

Abstract
Intermediate filament (IF) proteins and heat shock proteins (HSPs) are large multi-membered families that share several features. These features include protein abundance, significant up-regulation in response to a variety of stresses, function as cytoprotectors, and the phenocopying of several human diseases upon IF protein or HSP mutation. We are now coming to understand that these common elements point to IFs as important cellular stress proteins with some roles akin to those already well-characterized for HSPs. Unique functional roles for IFs include protection from mechanical stress while HSPs are characteristically involved in protein folding and as chaperones. Shared IF and HSP cytoprotective roles include inhibition of apoptosis, organelle homeostasis, and scaffolding. We review here recent data that corroborate the view that IFs function as highly-specialized cytoskeletal stress proteins that promote cellular organization and homeostasis.
The stress response
Tissues and cells are constantly subjected to stressful situations, meaning that components of the cell, including proteins, organelles and DNA, can be damaged. These stresses are both intrinsic, such as genetic and endoplasmic reticulum stress, and extrinsic/environmental stresses including heat, toxin or radiation, mechanical, wound and regenerative, infection, metabolic such as hypoxia and autophagy, osmotic and oxidative (Fig.1). The prototype response to stress, the heat stress response (HSR) is the best characterized and occurs via activation of heat shock transcription factors (HSFs), which drive the expression of multiple target genes containing the heat shock elements (HSEs) within their promoter sequence [1]. The heat shock proteins (HSPs) are the classical HSR targets and constitute a large family of proteins that are expressed constitutively and in response to stress [2] (Box 1). HSPs act as molecular chaperones assisting with normal folding, transport, and degradation of proteins and have an established stress-protective function, which extends far beyond the heat response [3, 4]. For example, HSPs have anti-apoptotic properties and their levels can be extensively modulated during inflammation, cancer and several other diseases [5, 6]. While HSPs are the classical stress proteins, the cellular stress response is far more complex and consists of up-regulation of many other less well-characterized genes or general down-regulation of mRNA splicing and protein translation [7, 8], and activation of diverse signaling cascades including phosphatases and kinases and anti-apoptotic proteins. Accumulating data also point to the family of intermediate filament (IF) proteins as being important contributors to cellular stress responses.
Fig.1. Multiple forms of stress modulate IFs.

Cells are exposed to various forms of stress that originate from within or from the environment. Depending on the type and duration of stress encountered, IFs may respond by (a) forming cell-specific inclusions (e.g., Mallory-Denk bodies in alcoholic and nonalcoholic steatohepatitis [21, 84]); (b) up-regulating or de-novo generation of filaments (e.g. K19/K20 following pancreatic injury, [17]; or (c) reorganizing filaments (e.g. K18 following shear stress [56]).
Our aim in this review is to highlight the stress-related features of IF proteins and provide a framework that compares them with the classical HSPs. We define stress proteins as proteins that (i) are upregulated in response to stress at least 3-fold at the mRNA and/or protein level, despite already being abundant proteins, (ii) serve a protective role in several biologic contexts and not only a single entity such as apoptosis, and (iii) upon overexpression prevent injury and/or whose absence promotes injury. Using this definition, and the similarities highlighted herein between HSPs and IFs, we review results from recent and previously published data to support the conclusion that IF proteins do indeed serve as stress proteins.
IFs and making the case for IFs as stress proteins
The cytoskeletal intermediate filament (IF) protein family consists of 73 gene products (www.interfil.org) [9] that are expressed in tissue-, cell-, differentiation-, and development-dependent manners (Table 1). IFs display a tripartite structure consisting of an α-helical central “rod” and flanking non-helical “head” and “tail” domains, and assemble to give rise to nuclear (lamins) or to different cytoplasmic IF networks [consisting of proteins such as keratins (K), vimentin, neurofilaments (NFs), peripherin, desmin, and glial fibrillary acidic protein (GFAP) depending on tissue and cell type] [10, 11] (Table 1). IFs are regulated through interactions with multiple IF-associated proteins (IFAPs) [12] that include signaling molecules, such as 14-3-3 proteins, apoptosis-related proteins, kinases and phosphatases [13] and by posttranslational modifications (PTMs). Prominent among IF PTMs is phosphorylation, which regulates IF solubility, IFAP binding, and provides essential cytoprotective functions [14]. IFs are degraded by caspases during apoptosis [15] and are targets for ubiquitination and proteosomal degradation as part of normal protein turnover [16].
Table 1. IF proteins, their tissue distribution and examples of stresses that cause protein or RNA upregulation.
| Type | IF protein[16] | Tissue/Cell distribution | Examples of stresses that induce IF overexpression | References* |
|---|---|---|---|---|
| I & II | K1-6, K9-17 | Stratified epithelia | Regeneration (skin wound healing), stretch, uv-light, virus, TNFα, IL-1, IFNγ, TGFα, TGFβ, irradiation | [17-23] |
| K7-8, K18-28 | Simple and other epithelia | Regenerative repair (pancreas), toxins, shear stress, oxidative stress, virus infection, hormones, IL-6, bile duct ligation, lithogenic diet, irradiation, aging | [20-22, 24-39] | |
| K31-40, K81-86 | Hair/nail | |||
| III | Desmin | Muscle | Regeneration, toxins | [40] [41] |
| GFAP | Astrocytes | Regeneration, infection (IL-1β), TNFα, LPS, aging, alcohol, ischemia | [42-47] | |
| Peripherin | Neurons, peripheral | Regeneration (neural) | [48-54] | |
| Vimentin | Mesenchymal cells and tissues | Regeneration (astrocytes, muscle, skin fibroblasts), toxins, age, electroporation, heat, oxidative stress | [18, 40-42, 55-60] | |
| Syncoilin | MuscleI | |||
| IV | NF-L, -M, -H | CNS neurons | Regeneration | [51, 61] |
| Nestin | Stem cells, neuroepithelia | Regeneration (astrocytes, spinal cord injury, muscle), toxins, aging | [42] [18] [40, 41, 45, 62] | |
| α-internexin | CNS neurons | Regeneration (neural) | [63] | |
| Synemin (α,β) splice variants | Muscle | Regeneration (astrocytes, muscle), scar formation (liver) | [64-67] | |
| V | Lamin A/C | All nuclei | Shear stress, starvation | [68-70] |
| Lamins B1, B2 | All nuclei | Heat | [71, 72] | |
| VI | Bfsp1, Bfsp2 | Fiber cells | ||
Bfsp, beaded filament structural protein; GFAP, glial fibrillary acidic protein; K, keratin; NF, neurofilaments (L, light; M, middle; and H, heavy chains). Note: The differential upregulation/expression of different IFs during embryogenesis, development and cancer is not included.
Several features suggest that IFs should be considered as stress proteins (Box 2). Despite their abundance, (keratins make up 0.2%-0.5% of total tissue protein in liver, pancreas and intestine, and 5% of HT29 cultured cell protein [17], and for vimentin 2-3% of total fibroblast protein [18]), IF RNA and/or proteins become induced several fold in response to a variety of stresses (Table 1). This upregulation involves several tissues and their corresponding IFs, and includes both constitutive and de novo IF RNA and protein synthesis (Supplementary Table S1; Fig.2). Enforced genetic overexpression of simple epithelial keratins are generally well tolerated as seen after modest but not supraphysiological overexpression in animal models [19]. However, forced expression of other IFs (e.g., neurofilaments, GFAP or peripherin) in mice leads to neurological disorders in the IF-relevant tissues [20]. Notably, IF overexpression in many chronic diseases is associated with cytoplasmic IF-containing aggregates that also include HSPs [21]. Interestingly, proteomic meta-analysis of the most altered proteins or protein families in several cancers and other biological systems of mouse, rat and human samples (based on an analysis of 188 articles published between 2004-2006 [22]) found that keratins and HSPs were the first and fifth most frequently altered protein families in the disease states analysed, respectively, potentially indicating an important role of IFs in responding to pathological processes. In some cases, IF upregulation in response to injury may reflect hyperproliferation of a specific cell type, such as increased K19 in bile duct proliferation (Fig.2), rather than an IF upregulation per se.
Fig.2. Keratin IFs and HSPs are upregulated after organ-specific stress in the liver and pancreas.
