by exploiting a RAd vector to drive gpUL18 expression in primary skin fibroblasts and in the context of HCMV productive infection.
Protection from LIR-1+ NK cell recognition was specifically associated with gpUL18 expression, both in the context of a productive HCMV infection and when expressed in isolation by using a RAd vector. LIR-1+ NK cell-mediated cytolysis or degranulation was inhibited by gpUL18 expression in both an allogeneic and an autologous setting using a range of effector cells: NKL, IFN-α-stimulated bulk cultures, and a single LIR-1high NK clone. LIR-1high NK cells were underrepresented in the high efficiency NK cloning system used (44), possibly due to suppressive signals received from MHC class I on feeder cells. Even when LIR-1high NK cells were specifically isolated by cell sorting, they failed to proliferate (data not shown). Protection was observed to correlate with the level of LIR-1 expression on the NK cell surface and could be inhibited with an anti-LIR-1-blocking Ab or F(ab’)2.
In assays using polyclonal NK cells, UL18-mediated inhibition of LIR-1+ NK cells was masked by an unexpected stimulatory effect of UL18 on LIR-1− cells. UL18-mediated NK activation constitutes a novel property of the glycoprotein. The effect became evident when a CD107a mobilization assay was used to correlate UL18-mediated functional NK cell inhibition with LIR-1 expression. The global NK cell response to gpUL18 in donors was dependent on several factors, variable among donors, 1) the frequency of LIR-1+ NK cells, 2) the level of LIR-1 expression (LIR-1high vs LIR-1low), and 3) the activation of a LIR-1− NK cell subset. The mechanism responsible for UL18-mediated activation has yet to be characterized: gpUL18 may be recognized directly by an activating NK receptor, may be acting by up-regulating a NK-activating ligand expression on the target cell surface, or suppressing expression of an inhibitory NK ligand. Because gpUL18 is activating a subset of LIR-1− NK cells, the effect is independent of the known gpUL18-LIR-1 interaction. The MCMV m157 protein interacts with an inhibitory receptor to suppress NK cell function in certain MCMV-susceptible mouse strains, but in C57BL/6 mice m157 binds Ly49H to both trigger NK cell function and elicit protection against infection (5). However, the situation with UL18 is quite distinct since both the activating and inhibitory properties of gpUL18 were observed in most donors. The net effect of UL18 on cytotoxicity assays using polyclonal NK cells was dependent on the composition of the NK repertoire.
gpUL18 is expressed at very low levels during productive HCMV infection, yet the results obtained in this study with the strain AD169 and the ΔUL18 mutant virus indicated that this level was sufficient to elicit protection. The exceptionally high affinity of the gpUL18-LIR-1 interaction is presumably responsible for the potency of this inhibition. It is not known whether the UL18 NK-activating effect is mediated by a low- or high-affinity interaction. The activation function was readily detected when gpUL18 was expressed efficiently using the RAd vector. HCMV may restrict gpUL18 expression levels so as to provide an overall bias against its NK cell-activating property. Alternatively, HCMV may be able to tolerate the UL18 NK activation function if the virus encodes an additional mechanism that counteracts this pathway. During lytic infection, HCMV tolerates or requires functions that provide for efficient activation of NKG2D ligands, but the virus counters this activation very effectively by sequestration of MICB, ULBP1. and ULBP2 by gpUL16 and MICA by gpUL142 (8, 11, 22). Although the UL18-activating function can be most readily detected in LIR-1− NK cells, that does not preclude it also effecting LIR-1+ NK cells. The overall signal received from gpUL18 may be balanced in LIR-1+ NK cell subsets or provide for inhibition, especially in LIR-1high NK cells.
