

Abstract
Insulin-like growth factors-I and -II (IGF-I and -II) are structurally related mitogenic polypeptides with potent growth promoting effects. These peptides and their corresponding IGF-I and -II receptors are selectively localized in the brain. To date, most of the effects of IGFs are believed to be mediated by IGF-I receptors whereas the significance of IGF-II receptor in mediating biological responses remains unclear. In the present study, we characterized the distribution of IGF-I and IGF-II receptor sites and investigated the effects of both factors on endogenous acetylcholine (ACh) release in adult rat hippocampus. [125I]IGF-I receptor binding sites are recognized by IGF-I> IGF-II> insulin, whereas [125I]IGF-II binding was competed potently by IGF-II> IGF-I but not by insulin. At the cellular level, IGF-I receptor sites were primarily noted in the molecular layer of the dentate gyrus and the CA2-CA3 subfields of the Ammon’s horn whereas IGF-II sites were localized predominantly in the pyramidal cell layer of the CA1-CA3 subfields and in the granular cell layer of the dentate gyrus. IGF-I (10−14–10−8 M) and des(1–3) IGF-I (10−10–10−8 M) were found to inhibit whereas IGF-II (10−14–10−8 M) potentiated K+-evoked ACh release from hippocampal slices. Tetrodotoxin altered the effects of IGF-I but not those of IGF-II suggesting that IGF-I acts indirectly via the release of other modulators whereas IGF-II acts directly on or in close proximity to the cholinergic terminals. The inhibitory effects of IGF-I were also observed in the frontal cortex but not in the striatum. In contrast, the stimulatory effects of IGF-II were evident both in the frontal cortex and striatum. Taken together, these results reveal the differential localization of IGF-I and IGF-II receptor sites in the hippocampal formation and the opposite role for these growth factors in the acute regulation of ACh release likely via two distinct mechanisms. Additionally, these data provide the first evidence for a direct role for IGF-II and its receptors in the regulation of transmitter release in the central nervous system.
Insulin-like growth factors-I and -II (IGF-I and -II) are pleiotropic polypeptides with structural and functional homologies to the insulin. These trophic factors have been shown to be selectively localized in various regions of the brain and their physiological responses are presumed to be mediated by specific interactions with cell surface receptors (1–4). The IGF-I (i.e., type I IGF) receptor that has higher affinity for IGF-I than for IGF-II or insulin, is a heterotetramer consisting of two α- and two β-subunits joined by disulfide bridges. The type II IGF/mannose-6-phosphate receptor (IGF-II receptor) has a higher affinity for IGF-II than IGF-I and does not bind insulin. It comprises a single polypeptide chain with a large extracellular domain and a short cytoplasmic tail (2, 5, 6). Although both IGF-I and IGF-II receptors and their mRNAs are widely distributed throughout the brain including the hippocampal formation (7–13), very little information is currently available on the pharmacological profile or cellular sites of localization of these receptors. Furthermore, while the role of the IGF-II receptor in transporting lysosomal enzymes and internalizing various compounds appears to be well established, its function in signal transduction, unlike that of IGF-I receptor, remains controversial (2, 5, 6).
A plethora of experimental approaches have shown that IGFs participate in the development of the nervous system by promoting neural growth, survival, and differentiation of neurones and glia (3, 14–18). In the adult nervous system, these growth factors are considered to have a role in the normal maintenance as well as activity-dependent functioning of the brain (9, 19–28). The evidence that IGF-I and IGF-II receptors have distinct distributional profiles (7, 10, 13) and respond differently to various manipulations (22, 23, 26) raises the possibility that these receptors may have selective roles in the regulation of certain brain functions. Paradoxically, most of the mitogenic and growth promoting effects of IGFs are believed to be mediated via activation of the IGF-I receptor whereas the IGF-II receptor is considered to have a role in the uptake and degradation of IGF-II (2, 5). Few studies from nonneuronal tissues, however, suggest that IGF-II acting via its own receptor is able to mediate certain cellular responses (29–31). The most extensively studied response is the stimulation of Ca2+ influx in primed BALB/c 3T3 fibroblast cells in which the IGF-II receptor appears to be coupled to a Ca2+ channel by a G protein (32–34). However, it is unclear whether IGF-I and IGF-II, acting via their respective receptors, can have distinct, unique role in the regulation of the normal or activity-dependent functioning of the brain. The present study shows that IGF-I and its analog des(1–3) IGF-I inhibit while IGF-II potentiates endogenous acetylcholine (ACh) release from selected regions of the brain by two distinct mechanisms. These results provide an evidence for differential roles of IGFs and their respective receptors in the regulation of cholinergic function. In addition, it suggests that IGF-II receptor, apart from internalization, may also participate in mediating certain biological responses of IGF-II in the brain.
MATERIALS AND METHODS
Materials.
Adult male Sprague-Dawley rats (2–3 month, 275–300 g) obtained from Charles River Breeding Laboratories were used in the study. All animals were housed according to guidelines of the Canadian Council on Animal Care and McGill University Policies, and given food and water ad libitum. Human recombinant IGF-I was purchased from ICN, human recombinant des(1–3) IGF-I from GroPep (Adelaide, Australia), human recombinant IGF-II from Sigma and porcine insulin from Calbiochem. Labeled [125I]IGF-I (2,000 Ci/mmol; 1 Ci = 37 GBq), [125I]IGF-II (2,000 Ci/mmol), and Hyperfilms were obtained from Amersham while [γ-32P]ATP was purchased from New England Nuclear. Tetraphenylboron and butyronitrile were obtained from Aldrich and AG 1-X8 Resin was purchased from Bio-Rad. All other chemicals were purchased either from Sigma or Fisher Scientific.