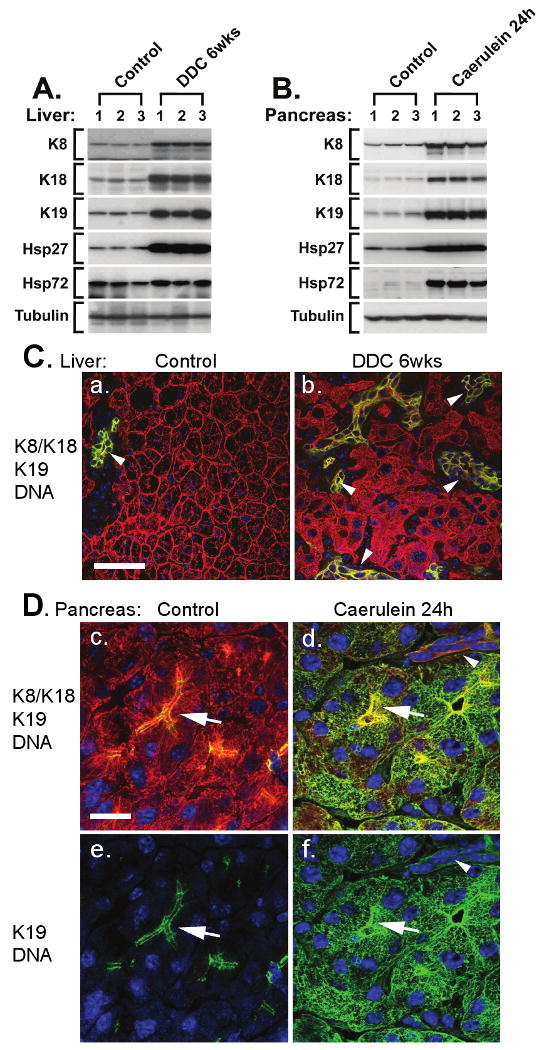
Livers from untreated mice or mice fed 0.1% 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) for 6 weeks (A, C) and, pancreata from untreated mice or mice injected hourly with 50 μg/kg caerulein 7 times, then recovered for 17 hours (B, D), were analyzed. (A-B) Total protein lysates from 3 different mice/treatment were probed with antibodies to K8, K18, K19, Hsp27, Hsp72, and tubulin (as a loading control). Alternatively, sections (1-2 μm) from fresh frozen livers (C) and pancreata (D) were fixed with cold acetone (10 minutes) then triple-stained to visualize K8/K18 (red), K19 (green), and nuclei (blue). Co-localization of K8/K18 and K19 appears yellow. Note the induction of K8, K18 and K19 (but not tubulin) upon liver stress (DDC; A, C) and pancreatic stress (caerulein; B, D). In the pancreas, K19 is dramatically upregulated upon recovery from caerulein-treatment (B) in acinar cells where it forms extensive cytoplasmic filaments with the resident K8/K18 filaments (Dd,f) compared to the preferential apico-lateral distribution (arrows) in untreated pancreata (Dc,e). In DDC-fed mice, hepatocyte K8/K18 upregulation (A) is reflected by the denser keratin networks (C) compared to control (Ca), while K19 upregulation is reflected by proliferation of K19-positive ductal cells (arrowheads, Cb). Scale bar in a for a-b is 50 m and in c for c-f is 20μm. This figure illustrates: (i) the upregulation of several keratin IFs in two different organs upon two unique and organ-specific toxic stresses; (ii) the joint upregulation of keratin IFs and two different HSPs during the two toxic treatments in the pancreas and liver, and (iii) that upregulation of the “stress protein” K19 is context dependent and may reflect either de-novo cytoplasmic expression (and overall increased expression) in acinar cells or hyperproliferation of ductal cells in the liver.
Several other findings support the importance of IFs as stress proteins. For example, human IF mutations cause diseases that are accentuated in response to stress such as mutations in keratin polypeptides 5 or 14 (K5 or K14) which cause epidermolysis bullosa simplex (EBS) (whereby skin pressure or rubbing causes blister formation) [23]. More pointedly, K8, K18 or K19 mutations are silent under normal circumstances yet predispose to liver injury and apoptosis (e.g., mediated by toxins), or to mechanical fragility of hepatocytes (upon isolation by liver perfusion in keratin-mutant or keratin-deficient mice) depending on the location of the mutation [11]. However, there are a few exceptions, including K8-null mice that are protected from coxsackievirus-induced pancreatitis [24], and double-null GFAP/vimentin mice which develop attenuated reactive gliosis after neurotrauma [25]. In summary, there is compelling evidence for an important role for IFs in the stress response in a context-dependent manner. Below we will discuss IFs and their role and modulation during different stress contexts.
Involvement of IFs in specific stress contexts
IFs in classical heat stress, and HSP-IF interactions
The HSR increases the levels of multiple stress-inducible genes with several HSPs being among the most prominent [4]. The heat-induced upregulation of HSPs is ubiquitous, conserved and is implicated in protection against a variety of otherwise lethal stresses [1]. Unlike HSP expression, changes in IF levels in response to heat stress are dependent on both the tissue and gene involved. For example, pancreatic K8, K18, K19 and the astrocytic GFAP expression remain unaltered after heat exposure [17, 26], while vimentin is heat-sensitive, in that it is elevated in non-neuronal cells and microvasculature in the hippocampus [27] as well as in human promonocytic cells [28], but diminished in heat-exposed rat embryos [29]. For lamins, both increased and decreased levels of lamin B upon heat stress have been reported [30, 31]. Heat stress also leads to K8 and vimentin hyperphosphorylation [32, 33] and to keratin, vimentin and GFAP filament reorganization [34-38]. The significance of stress-induced IF hyperphosphorylation and consequent filament reorganization are unclear but one hypothesis is that IF hyperphosphorylation functions, in part, as a phosphate sink or buffer that curtails unwarranted stress kinase activation [14]. This hypothesis has been tested in the context of apoptosis [39] but has not been tested in the context of other stresses.
A likely functional link between HSPs and IFs is their physical association, with different IFs having overlapping or selective HSP binding partners. For example, K18 and K8 preferentially associate with Mrj and HSP70, respectively [40, 41], while GFAP and vimentin both interact with HSP27 and αB-crystallin [42]. Addition of recombinant αB-crystallin to lens homogenates or exposure to mild in vitro heat shock results in binding of αB-crystallin to the IF proteins found in the lens: Bfsp1 (also called filensin) and Bfsp2 (also called CP49) and vimentin. [43]. The IF-HSP interaction increases upon heat exposure as noted for keratins and GFAP [41, 44], and there is some evidence (based on microinjection of anti-Mrj antibodies) that IF-HSP interaction contributes to normal IF filament organization [40]. However, IF-HSP interactions are likely to relate to other functional roles because such interaction occurs at relatively high stoichiometry [41] under non-stress basal conditions (thereby suggesting a non-chaperone/protein folding role) and IF filament assembly occurs in vitro without the need to include HSPs.
Another link between HSPs and IFs relates to the increased HSP levels in several IF mutation contexts including cells that express assembly-compromised GFAP variants [45], in mice harbouring mutant vimentin that have increased HSP70 expression [46], and in mice with altered GFAP or neurofilaments (NF)-L subunit whose disease phenotype is ameliorated by overexpression of the HSPs αB-crystallin and Hsp27, respectively [47, 48]. In addition, HSPs are observed by immune-staining within several types of human inclusion bodies which form as a result of IF mutation (e.g., GFAP, desmin) or independent of IF mutation such as Mallory-Denk bodies. In these situations, IF-HSP interactions are likely to be related to chaperone (HSP) and substrate (IF protein) physical associations that protects the misfolded proteins.
IFs during mechanical stress, wound healing and tissue repair
One clearly established function of IFs is their role in protecting cells and tissues from mechanical stress [49, 50] IFs are unique among cytoskeletal proteins in that their filaments can stretch 3 times their initial length before breaking [51, 52]. The best and first link of IFs to mechanical stress comes from the identification of K5 and K14 mutations in patients with EBS [23]. In addition, K8-null, K18-null, or K18 R90C mutation renders mouse hepatocytes markedly fragile upon liver perfusion [39]. In lung epithelial cells, shear stress (parallel or tangential force such as the stress created on endothelial cells during blood flow in vessels, Fig. 1) increases keratin solubility [53], degradation, aggregation [54], and K8 and K18 phosphorylation within the thickened keratin filaments and associated keratin particles [55]. In this context, keratin phosphorylation and filament reorganization appear to be 14-3-3 and PKCζ-dependent [56].
The importance of keratins in stress-signal transmission may occur at focal adhesions since the viscoelastic response of keratin-null rat hepatoma cells is reduced, as compared with K8- or K18-containing cells, when measured using fibronectin-coated polystyrene beads and mechanical stress generated by optical tweezers [57]. Whether shear stress affects IF expression has not been studied in detail, although stretch induces K6 and decreases K10 expression [49, 58]. Mechanical stress-sensing and signaling through IFs is not well understood but may involve nitric oxide synthase [59] and Rho-signaling and thus also involve actin and focal adhesion dynamics [60]. In contrast to IFs, the role of HSPs in protection from mechanical stress is not known.
Other IFs may play a role in the context of mechanical stress. For example, shear stress deforms vimentin networks in blood vessels [61]. Similar to keratins, the importance of vimentin in shear stress may relate to focal contacts which become smaller and less able to adhere to the substratum in vimentin-null cells [62]. Desmin, the major IF in smooth muscle, is also involved in vascular shear stress response, as supported by the decreased small artery reactivity in desmin-null mice [61]. In the case of lamins, nuclei exposed to shear stress change their shape and upregulate lamin A which becomes reorganized, and cells with lamin A mutation lack the ability to adapt to flow [63]. Furthermore, loss of intact lamin A leads to decreased viability and increased apoptosis upon mechanical strain, and to attenuated wound healing and nuclear stiffness [64].
Tissue repair processes such as wound healing [65] involve IF upregulation, such as K6, K16, and K17 de-novo transcription at the skin wound edge in activated keratinocytes within 3 hours of injury [65]. Recovery after pancreatitis is associated with upregulation of K19 and K20 as de novo dense acinar cell cytoplasmic filaments that are normally present only in the apicolateral membrane-proximal compartment (Fig.2 [17]). It is unknown what induces K19 and K20 to form cytoplasmic filaments and what role they play in the stress response. Similarly, there is rapid upregulation of nestin and vimentin, normally only present during embryonic myogenesis or early differentiation, during repair of damaged muscle [65]. To that end, vimentin-null fibroblasts are stiffer and vimentin-null embryos have slower wound healing [10]. Wound activated astrocytes also revert to an immature-astrocyte phenotype which includes de-novo expression of vimentin and nestin with the resident GFAP [66]. Similarly, peripherin, which is normally present in peripheral but not central brain neurons, is induced in some brain neurons after lesion injury or cerebral ischemia [67] and after alterations in synaptic plasticity in the hippocampus [68]. However, not all forms of injury are associated with IF induction. For example, neurofilaments and peripherin are down-regulated in peripheral dorsal root ganglia sensory neurons immediately after sensory fiber axotomy, though peripherin becomes upregulated a few days after the injury which suggests that peripherin may be important in the recovery process [69]. More detailed studies are required to elucidate the significance and mechanism of IF induction and the less common IF down-regulation in response to injury.