Controversy has surrounded the function of UL18 and its mechanism of action (30, 32, 33), although a recent study has tended to favor a role for NK cell evasion (31). This study has focused specifically on NK cells, although clearly LIR-1 is also expressed in B and T lymphocytes, dendritic cells, and myeloid cells. Having demonstrated that cell surface expression of gpUL18 during productive HCMV infection can directly affect the function of NK cells by its interaction with LIR-1, it appears logical to anticipate that it can also influence other cell types. Interestingly, the frequency of LIR-1+ NK and T cells is increased in HCMV-sero-positive individuals (51). This capacity of HCMV to impact on the composition of the NK repertoire is not unique to LIR-1, HCMV is also associated with an increased frequency of the NK-activating receptor CD94/NKG2C (51). HCMV UL40 inhibits NK recognition by up-regulation of HLA-E in infected cells. HLA-E is recognized by the heterodimeric inhibitory receptor CD94/NKG2A but can also bind the activating receptor CD94/NKG2C, albeit with reduced efficiency. Although an obvious hypothesis was that UL40 was selecting an expansion of CD94+NKG2C+ NK cells potentially capable of recognizing HCMV-infected cells, this subset proliferation can be driven by a HCMV UL40 deletion mutant in vitro (52). The expansion of LIR-1+ NK cells may be driven over time by gpUL18 expression or potentially could be induced independently to suppress autoimmunity in the face of chronic Ag stimulation. Irrespective of the mechanism controlling their expansion, the UL18 contribution to HCMV evasion of NK and T cell recognition may increase during a persistent lifelong infection with an increase in LIR-1+ cell frequency.
Acknowledgments
We are grateful to Siân Llewellyn-Lacey for provided technical support; Dr. Véronique Braud, Dr. Mark R. Wills, and Dr. Andrew J. Davison for helpful discussions; and Amgen for providing an anti-LIR-1 mAb.
Footnotes
This work was supported by funds from the Wellcome Trust and the Biotechnology and Biological Sciences Research Council. M.L.-B. is supported by the Spanish Ministry of Education and Science (SAF2004-07632).
- HCMV
- human CMV
- MCMV
- murine CMV
- MIC
- MHC class I-related chain
- ULBP
- U16-binding protein
- RAET
- retinoic acid early transport
- ORF
- open reading frame
- LIR-1
- leukocyte Ig-like receptor 1
- RAd
- replication-deficient adenovirus
- HFFF
- human fetal foreskin fibroblast
- MFI
- mean fluorescence intensity
- Ctrl
- control
Disclosures The authors have no financial conflict of interest.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 1989;320:1731–1735. doi: 10.1056/NEJM198906293202605. [DOI] [PubMed] [Google Scholar]
- 2.Gazit R, Garty BZ, Monselise Y, Hoffer V, Finkelstein Y, Markel G, Katz G, Hanna J, Achdout H, Gruda R, et al. Expression of KIR2DL1 on the entire NK cell population: a possible novel immunodeficiency syndrome. Blood. 2004;103:1965–1966. doi: 10.1182/blood-2003-11-3796. [DOI] [PubMed] [Google Scholar]
- 3.Bukowski JF, Warner JF, Dennert G, Welsh RM. Adoptive transfer studies demonstrating the antiviral effect of natural killer cells in vivo. J. Exp. Med. 1985;161:40–52. doi: 10.1084/jem.161.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farrell HE, Vally H, Lynch DM, Fleming P, Shellam GR, Scalzo AA, Davis-Poynter NJ. Inhibition of natural killer cells by a cytomega-lovirus MHC class I homologue in vivo. Nature. 1997;386:510–514. doi: 10.1038/386510a0. [DOI] [PubMed] [Google Scholar]
- 5.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296:1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- 6.Tortorella D, Gewurz B, Schust D, Furman M, Ploegh H. Down-regulation of MHC class I antigen presentation by HCMV; lessons for tumor immunology. Immunol. Invest. 2000;29:97–100. doi: 10.3109/08820130009062289. [DOI] [PubMed] [Google Scholar]