In Vitro Receptor Autoradiography.
Eight adult rats were killed by decapitation and their brains were cut serially (20 μm) through the hippocampal formation and processed for [125I]IGF-I and [125I]IGF-II receptor autoradiography as described earlier (13). For competition binding assays, consecutive brain sections were incubated with 50 pM [125I]IGF-I or 25 pM [125I]IGF-II containing either IGF-I, IGF-II, or insulin (10−12–10−6 M). For cellular localization of the respective binding sites, a batch of slides were incubated with [125I]IGF-I or [125I]IGF-II (in the presence or absence of corresponding unlabeled peptide), fixed in 5% glutaraldehyde solution, dehydrated in graded alcohols, dipped in Kodak NTB-2 emulsion, and then developed after 10–12 days. Autoradiograms were quantified densitometrically using a computerized image analysis system (MCID, Imaging Research, St Catharines, ON, Canada) as described earlier (13, 26). The binding data were then analyzed using the graphpad program (GraphPad Software, San Diego). All values are expressed as percent of specific binding and are represented as mean ± SEM.
In Vitro ACh Release.
Brain slices were prepared as described in detail elsewhere (35). Regions of interest (i.e., hippocampus, striatum, and frontal cortex) were first dissected out and then superfused with oxygenated Krebs buffer. After a 45 min stabilization period, effluents were collected every 20 min for 1 hr to establish the basal efflux. The tissues were then stimulated with high K+ Krebs buffer (25 mM KCl with equimolar reduction in NaCl to maintain isotonicity) for 1 hr either in the presence or absence of IGF-I, IGF-II, or their mixture. The analog des(1–3) IGF-I that exhibits more potent effects than IGF-I in a variety of biological system due to its low affinity for IGF binding proteins (28) has also been used, in parallel, to further establish the nature of the effect of IGF-I. At the end of the superfusion, tissue slices were removed and protein content was measured (36). The superfusates collected every 20 min throughout the experiment were then processed using radioenzymatic assay as described earlier (35, 37). Evoked transmitter release represents the net release above the basal efflux and is expressed as pmol ACh⋅min⋅mg protein. The data that are presented as mean ± SEM were analyzed using one-way ANOVA followed by Fisher’s post hoc test with level of significance set at P < 0.05.
RESULTS
Characterization and Distribution of [125I]IGF-I and [125I]IGF-II Receptor Binding Sites in Rat Hippocampal Formation.
[125I]IGF-I and [125I]IGF-II receptor binding sites exhibited different profiles of distribution throughout the hippocampal formation (Fig. 1). High levels of [125I]IGF-I binding sites were localized primarily in the CA2-CA3 subfields and the dentate gyrus (DG) (Fig. 1 A–H). Within the Ammon’s horn a relatively high density of silver grains was evident particularly in the stratum oriens and polymorphic neurones of the hilar region whereas the stratum radiatum and lacunosum moleculare showed only moderate amounts of grains. The pyramidal neurones of the Ammon’s horn exhibited relatively low density of silver grains (Fig. 1 B–E). In the DG, silver grains were predominantly localized in the molecular layer rather than the granular cell layer (Fig. 1 F, G). The pharmacological specificity of the observed [125I]IGF-I binding was determined by incubating consecutive sections in the absence or presence of increasing concentrations of IGF-I, IGF-II, and insulin (Fig. 2A). The rank order of potency of the competitors was IGF-I> IGF-II> insulin (Figs. 2A).
Figure 1.
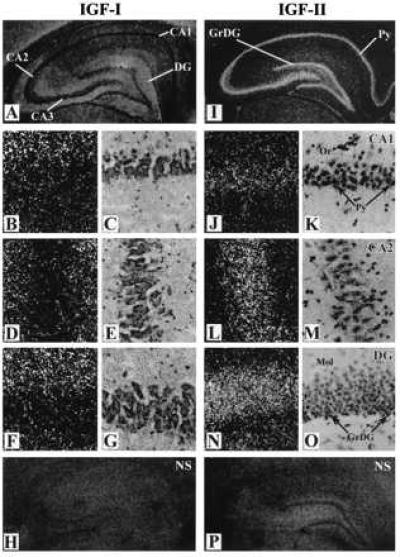
Photomicrographs showing the cellular distributions of [125I]IGF-I (A–G) and [125I]IGF-II (I–O) receptor sites in the hippocampus (A, I) and in the CA1 (B, C, J, K), CA2 (D, E, L, M) and the DG (F, G, N, O) regions of the adult rat. H and P represent nonspecific labeling in the presence of excess unlabeled IGF-I and IGF-II, respectively. C, E, and G are the bright field representative of B, D, and F whereas K, M, and O are the bright field representative of J, L, and N, respectively. Or, stratum oriens; Mol, molecular layers of the DG; Py, pyramidal neurones; GrDG, granule cells.
Figure 2.