IFs and apoptotic stress
Apart from being an important homeostatic process, apoptosis is often the end-result of many stress-situations [70] leading to caspase-directed degradation of cellular proteins, including many IFs which are caspase substrates [15, 71]. In fact one of the clearly established functions of IFs is to protect cells from apoptosis. For example, keratin-deficient or mutant mouse livers are highly susceptible to apoptosis through toxin-mediated injuries [15, 39, 72]. The role of K8 and K18 as anti-apoptotic proteins is likely to be multi-factorial and related to: K8 or K18 binding to the cytoplasmic domain of TNFR2 [73]; K18 binding to TRADD [74]; increased cell surface Fas ligand (FasL) receptors as noted in K8-null hepatocytes [15]; or an alteration in mitochondrial distribution, shape and function [75]. In addition, keratin phosphorylation plays a critical role in protection from liver injury and apoptosis [14], as exemplified by mutation of stress-induced K8 Ser74 phosphorylation in transgenic mice. K8 S74A mutation renders the mice highly susceptible to Fas ligand-induced apoptosis by shunting phosphorylation to other proteins that become pro-apoptotic upon phosphorylation [76]. Similarly, K17 binds and sequesters TRADD, and K17-null keratinocytes are also sensitive to TNF-mediated apoptosis [77].
Other mouse and cell culture models support the importance of IFs as anti-apoptotic proteins. For example, desmin-null mice, which develop dilated cardiomyopathy, have a mitochondrial defect and extensive cell death that can be ameliorated by overexpression of the anti-apoptotic protein Bcl-2 [71]. Similarly, double transgenic mice overexpressing TNF-α in the heart and a caspase-6 cleavage-resistant desmin mutant, showed protection against TNF-α mediated apoptosis indicating that desmin-cleavage is important for the progression of apoptosis [78]. In addition, protection against staurosporine-induced apoptosis was noted in cells expressing mutant vimentin [79]. One exception to the anti-apoptotic role of keratins is a paradoxical resistance to mechanically induced apoptosis in keratinocyte cell lines generated from humans with skin disease caused by K5 and K14 mutations [80]. This contrasting effect is also seen with HSP70 which can potentiate T-cell receptor linked apoptosis by associating with caspase-activated DNase [81] even if HSPs in general inhibit apoptosis [82].
IFs during toxin-related and oxidative stress
Toxin- and drug-induced stress has been extensively studied, particularly in livers of mice that lack K8 or K18 or overexpress K8-or K18-variants, which in turn are found to have increased susceptibility to injury [39]. Toxin exposure often increases keratin mRNA and protein, as well as phosphorylation levels [14, 39]. For example, thioacetamide-induced liver fibrosis upregulates K8 and K18 mRNA 4-5-fold as well as K8 or K18 and HSP27 protein [83]. DDC (Fig.2) and griseofulvin up-regulate keratin RNA and protein levels and upon chronic administration lead to inclusion body formation the significance of which is under study aside from the established role of their human orthologs as clinically useful markers of alcoholic and nonalcoholic steatohepatitis [21, 84, 85]. Furthermore, there is upregulation of nestin, vimentin and desmin in the puromycin amino-nucleoside nephrosis podocyte injurymodel [86], vimentin induction after exposure to 12-O-tetradecanoylphorbol-13-acetate [87], and increased GFAP after exposure to capsaicin [88] and ethanol [89]. The toxin-mediated upregulation of IFs may, at least in part, be related to oxidative injury given the observed upregulation of vimentin [90], and K18 [91] after oxidative injury.
Stress caused by increased age and high calorie intake has recently been linked to an HSP response [92] and may entail the upregulation of several IFs. For example, aging correlates with increased GFAP [93] and nestin [94] levels in astrocytes and rat-pituitary, respectively, while GFAP levels decrease after chronic food restriction related to decreased oxidation of proteins, lipids and increased life span [93]. Similarly, middle-aged mice upregulate K19 mRNA and protein significantly more than young mice after bone fracture and soft-tissue trauma [95], and vimentin protein and RNA increase 4-5 fold together with filament reorganization in senescent fibroblasts but far less so in young embryonic fibroblasts [96]. Furthermore, lipodystrophy-causing lamin A mutations result in the accumulation of farnesylated prelamin A and oxidative stress that trigger premature cellular senescence [97], and it has been shown that lamin levels in cell culture are related to the presence of serum [98].
IFs are also cytoprotective during oxidative-stress, whereby generation or exposure to cytotoxic oxidants and free radicals result in cellular degeneration. For example, nestin promotes survival during H2O2-induced oxidative injury via nestin association with Cdk5 [99] and nestin-depleted cells have increased predisposition to H2O2-triggered apoptosis [100]. Similarly, lamin A patient mutations and cells lacking full-length lamin B show significant increase in reactive oxygen species and sensitivity to oxidative stress [101, 102]. K8 and K18 may also protect by sequestering oxidized proteins such as the GTPase Ran [103]. In addition, livers that express a mutant K18 upregulate several oxidative-stress-related genes, leading to the formation of lipid and protein oxidation by-products [103]. However, knockdown of K8 in a tumor cell line protects the cells from peroxide-mediated injury [104] though this is likely to be a unique situation that is related to the transformed nature of the cells or to potential off-target knock-down effects. A common feature of IFs upon toxin and oxidative injury exposures is their hyperphosphorylation, which serves a protective role [14].
IFs and inflammatory signals
A potential role for IFs in modulating inflammatory signals is an exciting possibility that warrants further study particularly given the accumulating observations that inflammatory cytokines can modulate IF expression. For example, IL-1, IFNγ, TGFβ and TGFα induce K6, K16, and K17 [105], and IL-6 induces keratin expression in the intestine [106] and promote keratin reorganization in human epidermal keratinocytes [107]. TLR ligand (LPS) and certain (e.g., IL-1, TNFα) but not other (IL-4) cytokines induce GFAP in enteric glia [108]. Of note, HSPs modulate the immune response by playing important roles in antigen presentation and leukocyte activation [109] and are induced by several cytokines [110], but it is unknown whether HSPs have bona fide cytokine properties [111]. IFs may have direct immunomodulatory effects via their role in protein targeting as shown by reduced leukocyte homing to secondary lymphoid tissues associated with abnormal surface expression and distribution of adhesion molecules in vimentin-null leukocytes and endothelial cells [112].
IFs, organelle homeostasis and protein targeting
There is significant emerging evidence that IFs modulate organelle topography, shape and function as well as help target proteins in cells [71, 113, 114]. This is well-characterized for mitochondria, in particular, using knock-out mice for desmin in cardiac muscle [71], K19 in skeletal muscle [115] and K8 in liver [75] where mitochondria are found mislocalized, fragmented, or functionally altered. Abnormal mitochondrial morphology and organization have also been described in vimentin-depleted cells [116] and in K18-mutant (R90C) transfected cells [117]. In addition, transfected cells that express NF-L mutation that causes human Charcot-Marie-Tooth disease have defects in mitochondrial transport and fragmentation of the Golgi apparatus [118]. IFs also impact subcellular organelles other than mitochondria. For example, the positioning, sorting or movement of lysosomes or the crosstalk between lysosomes and endosomes is impacted by vimentin [114] desmin [71], and NF [119]; and vimentin and keratins are important for the transport of melanosomes [120, 121]. Furthermore, nuclear lamin A mutations cause a variety of organelle phenotypes, such as enlarged rough endoplasmic reticulum and mitochondria in mice, and gap junctional protein mislocalization in myocytes [122]. Lamin A/C-null mice have abnormally shaped skeletal and myocardial nuclei with redistribution of nuclear protein to cytoplasmic and ER locations [123, 124], a phenotype also seen in fibroblasts of patients with lamin A/C mutations [125]. How IFs target organelles and the molecular connections that may be involved are poorly understood but some candidate proteins are emerging. For example, disruption between keratins and their IFAPs Pirh2 [126] and plectin [127] results in abnormal mitochondrial distribution.
How specific diseases unmask the importance of IFs as stress proteins
The cytoprotective role of IFs is evidenced by the fact that IF variants cause or predispose to more than 80 human diseases [11]. Importantly, IF and HSP mutations can phenocopy the same disease, such as Charcot-Marie-Tooth which is caused by NF-L or Hspb1 mutations, desmin-related myopathy which is caused by desmin or αB-crystallin variants [48, 128], and cataracts which are caused by mutations in Bfsp1 and Bfsp2 IFs or αB-crystallin [129] (Box 1). Similar to HSPs [6], IFs are altered in several diseases, including neurological (elevated GFAP [20]), renal (de novo vimentin expression [130]) or liver disorders (elevated K8 and K18 in patients with primary biliary cirrhosis; de novo K7 and K19 expression in cholestatic diseases [11]). Also, IF-containing cytoplasmic inclusions are hallmarks of multiple muscle, neural and liver disorders [21, 39, 84]. Dysregulated IF expression and hyperphosphorylation, coupled with IF crosslinking by transglutaminase-2 (studied only in the case of keratin-containing inclusions), likely facilitate aggregate formation [21, 39, 84]. The interrelationship between HSPs and IFs in inclusion formation remains to be investigated.