- 7.Rolle A, Mousavi-Jazi M, Eriksson M, Odeberg J, Soderberg-Naucler C, Cosman D, Karre K, Cerboni C. Effects of human cytomegalovirus infection on ligands for the activating NKG2D receptor of NK cells: up-regulation of UL16-binding protein (ULBP)1 and ULBP2 is counteracted by the viral UL16 protein. J. Immunol. 2003;171:902–908. doi: 10.4049/jimmunol.171.2.902. [DOI] [PubMed] [Google Scholar]
- 8.Welte SA, Sinzger C, Lutz SZ, Singh-Jasuja H, Sampaio KL, Eknigk U, Rammensee HG, Steinle A. Selective intracellular retention of virally induced NKG2D ligands by the human cytomegalovirus UL16 glycoprotein. Eur. J. Immunol. 2003;33:194–203. doi: 10.1002/immu.200390022. [DOI] [PubMed] [Google Scholar]
- 9.Eagle RA, Traherne JA, Ashiru O, Wills MR, Trowsdale J. Regulation of NKG2D ligand gene expression. Hum. Immunol. 2006;67:159–169. doi: 10.1016/j.humimm.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 10.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727–729. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 11.Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, Kubin M, Chalupny NJ. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 2001;14:123–133. doi: 10.1016/s1074-7613(01)00095-4. [DOI] [PubMed] [Google Scholar]
- 12.Bacon L, Eagle RA, Meyer M, Easom N, Young NT, Trowsdale J. Two human ULBP/RAET1 molecules with transmembrane regions are ligands for NKG2D. J. Immunol. 2004;173:1078–1084. doi: 10.4049/jimmunol.173.2.1078. [DOI] [PubMed] [Google Scholar]
- 13.Tomasec P, Braud VM, Rickards C, Powell MB, McSharry BP, Gadola S, Cerundolo V, Borysiewicz LK, McMichael AJ, Wilkinson GW. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science. 2000;287:1031–1033. doi: 10.1126/science.287.5455.1031. [DOI] [PubMed] [Google Scholar]
- 14.Wang EC, McSharry B, Retiere C, Tomasec P, Williams S, Borysiewicz LK, Braud VM, Wilkinson GW. UL40-mediated NK evasion during productive infection with human cytomegalovirus. Proc. Natl. Acad. Sci. USA. 2002;99:7570–7575. doi: 10.1073/pnas.112680099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arnon TI, Achdout H, Levi O, Markel G, Saleh N, Katz G, Gazit R, Gonen-Gross T, Hanna J, Nahari E, et al. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat. Immunol. 2005;6:515–523. doi: 10.1038/ni1190. [DOI] [PubMed] [Google Scholar]
- 16.Cerboni C, Mousavi-Jazi M, Linde A, Soderstrom K, Brytting M, Wahren B, Karre K, Carbone E. Human cytomegalovirus strain-dependent changes in NK cell recognition of infected fibroblasts. J. Immunol. 2000;164:4775–4782. doi: 10.4049/jimmunol.164.9.4775. [DOI] [PubMed] [Google Scholar]
- 17.Tomasec P, Wang EC, Davison AJ, Vojtesek B, Armstrong M, Griffin C, McSharry BP, Morris RJ, Llewellyn-Lacey S, Rickards C, et al. Downregulation of natural killer cell-activating ligand CD155 by human cytomegalovirus UL141. Nat. Immunol. 2005;6:181–188. doi: 10.1038/ni1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C, Dargan DJ, McGeoch DJ, Gatherer D, Emery VC, Griffiths PD, Sinzger C, McSharry BP, Wilkinson GW, Davison AJ. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 2004;85:1301–1312. doi: 10.1099/vir.0.79888-0. [DOI] [PubMed] [Google Scholar]
- 19.Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, Cantoni C, Grassi J, Marcenaro S, Reymond N, et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J. Exp. Med. 2003;198:557–567. doi: 10.1084/jem.20030788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuchs A, Cella M, Giurisato E, Shaw AS, Colonna M. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155) J. Immunol. 2004;172:3994–3998. doi: 10.4049/jimmunol.172.7.3994. [DOI] [PubMed] [Google Scholar]
- 21.Wills MR, Ashiru O, Reeves MB, Okecha G, Trowsdale J, Tomasec P, Wilkinson GW, Sinclair J, Sissons JG. Human cytomegalovirus encodes an MHC class I-like molecule (UL142) that functions to inhibit NK cell lysis. J. Immunol. 2005;175:7457–7465. doi: 10.4049/jimmunol.175.11.7457. [DOI] [PubMed] [Google Scholar]
- 22.Chalupny NJ, Rein-Weston A, Dosch S, Cosman D. Down-regulation of the NKG2D ligand MICA by the human cytomegalovirus glycoprotein UL142. Biochem. Biophys. Res. Commun. 2006;346:175–181. doi: 10.1016/j.bbrc.2006.05.092. [DOI] [PubMed] [Google Scholar]