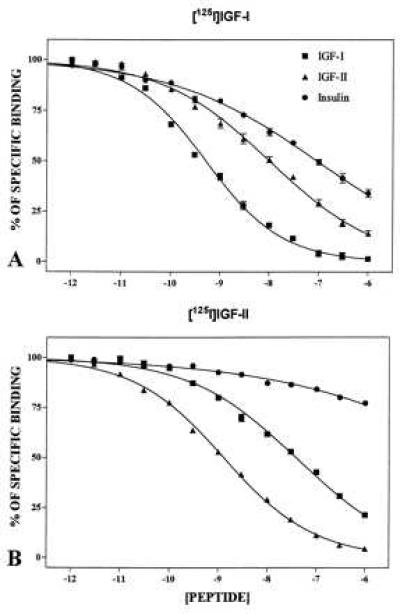
Competition binding profiles of IGF-I, IGF-II, and insulin against labeled [125I]IGF-I (A) and [125I]IGF-II (B) in adult rat hippocampal formation. The observed profiles clearly demonstrate [125I]IGF-I and [125I]IGF-II recognized the respective receptors at higher affinity than the related peptides. Unlabeled insulin did not compete for [125I]IGF-II binding. Each point represents the measurement of the mean ± SEM of data obtained from three to four determinations.
High amounts of [125I]IGF-II binding sites were evident primarily in the pyramidal cell layer of the CA1-CA3 subfields whereas other layers of the Ammon’s horn such as the strata oriens, radiatum, and lacunosum-moleculare showed apparently low levels of specific labeling (Fig. 1 I–P). The amounts of silver grains in the CA2 or CA3 pyramidal cell layer was higher than in the CA1 subfield (Fig. 1 J–M). In the DG, the granular cell layer, as opposed to the molecular layer, showed relatively high levels of [125I]IGF-II binding sites (Fig. 1 N, O). Relatively high amounts of silver grains were also evident in the polymorphic neurones of the hilar region. The observed [125I]IGF-II binding in various layers of the hippocampus was competed potently by IGF-II> IGF-I whereas insulin was found to be ineffective (up to 1 μM) (Fig. 2B).
Effects of IGF-I, Des(1–3) IGF-I, and IGF-II on Hippocampal ACh Release.
Depolarization-induced ACh release with 25 mM KCl is known to be optimal to reveal both drug-dependent attenuation as well as increase of transmitter release (35, 38). To determine the effects of IGF-I, des(1–3) IGF-I, and IGF-II on endogenous ACh release, hippocampal slices were stimulated with 25 mM K+ buffer either in the absence or presence of various concentrations of IGF-I (10−14–10−8 M), des(1–3) IGF-I (10−10–10−8 M), and IGF-II (10−14–10−8 M). The results clearly demonstrate that IGF-I, in a concentration-dependent manner, potently inhibited endogenous ACh release (Fig. 3A). The time dependency of the effects revealed that the inhibition of ACh release was apparent only during the last period of stimulation. Des(1–3) IGF-I also drastically reduced endogenous ACh release from hippocampal slices during the later phase of stimulation (Fig. 3B). This effect was found to be more potent than IGF-I as 10−10 M des(1–3) IGF-I, but not IGF-I, exhibited significant inhibition of ACh release (Fig. 3 A, B). IGF-II, in contrast to IGF-I or des(1–3) IGF-I, was found to potentiate evoked ACh release from hippocampal slices (Fig. 3C). This effect was also concentration-dependent (Fig. 3C) and evident during the first phase of stimulation. Interestingly, exposure of hippocampal slices to optimal concentrations of both IGF-I (10−8 M) and IGF-II (10−10 M) did not alter total ACh release over a 1 hr stimulation period thus indicating a possible neutralizing effects of these growth factors (Fig. 4). The time-dependency of the effects, as expected, showed a nonsignificant initial increase followed by a decrease in ACh release. Furthermore, unlike K+-evoked release, the spontaneous release of ACh from hippocampal slices was not altered by either 10−8 M IGF-I, 10−8 M des(1–3) IGF-I, or 10−10 M IGF-II.
Figure 3.
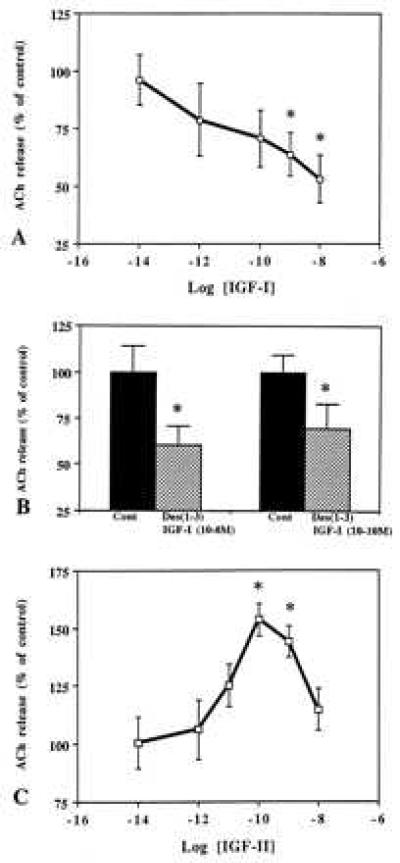
Effects of IGF-I (A), des(1–3) IGF-I (B), and IGF-II (C) on evoked ACh release from hippocampal slices. Slices were depolarized with 25 mM K+ buffer in the presence or absence of various concentrations of IGF-I, des(1–3) IGF-I, and IGF-II. Evoked release was significantly inhibited by IGF-I (A) and des(1–3) IGF-I (B) while potentiated by IGF-II (C). Results are expressed as the mean ± SEM of three experiments each performed quintuplicate for each concentration of peptide tested. cont, Control. *P < 0.05.