Regulation of IF induction
Stress induces IF transcription and translation (Table 1) and several transcription factors have been linked to the regulation of IF genes, either constitutively or after stress (Supplementary Material Table S2), but current molecular knowledge is limited when compared to the HSP family. The presence of traditional stress-induced transcription factor binding sites, such as AP1/AP2, Sp1/Sp2, c/EBP as well as NK-κB, upstream of most studied IF genes (Table S2) has been reported. However, IF gene transcription has not been linked to the heat-induced HSF/HSE (with the exception of HSF4 and Bfsp1 and Bfsp2, Table S2) which may be tightly regulated to control chaperone needs and not part of the IF stress response. IF regulation may entail different transcriptional machineries, even if parallel induction of some IFs and HSPs are observed (Fig.2). IF levels may also be regulated by microRNA (miRNA), as exemplified for vimentin and HSP20 (which belong to the mirR-30 family of miRNAs) that are upregulated in mice with proteinuria caused by selective podocyte inactivation of Dicer, the enzyme that generates miRNA, and consequently failed to activate vimentin and HSP20 mi-RNAs [131]. IF upregulation can also stem from IF stabilization and decreased IF degradation, as noted in patients with primary biliary chirrosis where increased IF protein, but not mRNA, was detected [132]. However, this finding may be related to transient mRNA induction and translation with remaining high IF protein levels due to the known long turnover of IFs except in cases where there is recovery from injury [17, 133]. In addition, epithelial and hair keratins are regulated by several endocrine signals such as glucocorticoids, vitamins and hormones. Accordingly, keratin promoter regions harbor response elements for thyroid responsive element in K5, vitamin D response elements in K15, androgen transactivatable-response element in hair K37, among others [134]. Clearly more work needs to focus on IF transcriptional regulation, as well as how IF mRNAs are targeted for translation [135] to different intracellular localization as might take place during the de novo expression of K19 and K20 in regenerating pancreata (Fig.2; though this hypothesis remains to be tested).
Utility of IFs as stress sensors and therapeutic targets
IFs serve as useful markers that can potentially tag the origin of metastatic tumors or serve as tumor/disease serum (or other fluid) markers [11]. Examples include GFAP in cerebrospinal fluid from patients with neuropathies or keratins in various liver disorders [11, 20]. In addition, IF caspase-generated cleavage products have potential clinical applications [11] and cell biologic utility as readily measurable markers of apoptosis (e.g., in media of cultured cells [11, 136]). IF phosphorylation-directed antibodies [14] also represent potential clinically-useful stress detection reagents, as shown for phospho-K8/K18 in IL-10-treated hepatitis C patients [137]. Similar to HSPs, whose antigenic properties are firmly established [111], IF auto-antibodies are detected in several diseases including rheumatoid arthritis (citrullinated vimentin) [138], tick-borne encephalitis (NFs) [139] and various liver diseases (K8/K18) [39].
The upregulation of IFs has potential therapeutic advantages. This possibility takes advantage of the relative functional redundancy of IFs, particularly keratins. To that end, induction by hormones or other readily available nontoxic natural products, such as the reported induction of K15 [140] or K17 [141], could compensate for mutant K14 in the blistering skin disease epidermolysis bullosa simplex and may hold promise as potential therapy of some dominant keratin disorders [134]. On the other hand, the commonly used anti-inflammatory glucocorticoids inhibit keratin expression, which in turn is upregulated during inflammatory skin response and wound healing [134]. Therefore, approaches aimed at upregulating keratin expression are attractive avenues to pursue for the treatment of some of the keratin disorders.
Overview of how IFs serve as stress proteins
The mechanism of cytoprotection offered by the IF cytoskeleton is likely to be multi-factorial and to be IF/tissue-specific. The IF stress-response is reflected by IF upregulation (Table 1, Table S1), IF PTMs (with phosphorylation being the best studied) and IFAPs, changes in IF solubility or organization, and IF degradation or aggregate formation (Fig.1,3). IF overexpression in response to stress leads to new filament formation and generation of additional soluble subunit pools available to buttress existing filaments and other IF subcellular compartments and protect from stress [55].
Fig.3. Specific stresses upregulate IFs and HSPs.

Environmental or internal stresses (see Fig.1) initiate stress signaling cascades, which activate the stress response and transcriptional machineries that induce the expression of the classical stress-induced HSP genes (constitutive HSP genes are not shown). Stress also induces the transcription and translation of constitutive/resident IF genes (shown in green circles) nonresident IF genes (shown in blue circles) and the nuclear lamin IFs (shown in orange). The increased expression of IFs provides pools for new filament assembly (not shown), binding to IFAPs (exemplified by the established binding with HSPs, red triangles). IF posttranslational modifications (e.g., phosphorylation (P)), serve important functions in IF filament dynamics, binding to IFAPs and targeting for degradation initially or after recovery from stress. Exposure to chronic stress, in the appropriate context, leads to inclusion body formation which also include HSPs and other chaperones. These alterations in IF homeostasis and dynamics help promote the cellular response which allows cells to migrate, divide, differentiate, or die.
The mechanisms by which the above alteration in IFs in response to stress impart a protective role include: (i) providing a mechanical support basally and during recovery from injury; (ii) serving as a scaffold that binds a variety of proteins including kinases, adaptor proteins such as 14-3-3, HSPs such as HSP70; (iii) serving as anti-apoptotic proteins; (iv) regulating organelle functions; and (v) providing a phosphate buffer or sponge to contain unwarranted phosphorylation of proapoptotic proteins that are activated by phosphorylation. Three of these five general mechanisms (scaffolding; anti-apoptosis; organelles) are shared by HSPs (Box 2) although the details of how HSPs carry out this role are generally different. For example, HSP70 inhibits apoptosis by preventing procaspases from assembling into a functional apoptosome, and by binding proapototic proteins such as p53 and c-myc [142] (see also IFs and apoptotic stress section). We consider HSPs regulators of organelle stress because of the unique HSPs that are found in mitochondria (Hsp60) and the ER (GRP78) (Box 1). We also consider HSPs as scaffolding proteins given their abundance and the numerous binding partners they interact with (e.g. [143]). The mechanical and phosphate sponge property of IFs is generally not shared by HSPs. In contrast, HSPs have the unique property of chaperoning and protein folding that is not shared by IFs.
Conclusions and open questions
Similar to HSPs, IF levels increase dramatically (3-4 fold despite their baseline abundance) in response to stress though it is unknown whether IF overexpression per se protects from stress (akin to HSPs) and/or promotes a regenerative response. However, it is clear that genetic ablation or mutation of IFs renders cells in most cases highly susceptible to a variety of stresses, serving as one line of evidence that normal wild type IFs are needed to cope with stress. In general terms, IF overexpression in the numerous mouse models that have been developed to date has either been well-tolerated (eg, K8, K18 or K19) [39] or has afforded a disease model (eg, NF or GFAP overexpression) [20] and is therefore toxic. Hence, it appears that IF overexpression in vivo needs to be context-dependent for it to be protective, and the molecular consequences for IF upregulation (be it forced or due physiologic/pathologic causes) remain to be defined.
IFs also likely provide a scaffold to promote recovery after injury by binding to a broad range of signaling proteins and to HSPs. In addition, IF phosphorylation plays a key role in cytoprotection by regulating IF-IFAP interactions and by serving as a sponge for excess undesired stress-associated cellular hyperphosphorylation. Little is known regarding IF-IFAP interactions during stress and the likely crosstalk between HSPs and IFs. In addition, the regulation of IF expression remains an understudied area though a better understanding of the regulation of IF expression is likely to have potential therapeutic benefit, particularly in the case of keratins where upregulation of an alternate normal keratin can mask the detrimental effects of a mutant keratin. It is possible that therapeutic upregulation of an HSP may be protective for at least some of the IF-pathies, given the shared cytoprotective roles of IFs and HSPs and the fact that some of their diseases phenocopy each other. In addition, careful experimental assessment of the time course of IF overexpression is warranted, including correlating the overexpression with HSP induction (both in time and space) in order to better understand whether IFs protect from injury and/or promote recovery from injury. Our aim in this review is to turn the spotlight on IFs as stress proteins and to highlight shared and unique IF features with HSPs. Considering the scaffolding and mechanical function of IFs, it is easy to opine that “adding structure to life helps when you are stressed out.”
Supplementary Material
Acknowledgments
We thank Kris Morrow for figure preparation and colleagues in Strnad laboratory for generating Figure 2. We also thank Roy Quinlan for sharing with us an unpublished review he is authoring and Lea Sistonen for helpful discussions and comments. Due to space limitations, we apologize for not being able to include as many original references as we would have liked. Our work is supported by The Academy of Finland, Juselius foundation, European Union FP7 IRG, Foundation “Liv och hälsa” (D.M.T.); the German Research Foundation (grants STR 1095/1-1 and 2-1 to P.S.); NIH grant DK069385 (A.H.); NIH grants DK47918 and DK52951, and a Department of Veteran Affairs Merit Award (M.B.O.).
Box 1
Major classes of mammalian heat shock proteins*
During their lifespan, cells are continuously subjected to processes leading to modification of the intracellular milieu and consequent damage of cellular components such as DNA or protein [1, 2]. While these potentially harmful conditions are manifold including mechanical, chemical and biological stressors, the cellular response to the damage is conserved and consists of transient down-regulation of housekeeping genes together with activation of stress-responsive genes [2]. The response to a heat shock exposure serves as a prototypical stress response and leads to a rapid induction of several heat shock proteins (HSP) described in Table I, which are considered the classic stress proteins. Given the universality of the stress response as well as the general cytoprotective function of most stress genes, an exposure to one form of the injury offers a cross-protective effect against other challenges [3].
Table I.