- 23.Beck S, Barrell BG. Human cytomegalovirus encodes a glycoprotein homologous to MHC class-I antigens. Nature. 1988;331:269–272. doi: 10.1038/331269a0. [DOI] [PubMed] [Google Scholar]
- 24.Browne H, Smith G, Beck S, Minson T. A complex between the MHC class I homologue encoded by human cytomegalovirus and β 2 microglobulin. Nature. 1990;347:770–772. doi: 10.1038/347770a0. [DOI] [PubMed] [Google Scholar]
- 25.Fahnestock ML, Johnson JL, Feldman RM, Neveu JM, Lane WS, Bjorkman PJ. The MHC class I homolog encoded by human cytomegalovirus binds endogenous peptides. Immunity. 1995;3:583–590. doi: 10.1016/1074-7613(95)90129-9. [DOI] [PubMed] [Google Scholar]
- 26.Griffin C, Wang EC, McSharry BP, Rickards C, Browne H, Wilkinson GW, Tomasec P. Characterization of a highly glycosylated form of the human cytomegalovirus HLA class I homologue gpUL18. J. Gen. Virol. 2005;86:2999–3008. doi: 10.1099/vir.0.81126-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park B, Oh H, Lee S, Song Y, Shin J, Sung YC, Hwang SY, Ahn K. The MHC class I homolog of human cytomegalovirus is resistant to down-regulation mediated by the unique short region protein (US)2, US3, US6, and US11 gene products. J. Immunol. 2002;168:3464–3469. doi: 10.4049/jimmunol.168.7.3464. [DOI] [PubMed] [Google Scholar]
- 28.Rawlinson WD, Farrell HE, Barrell BG. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 1996;70:8833–8849. doi: 10.1128/jvi.70.12.8833-8849.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. USA. 2002;99:8826–8831. doi: 10.1073/pnas.092258599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reyburn HT, Mandelboim O, Vales-Gomez M, Davis DM, Pazmany L, Strominger JL. The class I MHC homologue of human cytomegalovirus inhibits attack by natural killer cells. Nature. 1997;386:514–517. doi: 10.1038/386514a0. [DOI] [PubMed] [Google Scholar]
- 31.Kim JS, Choi SE, Yun IH, Kim JY, Ahn C, Kim SJ, Ha J, Hwang ES, Cha CY, Miyagawa S, Park CG. Human cytomegalovirus UL18 alleviated human NK-mediated swine endothelial cell lysis. Biochem. Biophys. Res. Commun. 2004;315:144–150. doi: 10.1016/j.bbrc.2004.01.027. [DOI] [PubMed] [Google Scholar]
- 32.Odeberg J, Cerboni C, Browne H, Karre K, Moller E, Carbone E, Soderberg-Naucler C. Human cytomegalovirus (HCMV)-infected endothelial cells and macrophages are less susceptible to natural killer lysis independent of the downregulation of classical HLA class I molecules or expression of the HCMV class I homologue, UL18. Scand. J. Immunol. 2002;55:149–161. doi: 10.1046/j.1365-3083.2002.01025.x. [DOI] [PubMed] [Google Scholar]
- 33.Leong CC, Chapman TL, Bjorkman PJ, Formankova D, Mocarski ES, Phillips JH, Lanier LL. Modulation of natural killer cell cytotoxicity in human cytomegalovirus infection: the role of endogenous class I major histocompatibility complex and a viral class I homolog. J. Exp. Med. 1998;187:1681–1687. doi: 10.1084/jem.187.10.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L, Hsu ML. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity. 1997;7:273–282. doi: 10.1016/s1074-7613(00)80529-4. [DOI] [PubMed] [Google Scholar]
- 35.Chapman TL, Heikeman AP, Bjorkman PJ. The inhibitory receptor LIR-1 uses a common binding interaction to recognize class I MHC molecules and the viral homolog UL18. Immunity. 1999;11:603–613. doi: 10.1016/s1074-7613(00)80135-1. [DOI] [PubMed] [Google Scholar]
- 36.Vitale M, Castriconi R, Parolini S, Pende D, Hsu M-L, Moretta L, Cosman D, Moretta A. The leukocyte Ig-like receptor (LIR)-1 for the cytomegalovirus UL18 protein displays a broad specificity for different HLA class I alleles: analysis of LIR-1+ NK cell clones. Int. Immunol. 1999;11:29–35. doi: 10.1093/intimm/11.1.29. [DOI] [PubMed] [Google Scholar]