Figure 4.
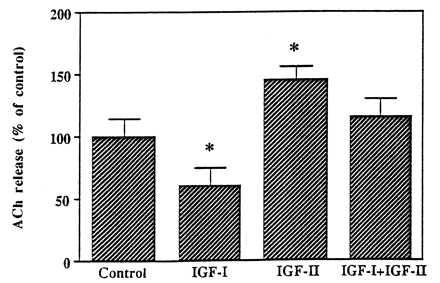
Comparative effects of IGF-I, IGF-II, and their mixture on evoked ACh release from hippocampal slices. Tissue slices were depolarized in the presence or absence of 10−8 M IGF-I or 10−10 M IGF-II alone and with both the peptides. Evoked ACh release was significantly inhibited by IGF-I while potentiated by IGF-II. Presence of both IGF-I and IGF-II did not significantly alter ACh release from hippocampal slices. Data are expressed as the mean ± SEM (n = 10–12). *P < 0.05.
Effects of Tetrodotoxin (TTX) on IGF-I and IGF-II-Modulated ACh Release.
Neuronal depolarization and firing are known to be suppressed by TTX due to ion fluxes through voltage-sensitive Na+-channels (39). By itself, TTX has been shown not to affect evoked ACh release from rat hippocampal slices (40). To determine whether the differential effects of IGF-I and IGF-II are being altered by TTX, hippocampal slices were superfused under similar conditions in the presence of 10 μM TTX (Fig. 5 A, B). While the inhibitory effects of 10−8 M IGF-I was found to be altered in the presence of TTX (Fig. 5A), the stimulatory response of 10−10 M IGF-II was found to be insensitive to the presence of TTX (Fig. 5B). This suggests that IGF-II probably acts directly on or in proximity to the hippocampal cholinergic terminals to potentiate ACh release whereas the inhibitory response of IGF-I possibly requires the initiation of impulses distal to cholinergic terminals.
Figure 5.
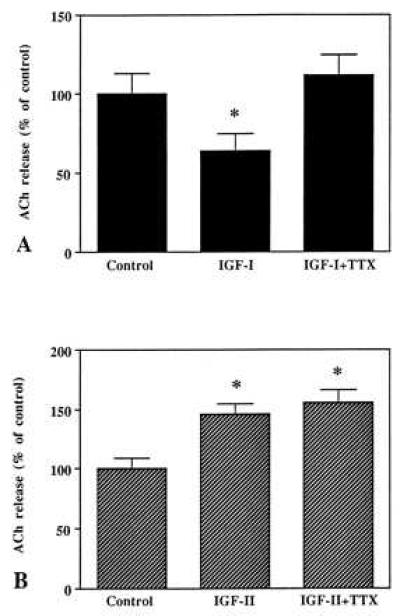
Effects of TTX on the IGF-I (A)- and IGF-II (B)-induced alterations of evoked ACh release from hippocampal slices. Tissue slices were depolarized with 25 mM K+ buffer in the presence or absence of 10−8 M IGF-I or 10−10 M IGF-II alone and with the peptide and 10 μM TTX. IGF-I-induced inhibition of evoked release was altered by TTX (A) whereas the effects of IGF-II remained unaffected in the presence of TTX (B). Data are expressed as the mean ± SEM (n = 10–12). *P < 0.05.
Regional Differences in the Effects of IGF-I and IGF-II.
Besides the hippocampal formation, cholinergic terminals are most concentrated in the cerebral cortex and in the striatum (41), regions that also express receptors for IGF-I and IGF-II (8, 10, 13). To evaluate regional specificity, slices of frontal cortex (Fig. 6 A, B) or striatum (Fig. 7 A, B) were superfused in the presence of 10−8 M IGF-I or 10−10 M IGF-II. IGF-I did not affect striatal ACh release (Fig. 7A) whereas release from the frontal cortex was found to be significantly decreased (Fig. 6A). Similar to the hippocampal formation, the inhibition was mostly apparent during the later period of stimulation. IGF-II, unlike IGF-I, augmented evoked ACh release from cortical (Fig. 6B) as well as striatal slices (Fig. 7B). Furthermore, consistent with the hippocampal formation, the potentiation of ACh release was evident during the early phase of stimulation in both the frontal cortex and striatum (Figs. 6B and 7B).
Figure 6.
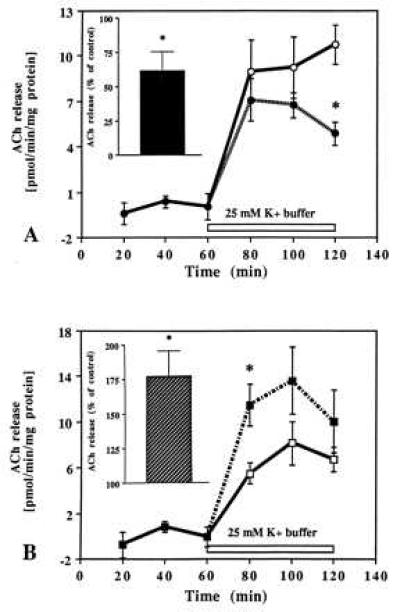
Time-course effects of IGF-I (A) and IGF-II (B) on evoked ACh release from slices of the frontal cortex. Tissue slices were stimulated with 25 mM K+ buffer in the presence (dotted lines) or absence (solid lines) of 10−8 M IGF-I and 10−10 M IGF-II. Evoked ACh release was significantly inhibited by IGF-I (A) while potentiated by IGF-II (B). A and B (Insets) represent total release as percentage of control for the respective peptides over 60 min stimulation period. Data are expressed as the mean ± SEM (n = 10–12). *P < 0.05.