Functions and disease associations of major classes of HSPs
| Class | HSP | Alternative names |
Functions | Human disease association |
|---|---|---|---|---|
| Small Hsp (HspB) family Unable to re-fold proteins, but holds them in non-aggregated state [4] | Hspb1 | HSP27; HSP25 | Inducible; protect from oxidative stress and protein aggregation [4] | Mutations lead to neuropathies (Charcot-Marie-Tooth); found in multiple human inclusion bodies [4] |
| Hspb5 | αB-Crystallin | Inducible; protect from heat/oxidative stress, apoptosis, and protein aggregation [4] | Mutations lead to (cardio)myopathies and cataracts; found in multiple human inclusion bodies [4] | |
| Hspb8 | HSP22; H11; E2IG1 | Inducibility cell type- and context-dependent, prevents aggregation of misfolded proteins, helps targeting them to macroautophagy [5, 6] | Mutations lead to neuropathies (e.g. Charcot-Marie-Tooth, Distal hereditary motor neuropathy)[5, 6] | |
| DNAJ (Hsp 40) family Co-chaperones, modify the interaction of Hsp70s with their substrates [7] | DNAJB1 | HSP40 | Inducible [7], involved in protein degradation [8] | Protects against neuronal inclusion body formation [8] |
| Hsp70 (HspA) family The only chaperones re-folding proteins on their own, central organizers of chaperone network, distribute proteins to downstream chaperones (Hsp60,90) [9] | Hspa1a | HSP70-1; HSP72 | Classical inducible Hsp70, protects from heat shock, UV, oxidative, osmotic stress, ischemia, pancreatitis, inclusion body formation [3, 10, 11]; extracellular Hsp72 acts as immunologic “danger signal” [12] | Increases after exercise [12]; Inducibility ameliorated during aging, Hsp72 is part of human inclusion bodies [1, 11] |
| Hspa5 | GRP78, Bip | Essential for survival, protein folding in ER, upregulated in ER stress response [10, 13] | Cleaved in haemolytic-uremic syndrome (HUS), function compromised in Marinesco-Sjörgen syndrome [13] | |
| Hspa8 | HSC70 | Constitutively expressed; essential for survival; involved in housekeeping functions as co-translational protein folding and translocation [7]; protection from apoptosis and protein aggregation [10] | ||
| Hsp90 (HspC) family “evolutionary capacitators”- assisting “difficult”(meta-stable) proteins to maintain correct conformation or organizing their degradation. Hsp90 uncovers “cryptic” phenotypes (genotoxic stress) [14] Assist in folding/production of viral proteins, increases virulence of pathogens, participates in degradation of human inclusions# | Hspc1 | HSP90AA1, HSP90A | Inducible, essential for survival [14] | |
| Hspc3 | HSP90AB1, HSP90B; HSP90-BETA | Constitutive, essential for survival [14] | ||
| Hsp110 (HspH) family co-chaperones, nucleotide exchange factor for Hsp70 [8] | HspH1 | Hsp105 | Suppresses protein aggregation (holding function) [8] | |
| Chaperonins Large cylindrical complexes which enclose their substrates during folding | Hspd1 | CPN60; GROEL; Hsp60 | Inducible, essential for cell survival, protection from stressful conditions including heat stress; extracellular Hsp60 acts as a danger signal [15] | Mutations lead to hereditary spastic paraplegia [1, 15]; strong immunogen, implicated in infectious, (auto)immune or prion diseases [15] |
See Suppl References for references related to Box 1.
Box 2
Characteristics of IFs in stress protection, and general cytoprotective mechanisms of HSPs and IFs
Characteristics that render IFs bona fide stress proteins:
IFs protect cells from mechanical stress. The role of HSPs in mechanical stress has not been formally tested.
Differential IF tissue expression provides specialized IFs that allow coping with stress in different tissues. Some of the HSPs have unique subcellular localization (Box 1).
IFs are highly abundant proteins (even more so than HSPs).
Resident/constitutive and de-novo IF proteins are upregulated (at RNA and/or protein levels) after a variety of stress conditions and during recovery from stress (similar to HSPs).
Modest IF overexpression in animal models and cell lines is generally not harmful. However, significant IF overexpression can be toxic to cells (this is different to HSPs).
IF mutations in humans cause numerous diseases some of which are phenocopied by HSP mutations.
IF knockout or mutation in animal models cause disease or render animals markedly susceptible to various types of stress. This feature is similar to what is seen with HSPs.
IF filament organization is highly dynamic and is often altered upon stress.
IFs bind to and scaffold several stress signaling molecules and regulate their availability and activity. This includes physical association with HSPs.
IFs and HSPs aggregate together as inclusions in several chronic human diseases.
IFs are major substrates for stress kinases. Ablation of IF phosphorylation by mutation renders cells highly susceptible to apoptosis. Both IFs and HSPs are anti-apoptotic.
IFs play an important role in the function of several organelles. Some of the HSPs also have unique organelle functions as shown in Table II.
Table II. Shared and unique cytoprotective general mechanisms of HSPs and IFs*.
| HSPs | IFs | |
|---|---|---|
| Inhibition of apoptosis | + | + |
| Organelle functions | + | + |
| Scaffolding | + | + |
| Mechanical | - | + |
| Phosphate sponge (or kinase buffer) | - | + |
| Chaperone/protein folding | + | - |
Some of the mechanisms may overlap (e.g., the role of mitochondria in apoptosis)
Footnotes
Please see separate files for: Supplemental Tables S1 and S2 and their references; and Supplemental References for Boxes 1 and 2.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Morimoto RI, Santoro MG. Stress-inducible responses and heat shock proteins: new pharmacologic targets for cytoprotection. Nat Biotechnol. 1998;16(9):833–8. doi: 10.1038/nbt0998-833. [DOI] [PubMed] [Google Scholar]
- 2.Kampinga HH, et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14(1):105–11. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akerfelt M, et al. Heat shock factors at a crossroad between stress and development. Ann N Y Acad Sci. 2007;1113:15–27. doi: 10.1196/annals.1391.005. [DOI] [PubMed] [Google Scholar]
- 4.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22(11):1427–38. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Noort JM. Stress proteins in CNS inflammation. J Pathol. 2008;214(2):267–75. doi: 10.1002/path.2273. [DOI] [PubMed] [Google Scholar]
- 6.Calderwood SK, et al. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci. 2006;31(3):164–72. doi: 10.1016/j.tibs.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Biamonti G, Caceres JF. Cellular stress and RNA splicing. Trends Biochem Sci. 2009;34(3):146–53. doi: 10.1016/j.tibs.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 8.Cully M, Downward J. Translational responses to growth factors and stress. Biochem Soc Trans. 2009;37(Pt 1):284–8. doi: 10.1042/BST0370284. [DOI] [PubMed] [Google Scholar]
- 9.Szeverenyi I, et al. The Human Intermediate Filament Database: comprehensive information on a gene family involved in many human diseases. Hum Mutat. 2008;29(3):351–60. doi: 10.1002/humu.20652. [DOI] [PubMed] [Google Scholar]
- 10.Herrmann H, et al. Intermediate filaments: primary determinants of cell architecture and plasticity. J Clin Invest. 2009;119(7):1772–83. doi: 10.1172/JCI38214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Omary MB. “IF-pathies”: a broad spectrum of intermediate filament-associated diseases. J Clin Invest. 2009;119(7):1756–62. doi: 10.1172/JCI39894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Green KJ, et al. Intermediate filament associated proteins. Adv Protein Chem. 2005;70:143–202. doi: 10.1016/S0065-3233(05)70006-1. [DOI] [PubMed] [Google Scholar]
- 13.Pallari HM, Eriksson JE. Intermediate filaments as signaling platforms. Sci STKE. 2006;2006(366):pe53. doi: 10.1126/stke.3662006pe53. [DOI] [PubMed] [Google Scholar]
- 14.Omary MB, et al. “Heads and tails” of intermediate filament phosphorylation: multiple sites and functional insights. Trends Biochem Sci. 2006;31(7):383–94. doi: 10.1016/j.tibs.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 15.Marceau N, et al. Dual roles of intermediate filaments in apoptosis. Exp Cell Res. 2007;313(10):2265–81. doi: 10.1016/j.yexcr.2007.03.038. [DOI] [PubMed] [Google Scholar]
- 16.Ku NO, Omary MB. Keratins turn over by ubiquitination in a phosphorylation-modulated fashion. Journal of Cell Biology. 2000;149(3):547–52. doi: 10.1083/jcb.149.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong B, et al. Organ-specific stress induces mouse pancreatic keratin overexpression in association with NF-kappaB activation. J Cell Sci. 2004;117(Pt 9):1709–19. doi: 10.1242/jcs.01016. [DOI] [PubMed] [Google Scholar]
- 18.Goldman RD, et al. Intermediate filaments: versatile building blocks of cell structure. Curr Opin Cell Biol. 2008;20(1):28–34. doi: 10.1016/j.ceb.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toivola DM, et al. Keratin overexpression levels correlate with the extent of spontaneous pancreatic injury. Am J Pathol. 2008;172(4):882–92. doi: 10.2353/ajpath.2008.070830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liem RK, Messing A. Dysfunctions of neuronal and glial intermediate filaments in disease. J Clin Invest. 2009;119(7):1814–24. doi: 10.1172/JCI38003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zatloukal K, et al. From Mallory to Mallory-Denk bodies: what, how and why? Exp Cell Res. 2007;313(10):2033–49. doi: 10.1016/j.yexcr.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 22.Petrak J, et al. Deja vu in proteomics. A hit parade of repeatedly identified differentially expressed proteins. Proteomics. 2008;8(9):1744–9. doi: 10.1002/pmic.200700919. [DOI] [PubMed] [Google Scholar]