- 37.Lepin EJ, Bastin JM, Allan DS, Roncador G, Braud VM, Mason DY, van der Merwe PA, McMichael AJ, Bell JI, Powis SH, O’Callaghan CA. Functional characterization of HLA-F and binding of HLA-F tetramers to ILT2 and ILT4 receptors. Eur. J. Immunol. 2000;30:3552–3561. doi: 10.1002/1521-4141(200012)30:12<3552::AID-IMMU3552>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 38.Shiroishi M, Tsumoto K, Amano K, Shirakihara Y, Colonna M, Braud VM, Allan DS, Makadzange A, Rowland-Jones S, Willcox B, et al. Human inhibitory receptors Ig-like transcript 2 (ILT2) and ILT4 compete with CD8 for MHC class I binding and bind preferentially to HLA-G. Proc. Natl. Acad. Sci. USA. 2003;100:8856–8861. doi: 10.1073/pnas.1431057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colonna M, Navarro F, Bellon T, Llano M, Garcia P, Samaridis J, Angman L, Cella M, Lopez-Botet M. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J. Exp. Med. 1997;186:1809–1818. doi: 10.1084/jem.186.11.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berg L, Riise GC, Cosman D, Bergstrom T, Olofsson S, Karre K, Carbone E. LIR-1 expression on lymphocytes, and cytomegalovirus disease in lung-transplant recipients. Lancet. 2003;361:1099–1101. doi: 10.1016/S0140-6736(03)12855-3. [DOI] [PubMed] [Google Scholar]
- 41.Antrobus RD, Khan N, Hislop AD, Montamat-Sicotte D, Garner LI, Rickinson AB, Moss PA, Willcox BE. Virus-specific cytotoxic T lymphocytes differentially express cell-surface leukocyte immunoglobulin-like receptor-1, an inhibitory receptor for class I major histocompatibility complex molecules. J. Infect. Dis. 2005;191:1842–1853. doi: 10.1086/429927. [DOI] [PubMed] [Google Scholar]
- 42.McSharry BP, Jones CJ, Skinner JW, Kipling D, Wilkinson GW. Human telomerase reverse transcriptase-immortalized MRC-5 and HCA2 human fibroblasts are fully permissive for human cytomegalovirus. J. Gen. Virol. 2001;82:855–863. doi: 10.1099/0022-1317-82-4-855. [DOI] [PubMed] [Google Scholar]
- 43.Robertson MJ, Cochran KJ, Cameron C, Le JM, Tantravahi R, Ritz J. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. Exp. Hematol. 1996;24:406–415. [PubMed] [Google Scholar]
- 44.Morris RJ, Chong LK, Wilkinson GW, Wang EC. A high-efficiency system of natural killer cell cloning. J. Immunol. Methods. 2005;307:24–33. doi: 10.1016/j.jim.2005.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Browne H, Churcher M, Minson T. Construction and characterization of a human cytomegalovirus mutant with the UL18 (class I homolog) gene deleted. J. Virol. 1992;66:6784–6787. doi: 10.1128/jvi.66.11.6784-6787.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lilley BN, Ploegh HL, Tirabassi RS. Human cytomegalovirus open reading frame TRL11/IRL11 encodes an immunoglobulin G Fc-binding protein. J. Virol. 2001;75:11218–11221. doi: 10.1128/JVI.75.22.11218-11221.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Atalay R, Zimmermann A, Wagner M, Borst E, Benz C, Messerle M, Hengel H. Identification and expression of human cytomegalovirus transcription units coding for two distinct Fcγ receptor homologs. J. Virol. 2002;76:8596–8608. doi: 10.1128/JVI.76.17.8596-8608.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davison AJ, Dolan A, Akter P, Addison C, Dargan DJ, Alcendor DJ, McGeoch DJ, Hayward GS. The human cytomegalovirus genome revisited: comparison with the chimpanzee cytomegalovirus genome. J. Gen. Virol. 2003;84:17–28. doi: 10.1099/vir.0.18606-0. [DOI] [PubMed] [Google Scholar]
- 49.Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002;419:734–738. doi: 10.1038/nature01112. [DOI] [PubMed] [Google Scholar]
- 50.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA. 2003;100:14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guma M, Angulo A, Vilches C, Gomez-Lozano N, Malats N, Lopez-Botet M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood. 2004;104:3664–3671. doi: 10.1182/blood-2004-05-2058. [DOI] [PubMed] [Google Scholar]
- 52.Guma M, Budt M, Saez A, Brckalo T, Hengel H, Angulo A, Lopez-Botet M. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood. 2006;107:3624–3631. doi: 10.1182/blood-2005-09-3682. [DOI] [PubMed] [Google Scholar]