Figure 7.
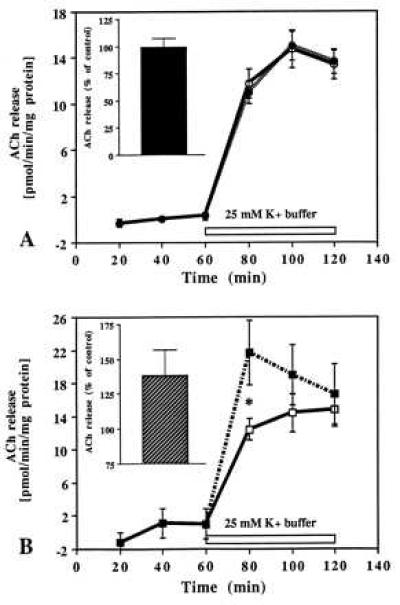
Time-course effects of IGF-I (A) and IGF-II (B) on evoked ACh release from slices of the striatum. Tissue slices were stimulated with 25 mM K+ buffer in the presence (dotted lines) or absence (solid lines) of 10−8 M IGF-I and 10−10 M IGF-II. Evoked ACh release was unaffected by IGF-I (A) while potentiated significantly at the early phase of stimulation by IGF-II (B). A and B (Insets) represent total release as percentage of control for the respective peptides over 60 min stimulation period. Data are expressed as the mean ± SEM (n = 10–12). *P < 0.05.
DISCUSSION
The present study clearly indicates that IGF-I and IGF-II differentially regulate endogenous ACh release from selected regions of the adult rat brain. The effects of both peptides are concentration dependent, region specific, and are likely mediated by different mechanisms since one is TTX-sensitive while other is not. IGF-I inhibited K+-evoked ACh release from hippocampal and cortical slices but not from the striatum whereas IGF-II potentiated evoked release from all three regions. Des(1–3) IGF-I, an analog of IGF-I that exhibit low affinity for IGF binding proteins (2, 18), also potently inhibited endogenous ACh release from hippocampal slices. These results, together with the findings that each growth factor recognizes its cognate receptor with high affinity, provide evidence in favor of contrasting roles for these growth factors/receptors in the regulation of brain cholinergic function in addition to a unique role for the IGF-II receptor in the rat central nervous system.
The widespread distribution of [125I]IGF-I and [125I]IGF-II receptor binding sites, in keeping with earlier reports (7, 8, 10, 13), was noted throughout the hippocampal formation. These sites, as evident by competition experiments, demonstrate binding affinity characteristics of the IGF-I and IGF-II receptors. Furthermore, as insulin does not bind IGF binding proteins or IGF-II receptor, competition for [125I]IGF-I but not [125I]IGF-II binding sites by high concentration of insulin indicate that both radioligands under our assay conditions identified genuine IGF-I and IGF-II receptor sites (7, 10, 13). This is further reiterated by emulsion autoradiographic studies that clearly showed that binding sites recognized by these two ligands are concentrated in anatomically distinct regions of the hippocampal formation.
The in vitro release of ACh from neuronal tissues is known to be Ca2+-independent for basal, unstimulated release but Ca2+-dependent for evoked release (42). High K+ Krebs buffer activates voltage-sensitive calcium channels that results in an influx of Ca2+ followed by transmitter release (43). The present results that IGF-I and des(1–3) IGF-I inhibited while IGF-II potentiated K+-evoked but not basal ACh release indicate the involvement of Ca2+-sensitive mechanisms in the regulation of ACh release by the IGFs. The concentrations required to modulate 50% of the evoked ACh release (i.e., EC50) from hippocampal slices are apparently 0.1 nM for IGF-I and 0.01 nM for IGF-II. Interestingly, 10−8 M IGF-II, in contrast to the lower concentrations (10−11–10−9 M), did not significantly alter ACh release over the 1 hr stimulation period. Given the evidence that higher concentrations of IGF-II can recognize the IGF-I receptor, it is likely that the stimulatory effects of 10−8 M IGF-II is being neutralized by simultaneous activation of IGF-I receptor inhibiting ACh release. It is also of interest to note that combined administration of optimal concentrations of IGF-I and IGF-II did not significantly alter hippocampal ACh release thus suggesting a possible neutralizing effect of these opposing factors. Furthermore, the differential effects of IGFs were found to be likely mediated by two distinct mechanisms, since the action of IGF-I was blocked by TTX while that of IGF-II was not. These results, together with the observation that striatal cholinergic interneurones are sensitive to IGF-II but not IGF-I, provide an evidence that the contrasting effects of IGF-I and IGF-II on evoked hippocampal ACh release are most likely mediated by activation of the respective receptors.