- 23.Coulombe PA, Kerns ML, Fuchs E. Epidermolysis bullosa simplex: a paradigm for disorders of tissue fragility. J Clin Invest. 2009;119(7):1784–93. doi: 10.1172/JCI38177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toivola DM, et al. Keratins provide virus-dependent protection or predisposition to injury in coxsackievirus-induced pancreatitis. Cell Health and Cytoskeleton. 2009;1:51–64. [Google Scholar]
- 25.Widestrand A, et al. Increased neurogenesis and astrogenesis from neural progenitor cells grafted in the hippocampus of GFAP-/- Vim-/- mice. Stem Cells. 2007;25(10):2619–27. doi: 10.1634/stemcells.2007-0122. [DOI] [PubMed] [Google Scholar]
- 26.Goldbaum O, et al. The small heat shock protein HSP25 protects astrocytes against stress induced by proteasomal inhibition. Glia. 2009 doi: 10.1002/glia.20870. [DOI] [PubMed] [Google Scholar]
- 27.Bechtold DA, Brown IR. Induction of Hsp27 and Hsp32 stress proteins and vimentin in glial cells of the rat hippocampus following hyperthermia. Neurochem Res. 2003;28(8):1163–73. doi: 10.1023/a:1024268126310. [DOI] [PubMed] [Google Scholar]
- 28.Vilaboa NE, et al. Heat-shock and cadmium chloride increase the vimentin mRNA and protein levels in U-937 human promonocytic cells. J Cell Sci. 1997;110(Pt 2):201–7. doi: 10.1242/jcs.110.2.201. [DOI] [PubMed] [Google Scholar]
- 29.Fisher BR, Heredia DJ, Brown KM. Heat-induced alterations in embryonic cytoskeletal and stress proteins precede somite malformations in rat embryos. Teratog Carcinog Mutagen. 1996;16(1):49–64. doi: 10.1002/(SICI)1520-6866(1996)16:1<49::AID-TCM6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 30.Dynlacht JR, et al. Lamin B is a prompt heat shock protein. J Cell Physiol. 1999;178(1):28–34. doi: 10.1002/(SICI)1097-4652(199901)178:1<28::AID-JCP4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 31.Haddad N, Paulin-Levasseur M. Effects of heat shock on the distribution and expression levels of nuclear proteins in HeLa S3 cells. J Cell Biochem. 2008;105(6):1485–500. doi: 10.1002/jcb.21968. [DOI] [PubMed] [Google Scholar]
- 32.Cheng TJ, Lai YK. Transient increase in vimentin phosphorylation and vimentin-HSC70 association in 9L rat brain tumor cells experiencing heat-shock. J Cell Biochem. 1994;54(1):100–9. doi: 10.1002/jcb.240540111. [DOI] [PubMed] [Google Scholar]
- 33.Liao J, Lowthert LA, Omary MB. Heat stress or rotavirus infection of human epithelial cells generates a distinct hyperphosphorylated form of keratin 8. Experimental Cell Research. 1995;219(2):348–57. doi: 10.1006/excr.1995.1238. [DOI] [PubMed] [Google Scholar]
- 34.Shyy TT, Asch BB, Asch HL. Concurrent collapse of keratin filaments, aggregation of organelles, and inhibition of protein synthesis during the heat shock response in mammary epithelial cells. J Cell Biol. 1989;108(3):997–1008. doi: 10.1083/jcb.108.3.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tseng WC, et al. Redistribution of GFAP and alphaB-crystallin after thermal stress in C6 glioma cell line. J Biomed Sci. 2006;13(5):681–94. doi: 10.1007/s11373-006-9091-9. [DOI] [PubMed] [Google Scholar]
- 36.Welch WJ, Suhan JP. Morphological study of the mammalian stress response: characterization of changes in cytoplasmic organelles, cytoskeleton, and nucleoli, and appearance of intranuclear actin filaments in rat fibroblasts after heat-shock treatment. J Cell Biol. 1985;101(4):1198–211. doi: 10.1083/jcb.101.4.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee SY, et al. Rac1 regulates heat shock responses by reorganization of vimentin filaments: identification using MALDI-TOF MS. Cell Death Differ. 2001;8(11):1093–102. doi: 10.1038/sj.cdd.4400923. [DOI] [PubMed] [Google Scholar]
- 38.Lee YJ, et al. Heat-resistant variants of the Chinese hamster ovary cell: alteration of cellular structure and expression of vimentin. J Cell Physiol. 1992;151(1):138–46. doi: 10.1002/jcp.1041510118. [DOI] [PubMed] [Google Scholar]
- 39.Omary MB, et al. Toward unraveling the complexity of simple epithelial keratins in human disease. J Clin Invest. 2009;119(7):1794–805. doi: 10.1172/JCI37762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Izawa I, et al. Identification of Mrj, a DnaJ/Hsp40 family protein, as a keratin 8/18 filament regulatory protein. J Biol Chem. 2000;275(44):34521–7. doi: 10.1074/jbc.M003492200. [DOI] [PubMed] [Google Scholar]
- 41.Liao J, et al. The 70-kDa heat shock proteins associate with glandular intermediate filaments in an ATP-dependent manner. Journal of Biological Chemistry. 1995;270(2):915–22. doi: 10.1074/jbc.270.2.915. [DOI] [PubMed] [Google Scholar]
- 42.Perng MD, et al. Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J Cell Sci. 1999;112(Pt 13):2099–112. doi: 10.1242/jcs.112.13.2099. [DOI] [PubMed] [Google Scholar]
- 43.Muchowski PJ, Valdez MM, Clark JI. AlphaB-crystallin selectively targets intermediate filament proteins during thermal stress. Invest Ophthalmol Vis Sci. 1999;40(5):951–8. [PubMed] [Google Scholar]
- 44.Djabali K, et al. AlphaB-crystallin interacts with intermediate filaments in response to stress. J Cell Sci. 1997;110(Pt 21):2759–69. doi: 10.1242/jcs.110.21.2759. [DOI] [PubMed] [Google Scholar]
- 45.Perng MD, et al. Glial fibrillary acidic protein filaments can tolerate the incorporation of assembly-compromised GFAP-delta, but with consequences for filament organization and alphaB-crystallin association. Mol Biol Cell. 2008;19(10):4521–33. doi: 10.1091/mbc.E08-03-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bornheim R, et al. A dominant vimentin mutant upregulates Hsp70 and the activity of the ubiquitin-proteasome system, and causes posterior cataracts in transgenic mice. J Cell Sci. 2008;121(Pt 22):3737–46. doi: 10.1242/jcs.030312. [DOI] [PubMed] [Google Scholar]
- 47.Hageman J, Kampinga HH. Computational analysis of the human HSPH/HSPA/DNAJ family and cloning of a human HSPH/HSPA/DNAJ expression library. Cell Stress Chaperones. 2009;14(1):1–21. doi: 10.1007/s12192-008-0060-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhai J, et al. Disruption of neurofilament network with aggregation of light neurofilament protein: a common pathway leading to motor neuron degeneration due to Charcot-Marie-Tooth disease-linked mutations in NFL and HSPB1. Hum Mol Genet. 2007;16(24):3103–16. doi: 10.1093/hmg/ddm272. [DOI] [PubMed] [Google Scholar]
- 49.Pekny M, Lane EB. Intermediate filaments and stress. Exp Cell Res. 2007;313(10):2244–54. doi: 10.1016/j.yexcr.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 50.Kim S, Coulombe PA. Intermediate filament scaffolds fulfill mechanical, organizational, and signaling functions in the cytoplasm. Genes Dev. 2007;21(13):1581–97. doi: 10.1101/gad.1552107. [DOI] [PubMed] [Google Scholar]
- 51.Wagner OI, et al. Softness, strength and self-repair in intermediate filament networks. Exp Cell Res. 2007;313(10):2228–35. doi: 10.1016/j.yexcr.2007.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sivaramakrishnan S, et al. Micromechanical properties of keratin intermediate filament networks. Proc Natl Acad Sci U S A. 2008;105(3):889–94. doi: 10.1073/pnas.0710728105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ridge KM, et al. Keratin 8 phosphorylation by protein kinase C delta regulates shear stress-mediated disassembly of keratin intermediate filaments in alveolar epithelial cells. J Biol Chem. 2005;280(34):30400–5. doi: 10.1074/jbc.M504239200. [DOI] [PubMed] [Google Scholar]
- 54.Jaitovich A, et al. Ubiquitin-proteasome-mediated degradation of keratin intermediate filaments in mechanically stimulated A549 cells. J Biol Chem. 2008;283(37):25348–55. doi: 10.1074/jbc.M801635200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flitney EW, et al. Insights into the mechanical properties of epithelial cells: the effects of shear stress on the assembly and remodeling of keratin intermediate filaments. FASEB J. 2009;23(7):2110–9. doi: 10.1096/fj.08-124453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sivaramakrishnan S, et al. Shear stress induced reorganization of the keratin intermediate filament network requires phosphorylation by protein kinase C zeta. Mol Biol Cell. 2009;20(11):2755–65. doi: 10.1091/mbc.E08-10-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bordeleau F, et al. Keratin contribution to cellular mechanical stress response at focal adhesions as assayed by laser tweezers. Biochem Cell Biol. 2008;86(4):352–9. doi: 10.1139/o08-076. [DOI] [PubMed] [Google Scholar]
- 58.Yano S, et al. Mechanical stretching in vitro regulates signal transduction pathways and cellular proliferation in human epidermal keratinocytes. J Invest Dermatol. 2004;122(3):783–90. doi: 10.1111/j.0022-202X.2004.22328.x. [DOI] [PubMed] [Google Scholar]
- 59.Su Y, Kondrikov D, Block ER. Cytoskeletal regulation of nitric oxide synthase. Cell Biochem Biophys. 2005;43(3):439–49. doi: 10.1385/CBB:43:3:439. [DOI] [PubMed] [Google Scholar]
- 60.Eriksson JE, et al. Introducing intermediate filaments: from discovery to disease. J Clin Invest. 2009;119(7):1763–71. doi: 10.1172/JCI38339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Loufrani L, Henrion D. Role of the cytoskeleton in flow (shear stress)-induced dilation and remodeling in resistance arteries. Med Biol Eng Comput. 2008;46(5):451–60. doi: 10.1007/s11517-008-0306-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsuruta D, Jones JC. The vimentin cytoskeleton regulates focal contact size and adhesion of endothelial cells subjected to shear stress. J Cell Sci. 2003;116(Pt 24):4977–84. doi: 10.1242/jcs.00823. [DOI] [PubMed] [Google Scholar]
- 63.Philip JT, Dahl KN. Nuclear mechanotransduction: response of the lamina to extracellular stress with implications in aging. J Biomech. 2008;41(15):3164–70. doi: 10.1016/j.jbiomech.2008.08.024. [DOI] [PubMed] [Google Scholar]
- 64.Worman HJ, et al. Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest. 2009;119(7):1825–36. doi: 10.1172/JCI37679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.DePianto D, Coulombe PA. Intermediate filaments and tissue repair. Exp Cell Res. 2004;301(1):68–76. doi: 10.1016/j.yexcr.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 66.Shibuya S, et al. Embryonic intermediate filament, nestin, expression following traumatic spinal cord injury in adult rats. Neuroscience. 2002;114(4):905–16. doi: 10.1016/s0306-4522(02)00323-8. [DOI] [PubMed] [Google Scholar]
- 67.Beaulieu JM, Kriz J, Julien JP. Induction of peripherin expression in subsets of brain neurons after lesion injury or cerebral ischemia. Brain Res. 2002;946(2):153–61. doi: 10.1016/s0006-8993(02)02830-5. [DOI] [PubMed] [Google Scholar]
- 68.Kriz J, et al. Up-regulation of peripherin is associated with alterations in synaptic plasticity in CA1 and CA3 regions of hippocampus. Neurobiol Dis. 2005;18(2):409–20. doi: 10.1016/j.nbd.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 69.Fornaro M, et al. Neuronal intermediate filament expression in rat dorsal root ganglia sensory neurons: an in vivo and in vitro study. Neuroscience. 2008;153(4):1153–63. doi: 10.1016/j.neuroscience.2008.02.080. [DOI] [PubMed] [Google Scholar]
- 70.Franco R, et al. Environmental toxicity, oxidative stress and apoptosis: menage a trois. Mutat Res. 2009;674(12):3–22. doi: 10.1016/j.mrgentox.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 71.Capetanaki Y, et al. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp Cell Res. 2007;313(10):2063–76. doi: 10.1016/j.yexcr.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 72.Leifeld L, et al. Keratin 18 provides resistance to Fas-mediated liver failure in mice. Eur J Clin Invest. 2009;39(6):481–8. doi: 10.1111/j.1365-2362.2009.02133.x. [DOI] [PubMed] [Google Scholar]
- 73.Caulin C, et al. Keratin-dependent, epithelial resistance to tumor necrosis factor- induced apoptosis. J Cell Biol. 2000;149(1):17–22. doi: 10.1083/jcb.149.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Inada H, et al. Keratin attenuates tumor necrosis factor-induced cytotoxicity through association with TRADD. J Cell Biol. 2001;155(3):415–26. doi: 10.1083/jcb.200103078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tao GZ, et al. Keratins modulate the shape and function of hepatocyte mitochondria: a mechanism for protection from apoptosis. 2009 doi: 10.1242/jcs.051862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ku NO, Omary MB. A disease- and phosphorylation-related nonmechanical function for keratin 8. J Cell Biol. 2006;174(1):115–25. doi: 10.1083/jcb.200602146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tong X, Coulombe PA. Keratin 17 modulates hair follicle cycling in a TNFalpha-dependent fashion. Genes Dev. 2006;20(10):1353–64. doi: 10.1101/gad.1387406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Panagopoulou P, et al. Desmin mediates TNF-alpha-induced aggregate formation and intercalated disk reorganization in heart failure. J Cell Biol. 2008;181(5):761–75. doi: 10.1083/jcb.200710049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schietke R, et al. Mutations in vimentin disrupt the cytoskeleton in fibroblasts and delay execution of apoptosis. Eur J Cell Biol. 2006;85(1):1–10. doi: 10.1016/j.ejcb.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 80.Russell D, Ross H, Lane EB. ERK Involvement in Resistance to Apoptosis in Keratinocytes with Mutant Keratin. J Invest Dermatol. 2009 doi: 10.1038/jid.2009.327. [DOI] [PubMed] [Google Scholar]
- 81.Liu QL, et al. Heat shock protein 70 binds caspase-activated DNase and enhances its activity in TCR-stimulated T cells. Blood. 2003;102(5):1788–96. doi: 10.1182/blood-2002-11-3499. [DOI] [PubMed] [Google Scholar]
- 82.Garrido C, et al. Heat shock proteins: endogenous modulators of apoptotic cell death. Biochem Biophys Res Commun. 2001;286(3):433–42. doi: 10.1006/bbrc.2001.5427. [DOI] [PubMed] [Google Scholar]
- 83.Strnad P, et al. Keratin mutation predisposes to mouse liver fibrosis and unmasks differential effects of the carbon tetrachloride and thioacetamide models. Gastroenterology. 2008;134(4):1169–79. doi: 10.1053/j.gastro.2008.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Strnad P, et al. Mallory-Denk-bodies: lessons from keratin-containing hepatic inclusion bodies. Biochim Biophys Acta. 2008;1782(12):764–74. doi: 10.1016/j.bbadis.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 85.Cadrin M, et al. Early perturbations in keratin and actin gene expression and fibrillar organisation in griseofulvin-fed mouse liver. J Hepatol. 2000;33(2):199–207. doi: 10.1016/s0168-8278(00)80360-8. [DOI] [PubMed] [Google Scholar]
- 86.Zou J, et al. Upregulation of nestin, vimentin, and desmin in rat podocytes in response to injury. Virchows Arch. 2006;448(4):485–92. doi: 10.1007/s00428-005-0134-9. [DOI] [PubMed] [Google Scholar]
- 87.Rauscher A, et al. Similar effects of electroporational stress and treatment with the phorbol ester 12-O-tetradecanoylphorbol-13-acetate on vimentin expression in mouse plasmacytoma cells. Biochim Biophys Acta. 2000;1493(12):170–9. doi: 10.1016/s0167-4781(00)00184-6. [DOI] [PubMed] [Google Scholar]
- 88.Okere CO, Waterhouse BD. Capsaicin increases GFAP and glutamine synthetase immunoreactivity in rat arcuate nucleus and median eminence. Neuroreport. 2004;15(2):255–8. doi: 10.1097/00001756-200402090-00008. [DOI] [PubMed] [Google Scholar]
- 89.Gonzalez A, Pariente JA, Salido GM. Ethanol stimulates ROS generation by mitochondria through Ca2+ mobilization and increases GFAP content in rat hippocampal astrocytes. Brain Res. 2007;1178:28–37. doi: 10.1016/j.brainres.2007.08.040. [DOI] [PubMed] [Google Scholar]
- 90.Zamoner A, et al. Hyperthyroidism in the developing rat testis is associated with oxidative stress and hyperphosphorylated vimentin accumulation. Mol Cell Endocrinol. 2007;267(12):116–26. doi: 10.1016/j.mce.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 91.Gonsebatt ME, et al. Arsenite induced oxidative damage in mouse liver is associated with increased cytokeratin 18 expression. Arch Toxicol. 2007;81(9):619–26. doi: 10.1007/s00204-007-0192-7. [DOI] [PubMed] [Google Scholar]
- 92.Westerheide SD, et al. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323(5917):1063–6. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morgan TE, et al. Increased transcription of the astrocyte gene GFAP during middle-age is attenuated by food restriction: implications for the role of oxidative stress. Free Radic Biol Med. 1997;23(3):524–8. doi: 10.1016/s0891-5849(97)00120-2. [DOI] [PubMed] [Google Scholar]
- 94.Gautron L, De-Smedt V, Laye S. Age-related changes in nestin immunoreactivity in the rat pituitary gland. Neuroendocrinology. 2009;90(1):19–30. doi: 10.1159/000220994. [DOI] [PubMed] [Google Scholar]
- 95.Matsutani T, et al. Young and middle-age associated differences in cytokeratin expression after bone fracture, tissue trauma, and hemorrhage. Am J Surg. 2007;193(1):61–8. doi: 10.1016/j.amjsurg.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 96.Nishio K, et al. Senescence and cytoskeleton: overproduction of vimentin induces senescent-like morphology in human fibroblasts. Histochem Cell Biol. 2001;116(4):321–7. doi: 10.1007/s004180100325. [DOI] [PubMed] [Google Scholar]
- 97.Caron M, et al. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007;14(10):1759–67. doi: 10.1038/sj.cdd.4402197. [DOI] [PubMed] [Google Scholar]
- 98.Pugh GE, et al. Distinct nuclear assembly pathways for lamins A and C lead to their increase during quiescence in Swiss 3T3 cells. J Cell Sci. 1997;110(Pt 19):2483–93. doi: 10.1242/jcs.110.19.2483. [DOI] [PubMed] [Google Scholar]
- 99.Sahlgren CM, et al. A nestin scaffold links Cdk5/p35 signaling to oxidant-induced cell death. EMBO J. 2006;25(20):4808–19. doi: 10.1038/sj.emboj.7601366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huang YL, et al. Nestin serves as a prosurvival determinant that is linked to the cytoprotective effect of epidermal growth factor in rat vascular smooth muscle cells. J Biochem. 2009 doi: 10.1093/jb/mvp070. [DOI] [PubMed] [Google Scholar]