While the transmembrane signaling role of IGF-I receptor is well established (via phosphatidylinositol-3 kinase), the significance of the IGF-II receptor in mediating genuine biological responses is still a matter of intense debate. A series of in vitro studies have shown that IGF-II binding to its receptor stimulates glycogen synthesis in rat hepatoma cells (44) and amino acid uptake in human myoblasts (29), promotes Na+/H+ exchange and inositol triphosphate production in canine kidney proximal tubular cells (45, 46) and stimulates Ca2+ influx and DNA synthesis in primed BALB/c 3T3 cells (32, 34). There is also evidence that IGF-II by interacting with the IGF-II receptor stimulates the motility of human rhabdomyosarcoma cells (30) and the differentiation of muscle cells (31). In keeping with these observations, it seems likely that the IGF-II receptor, apart from its role in protein trafficking, might be involved in mediating certain functional responses such as the regulation of ACh release in selected regions of the brain.
The use of TTX clearly demonstrated that the inhibitory effects of IGF-I required the initiation of impulse activity while the potentiating effects of IGF-II were due to an action at or in close proximity to the cholinergic terminals. Supporting a direct action of IGF-II, it has recently been shown that IGF-II receptors are located on septal cholinergic neurones and are involved in IGF-II-mediated increase in choline acetyltransferase activity (47), a result that may be associated with increased ACh release. In contrast, the TTX-sensitive effect of IGF-I is possibly being mediated via the release of transmitter that subsequently inhibits ACh release from cholinergic terminals. Given the evidence that IGF-I modulates γ-aminobutyric acid release (21) that is known to inhibit ACh release from the hippocampus (48), it is possible that γ-aminobutyric acid may be involved in regulating the inhibition of ACh release by IGF-I. Interestingly, striatal cholinergic interneurones, unlike the hippocampus or cortex, were found to be insensitive to IGF-I, suggesting IGF-I receptors in this region are not appropriately positioned and/or coupled to effector mechanisms to modulate the exocytotic process involved in the release of ACh. Alternatively, it is possible that higher amounts of binding proteins in this region may play a role in altering the inhibitory effects of IGF-I.
The intracellular events associated with IGF-I and IGF-II regulated endogenous ACh release are unknown. Earlier reports have shown that IGF-I effects at the cellular level are initiated by the activation of intrinsic tyrosine kinase present in the β-subunit of the IGF-I receptor. This subsequently induces a cascade of phosphorylation events including the activation of the insulin receptor substrates (IRS-1 and IRS-2), phosphatidylinositol-3 kinase, and protein kinase C leading to alterations in Ca2+ influx and its mobilization from intracellular stores (2, 6). It has recently been shown that IGF-I modulates glutamate-induced γ-aminobutyric acid release from cerebellum by simultaneous activation of protein kinase C and NO signaling pathways (49). In the context of ACh release, it remains to be established if protein kinase C and/or NO signaling pathways is involved in mediating the attenuation of ACh release by IGF-I or des(1–3) IGF-I. As for the action of IGF-II, some evidence suggests that this factor (0.1 nM) can stimulate Ca2+ influx in primed BALB/c 3T3 fibroblasts possibly by coupling to some GTP binding proteins (32–34). Recently, it is reported that the IGF-II receptor can activate G proteins through its cytoplasmic domains that subsequently alter adenyl cyclase activity in transfected COS cells (50). It would of interest to determine if a similar mechanism is involved in the regulation of ACh release.
Accumulated evidence (47, 51–53) and the present data suggest that IGF-I and IGF-II act both as trophic factors as well as fast acting neuromodulators for selected populations of cholinergic neurones and thus may be of relevance to certain degenerative diseases, particularly Alzheimer disease—where decreased levels of cholinergic markers are associated with impairments in cognitive functions (54–56). Interestingly, in support of a possible role for the IGFs in Alzheimer disease, it has recently been shown that IGF-I binding sites are increased in the cortical areas in this disease (57), IGF-I can protect/rescue cultured hippocampal neurones against β-amyloid mediated toxicity (58) and immunoreactive IGF-II receptors are localized in β-amyloid containing neuritic plaques (4). Whether decreased levels of ACh and/or selective loss of basal forebrain cholinergic neurones in Alzheimer disease are directly associated with altered trophic and/or neuromodulatory roles of IGFs remain to be established.
Acknowledgments
This work was supported by Medical Research Council of Canada. R.Q. is a recipient of Chercheur Boursier de merite exceptionel and S.K. is a Chercheur-Boursier Junior 1 of the Fonds de la recherche en Sante du Quebec. S.D. is a postdoctoral fellow of the Alzheimer Society of Canada and D.S. is a recipient of studentship award from the Fonds de la recherche en Sante du Quebec.
ABBREVIATIONS
- ACh
acetylcholine
- DG
dentate gyrus
- IGF-I and -II
insulin-like growth factor-I and -II
- TTX
tetrodotoxin
References
- 1.Baskin D G, Wilcox B J, Figlewicz D P, Dorsa D M. Trends Neurosci. 1988;11:107–111. doi: 10.1016/0166-2236(88)90155-5. [DOI] [PubMed] [Google Scholar]
- 2.Jones J I, Clemmons R. Endocr Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 3.Pablo F D, de la Rosa E J. Trends Neurosci. 1995;18:143–150. doi: 10.1016/0166-2236(95)93892-2. [DOI] [PubMed] [Google Scholar]
- 4.Doré S, Kar S, Quirion R. Trends Neurosci. 1997;20:326–331. doi: 10.1016/s0166-2236(96)01036-3. [DOI] [PubMed] [Google Scholar]
- 5.Kornfeld S. Annu Rev Biochem. 1992;61:307–330. doi: 10.1146/annurev.bi.61.070192.001515. [DOI] [PubMed] [Google Scholar]
- 6.LeRoith D, Werner H, Beitner-Johnson D, Roberts C T. Endocr Rev. 1995;16:143–163. doi: 10.1210/edrv-16-2-143. [DOI] [PubMed] [Google Scholar]
- 7.Lesniak M A, Hill J M, Keiss W, Rojeski M, Pert C B, Roth J. Endocrinology. 1988;123:2089–2099. doi: 10.1210/endo-123-4-2089. [DOI] [PubMed] [Google Scholar]
- 8.Smith M, Clemens J, Kerchner G A, Mendelsohn L G. Brain Res. 1988;445:241–246. doi: 10.1016/0006-8993(88)91185-7. [DOI] [PubMed] [Google Scholar]
- 9.Araujo D M, Lapchak P A, Collier B, Chabot J G, Quirion R. Brain Res. 1989;484:130–138. doi: 10.1016/0006-8993(89)90355-7. [DOI] [PubMed] [Google Scholar]
- 10.Werther G A, Hogg A, Oldfield B J, McKinley M J, Figdor R, Mendelsohn F A O. J Neuroendocrinol. 1990;1:369–377. doi: 10.1111/j.1365-2826.1989.tb00131.x. [DOI] [PubMed] [Google Scholar]
- 11.Bondy C, Werner H, Roberts C T, Jr, LeRoith D. Neuroscience. 1992;46:909–924. doi: 10.1016/0306-4522(92)90193-6. [DOI] [PubMed] [Google Scholar]
- 12.Couce M E, Weatherington A J, McGinty J F. Endocrinology. 1992;131:1636–1642. doi: 10.1210/endo.131.4.1396308. [DOI] [PubMed] [Google Scholar]
- 13.Kar S, Chabot J G, Quirion R. J Comp Neurol. 1993;333:375–397. doi: 10.1002/cne.903330306. [DOI] [PubMed] [Google Scholar]
- 14.Lenoir D, Honegger P. Dev Brain Res. 1983;7:205–213. doi: 10.1016/0165-3806(83)90177-3. [DOI] [PubMed] [Google Scholar]
- 15.Aizeman Y, De Vellis J. Brain Res. 1987;406:32–42. doi: 10.1016/0006-8993(87)90766-9. [DOI] [PubMed] [Google Scholar]
- 16.Shemer J, Raizada M K, Masters B A, Ota A, LeRoith D. J Biol Chem. 1987;262:7693–7699. [PubMed] [Google Scholar]
- 17.Baker J, Liu J P, Robertson E J, Efstratiadis A. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- 18.Carson M J, Behringer R R, Brinster R L, McMorris F A. Neuron. 1993;10:729–740. doi: 10.1016/0896-6273(93)90173-o. [DOI] [PubMed] [Google Scholar]
- 19.Berelowitz M, Szabo M, Frohman L A, Firestone S, Chu L, Hintz R L. Science. 1981;212:1279–1281. doi: 10.1126/science.6262917. [DOI] [PubMed] [Google Scholar]
- 20.Nilsson L, Sara V R, Nordberg A. Neurosci Lett. 1988;88:221–226. doi: 10.1016/0304-3940(88)90130-9. [DOI] [PubMed] [Google Scholar]
- 21.Castro-Alamancos M A, Torres-Aleman I. Proc Natl Acad Sci USA. 1993;90:7386–7390. doi: 10.1073/pnas.90.15.7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee W H, Clemens J A, Bondy C A. Molec Cell Neurosci. 1992;3:36–43. doi: 10.1016/1044-7431(92)90006-n. [DOI] [PubMed] [Google Scholar]
- 23.Beilharz E J, Bassett N S, Sirimanne E S, Williams C E, Gluckman P D. Mol Brain Res. 1995;29:81–91. doi: 10.1016/0169-328x(94)00232-4. [DOI] [PubMed] [Google Scholar]
- 24.Guan J, Williams C, Gunning M, Mallard E C, Gluckman P D. J Cereb Blood Flow Metab. 1995;13:609–616. doi: 10.1038/jcbfm.1993.79. [DOI] [PubMed] [Google Scholar]
- 25.Breese C R, D’Costa A, Rollins Y D, Adams C, Booze R M, Sonntag W E, Leonard S. J Comp Neurol. 1996;369:388–404. doi: 10.1002/(SICI)1096-9861(19960603)369:3<388::AID-CNE5>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 26.Kar S, Baccichet A, Quirion R, Poirier J. Neuroscience. 1993;55:69–80. doi: 10.1016/0306-4522(93)90455-o. [DOI] [PubMed] [Google Scholar]
- 27.Guthrie K M, Nguyen T, Gall C M. J Comp Neurol. 1995;352:147–160. doi: 10.1002/cne.903520111. [DOI] [PubMed] [Google Scholar]
- 28.Ballard F J, Wallace J C, Francis G L, Read L C, Tomas F M. Int J Biochem Cell Biol. 1996;28:1085–1087. doi: 10.1016/1357-2725(96)00056-8. [DOI] [PubMed] [Google Scholar]