- 101.Verstraeten VL, et al. The R439C mutation in LMNA causes lamin oligomerization and susceptibility to oxidative stress. J Cell Mol Med. 2009;13(5):959–71. doi: 10.1111/j.1582-4934.2009.00690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Malhas AN, Lee CF, Vaux DJ. Lamin B1 controls oxidative stress responses via Oct-1. J Cell Biol. 2009;184(1):45–55. doi: 10.1083/jcb.200804155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tao GZ, et al. Human Ran cysteine 112 oxidation by pervanadate regulates its binding to keratins. J Biol Chem. 2005;280(13):12162–7. doi: 10.1074/jbc.M412505200. [DOI] [PubMed] [Google Scholar]
- 104.Mathew J, et al. Keratin-protein kinase C interaction in reactive oxygen species-induced hepatic cell death through mitochondrial signaling. Free Radic Biol Med. 2008;45(4):413–24. doi: 10.1016/j.freeradbiomed.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 105.Freedberg IM, et al. Keratins and the keratinocyte activation cycle. J Invest Dermatol. 2001;116(5):633–40. doi: 10.1046/j.1523-1747.2001.01327.x. [DOI] [PubMed] [Google Scholar]
- 106.Wang L, et al. Interleukin-6 induces keratin expression in intestinal epithelial cells: potential role of keratin-8 in interleukin-6-induced barrier function alterations. J Biol Chem. 2007;282(11):8219–27. doi: 10.1074/jbc.M604068200. [DOI] [PubMed] [Google Scholar]
- 107.Hernandez-Quintero M, et al. Interleukin-6 promotes human epidermal keratinocyte proliferation and keratin cytoskeleton reorganization in culture. Cell Tissue Res. 2006;325(1):77–90. doi: 10.1007/s00441-006-0173-9. [DOI] [PubMed] [Google Scholar]
- 108.von Boyen GB, et al. Proinflammatory cytokines increase glial fibrillary acidic protein expression in enteric glia. Gut. 2004;53(2):222–8. doi: 10.1136/gut.2003.012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Osterloh A, Breloer M. Heat shock proteins: linking danger and pathogen recognition. Med Microbiol Immunol. 2008;197(1):1–8. doi: 10.1007/s00430-007-0055-0. [DOI] [PubMed] [Google Scholar]
- 110.D'Souza SD, Antel JP, Freedman MS. Cytokine induction of heat shock protein expression in human oligodendrocytes: an interleukin-1-mediated mechanism. J Neuroimmunol. 1994;50(1):17–24. doi: 10.1016/0165-5728(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 111.Tsan MF, Gao B. Heat shock proteins and immune system. J Leukoc Biol. 2009;85(6):905–10. doi: 10.1189/jlb.0109005. [DOI] [PubMed] [Google Scholar]
- 112.Nieminen M, et al. Vimentin function in lymphocyte adhesion and transcellular migration. Nat Cell Biol. 2006;8(2):156–62. doi: 10.1038/ncb1355. [DOI] [PubMed] [Google Scholar]
- 113.Toivola DM, et al. Cellular integrity plus: organelle-related and protein-targeting functions of intermediate filaments. Trends Cell Biol. 2005;15(11):608–17. doi: 10.1016/j.tcb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 114.Styers ML, Kowalczyk AP, Faundez V. Intermediate Filaments and Vesicular Membrane Traffic: The Odd Couple's First Dance? Traffic. 2005;6(5):359–65. doi: 10.1111/j.1600-0854.2005.00286.x. [DOI] [PubMed] [Google Scholar]
- 115.Stone MR, et al. Absence of keratin 19 in mice causes skeletal myopathy with mitochondrial and sarcolemmal reorganization. J Cell Sci. 2007;120(Pt 22):3999–4008. doi: 10.1242/jcs.009241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tang HL, et al. Vimentin supports mitochondrial morphology and organization. Biochem J. 2008;410(1):141–6. doi: 10.1042/BJ20071072. [DOI] [PubMed] [Google Scholar]
- 117.Kumemura H, et al. Mutation in keratin 18 induces mitochondrial fragmentation in liver-derived epithelial cells. Biochem Biophys Res Commun. 2008;367(1):33–40. doi: 10.1016/j.bbrc.2007.12.116. [DOI] [PubMed] [Google Scholar]
- 118.Perez-Olle R, et al. Mutations in the neurofilament light gene linked to Charcot-Marie-Tooth disease cause defects in transport. J Neurochem. 2005;93(4):861–74. doi: 10.1111/j.1471-4159.2005.03095.x. [DOI] [PubMed] [Google Scholar]
- 119.Perrot R, Julien JP. Real-time imaging reveals defects of fast axonal transport induced by disorganization of intermediate filaments. FASEB J. 2009 doi: 10.1096/fj.09-129585. [DOI] [PubMed] [Google Scholar]
- 120.Chang L, et al. The dynamic properties of intermediate filaments during organelle transport. J Cell Sci. 2009 doi: 10.1242/jcs.046789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Planko L, et al. Identification of a keratin-associated protein with a putative role in vesicle transport. Eur J Cell Biol. 2007;86(1112):827–39. doi: 10.1016/j.ejcb.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 122.Mounkes LC, et al. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum Mol Genet. 2005;14(15):2167–80. doi: 10.1093/hmg/ddi221. [DOI] [PubMed] [Google Scholar]
- 123.Lammerding J, et al. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113(3):370–8. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nikolova V, et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004;113(3):357–69. doi: 10.1172/JCI19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Muchir A, et al. Nuclear envelope alterations in fibroblasts from LGMD1B patients carrying nonsense Y259X heterozygous or homozygous mutation in lamin A/C gene. Exp Cell Res. 2003;291(2):352–62. doi: 10.1016/j.yexcr.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 126.Duan S, et al. The Pirh2-keratin 8/18 interaction modulates the cellular distribution of mitochondria and UV-induced apoptosis. Cell Death Differ. 2009;16(6):826–37. doi: 10.1038/cdd.2009.12. [DOI] [PubMed] [Google Scholar]
- 127.Winter L, Abrahamsberg C, Wiche G. Plectin isoform 1b mediates mitochondrion-intermediate filament network linkage and controls organelle shape. J Cell Biol. 2008;181(6):903–11. doi: 10.1083/jcb.200710151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Goldfarb LG, Dalakas MC. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest. 2009;119(7):1806–13. doi: 10.1172/JCI38027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Song S, et al. Functions of the intermediate filament cytoskeleton in the eye lens. J Clin Invest. 2009;119(7):1837–48. doi: 10.1172/JCI38277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Runembert I, et al. Recovery of Na-glucose cotransport activity after renal ischemia is impaired in mice lacking vimentin. Am J Physiol Renal Physiol. 2004;287(5):F960–8. doi: 10.1152/ajprenal.00064.2004. [DOI] [PubMed] [Google Scholar]
- 131.Shi S, et al. Podocyte-selective deletion of dicer induces proteinuria and glomerulosclerosis. J Am Soc Nephrol. 2008;19(11):2159–69. doi: 10.1681/ASN.2008030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fickert P, et al. Mallory body formation in primary biliary cirrhosis is associated with increased amounts and abnormal phosphorylation and ubiquitination of cytokeratins. J Hepatol. 2003;38(4):387–94. doi: 10.1016/s0168-8278(02)00439-7. [DOI] [PubMed] [Google Scholar]
- 133.Zhong B, Omary MB. Actin overexpression parallels severity of pancreatic injury. Exp Cell Res. 2004;299(2):404–14. doi: 10.1016/j.yexcr.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 134.Ramot Y, et al. Endocrine controls of keratin expression. Bioessays. 2009;31(4):389–99. doi: 10.1002/bies.200800121. [DOI] [PubMed] [Google Scholar]
- 135.Chou YH, et al. The motility and dynamic properties of intermediate filaments and their constituent proteins. Exp Cell Res. 2007;313(10):2236–43. doi: 10.1016/j.yexcr.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 136.Tao GZ, et al. Monitoring of epithelial cell caspase activation via detection of durable keratin fragment formation. J Pathol. 2008;215(2):164–74. doi: 10.1002/path.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Toivola DM, et al. Keratin 8 and 18 hyperphosphorylation is a marker of progression of human liver disease. Hepatology. 2004;40(2):459–66. doi: 10.1002/hep.20277. [DOI] [PubMed] [Google Scholar]
- 138.Steiner G. Auto-antibodies and autoreactive T-cells in rheumatoid arthritis: pathogenetic players and diagnostic tools. Clin Rev Allergy Immunol. 2007;32(1):23–36. doi: 10.1007/BF02686079. [DOI] [PubMed] [Google Scholar]
- 139.Fokina GI, et al. Development of antibodies to axonal neurofilaments in the progression of chronic tick-borne encephalitis. Acta Virol. 1991;35(5):458–63. [PubMed] [Google Scholar]
- 140.Radoja N, et al. Thyroid hormones and gamma interferon specifically increase K15 keratin gene transcription. Mol Cell Biol. 2004;24(8):3168–79. doi: 10.1128/MCB.24.8.3168-3179.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kerns ML, et al. Reprogramming of keratin biosynthesis by sulforaphane restores skin integrity in epidermolysis bullosa simplex. Proc Natl Acad Sci U S A. 2007;104(36):14460–5. doi: 10.1073/pnas.0706486104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Beere HM, Green DR. Stress management - heat shock protein-70 and the regulation of apoptosis. Trends Cell Biol. 2001;11(1):6–10. doi: 10.1016/s0962-8924(00)01874-2. [DOI] [PubMed] [Google Scholar]
- Dudek J, et al. Functions and pathologies of BiP and its interaction partners. Cell Mol Life Sci. 2009;66(9):1556–69. doi: 10.1007/s00018-009-8745-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.