- 29.Shimizu M, Webster C, Morgan D O, Blau H M, Roth R. Am J Physiol. 1986;251:E611–E615. doi: 10.1152/ajpendo.1986.251.5.E611. [DOI] [PubMed] [Google Scholar]
- 30.Minniti C P, Kohn E C, Grubb J H, Sly W S, Oh Y, Muller H L, Rosenfeld R G, Helman L J. J Biol Chem. 1992;267:9000–9004. [PubMed] [Google Scholar]
- 31.Rosenthal S M, Hsiao D, Silverman L A. Endocrinology. 1994;135:38–44. doi: 10.1210/endo.135.1.8013373. [DOI] [PubMed] [Google Scholar]
- 32.Nishimoto I, Hata Y, Ogata E, Kojima I. J Biol Chem. 1987;262:12120–12126. [PubMed] [Google Scholar]
- 33.Nishimoto I. Mol Reprod Dev. 1993;35:398–407. doi: 10.1002/mrd.1080350414. [DOI] [PubMed] [Google Scholar]
- 34.Kojima I, Nishimoto I, Iiri T, Ogata E, Rosenfeld R G. Biochem Biophys Res Commun. 1988;154:9–19. doi: 10.1016/0006-291x(88)90642-0. [DOI] [PubMed] [Google Scholar]
- 35.Kar S, Seto D, Gaudreau P, Quirion R. J Neurosci. 1996;16:1034–1040. doi: 10.1523/JNEUROSCI.16-03-01034.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lowery O H, Rosebrough N J, Farr A L, Randall R J. J Biol Chem. 1952;193:265–275. [PubMed] [Google Scholar]
- 37.Goldberg A M, McCaman R E. J Neurochem. 1973;20:1–8. doi: 10.1111/j.1471-4159.1973.tb12097.x. [DOI] [PubMed] [Google Scholar]
- 38.Pearce L B, Buck T, Adamec E. J Neurochem. 1991;57:636–647. doi: 10.1111/j.1471-4159.1991.tb03795.x. [DOI] [PubMed] [Google Scholar]
- 39.Narahashi T. Physiol Rev. 1974;54:813–889. doi: 10.1152/physrev.1974.54.4.813. [DOI] [PubMed] [Google Scholar]
- 40.Araujo D M, Lapchak P A, Collier B, Quirion R. J Neurochem. 1990;55:1546–1555. doi: 10.1111/j.1471-4159.1990.tb04937.x. [DOI] [PubMed] [Google Scholar]
- 41.Fibiger H C. Brain Res Rev. 1982;4:327–388. doi: 10.1016/0165-0173(82)90011-x. [DOI] [PubMed] [Google Scholar]
- 42.MacIntosh F C, Collier B. In: Neuromuscular Junction. Zaimis E, editor. Berlin: Springer; 1976. pp. 99–228. [Google Scholar]
- 43.Turner T J, Goldin S M. J Neurosci. 1985;5:841–849. doi: 10.1523/JNEUROSCI.05-03-00841.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hari J, Pearce S B, Morgan D O, Sara V, Smith M C, Roth R A. EMBO J. 1987;6:3367–3371. doi: 10.1002/j.1460-2075.1987.tb02658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rogers S A, Hammerman M R. J Biol Chem. 1989;265:9722–9727. [PubMed] [Google Scholar]
- 46.Mellas J, Gavin J R, III, Hammerman M R. J Biol Chem. 1986;261:14437–14442. [PubMed] [Google Scholar]
- 47.Konishi Y, Takahashi K, Chui D-H, Rosenfeld R G, Himeno M, Tabira T. Brain Res. 1994;649:53–61. doi: 10.1016/0006-8993(94)91048-0. [DOI] [PubMed] [Google Scholar]
- 48.Thompson S M. Prog Neurobiol. 1994;42:575–609. doi: 10.1016/0301-0082(94)90044-2. [DOI] [PubMed] [Google Scholar]
- 49.Castro-Alamancos M A, Arevalo M A, Torres-Aleman I. Neuroscience. 1996;70:843–847. doi: 10.1016/0306-4522(95)00472-6. [DOI] [PubMed] [Google Scholar]
- 50.Ikezu T, Okamoto T, Giambarella U, Yokota T. J Biol Chem. 1995;270:29224–29228. doi: 10.1074/jbc.270.49.29224. [DOI] [PubMed] [Google Scholar]
- 51.Knusel B, Michel P P, Schwaber J S, Hefti F. J Neurosci. 1990;10:558–570. doi: 10.1523/JNEUROSCI.10-02-00558.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng B, Mattson M P. J Neurosci. 1992;12:1558–1566. doi: 10.1523/JNEUROSCI.12-04-01558.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gage S L, Keim S R, Low W C. Prog Brain Res. 1990;82:73–80. doi: 10.1016/s0079-6123(08)62592-3. [DOI] [PubMed] [Google Scholar]
- 54.Perry E K. Br Med Bull. 1986;42:63–69. doi: 10.1093/oxfordjournals.bmb.a072100. [DOI] [PubMed] [Google Scholar]
- 55.Quirion R. J Psychiatr Neurosci. 1993;18:226–234. [PMC free article] [PubMed] [Google Scholar]
- 56.Selkoe D. Annu Rev Neurosci. 1994;17:489–517. doi: 10.1146/annurev.ne.17.030194.002421. [DOI] [PubMed] [Google Scholar]
- 57.Crews F T, McElhaney R, Freund G, Ballinger W E, Jr, Raizada M K. J Neurochem. 1992;58:1205–1210. doi: 10.1111/j.1471-4159.1992.tb11330.x. [DOI] [PubMed] [Google Scholar]
- 58.Doré S, Kar S, Quirion R. Proc Natl Acad Sci USA. 1997;94:4772–4777. doi: 10.1073/pnas.94.9.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]