

Abstract
P2X receptors are ligand-gated cation channels that transition from closed to open states upon binding ATP. The crystal structure of the closed zebrafish P2X4.1 receptor directly reveals that the ion-conducting pathway is formed by three transmembrane domain 2 (TM2) α-helices, each being provided by the three subunits of the trimer. However, the transitions in TM2 that accompany channel opening are incompletely understood and remain unresolved. In this study, we quantified gated access to Cd2+ at substituted cysteines in TM2 of P2X2 receptors in the open and closed states. Our data for the closed state are consistent with the zebrafish P2X4.1 structure, with isoleucines and threonines (Ile-332 and Thr-336) positioned one helical turn apart lining the channel wall on approach to the gate. Our data for the open state reveal gated access to deeper parts of the pore (Thr-339, Val-343, Asp-349, and Leu-353), suggesting the closed channel gate is between Thr-336 and Thr-339. We also found unexpected interactions between native Cys-348 and D349C that result in tight Cd2+ binding deep within the intracellular vestibule in the open state. Interpreted with a P2X2 receptor structural model of the closed state, our data suggest that the channel gate opens near Thr-336/Thr-339 and is accompanied by movement of the pore-lining regions, which narrow toward the cytosolic end of TM2 in the open state. Such transitions would relieve the barrier to ion flow and render the intracellular vestibule less splayed during channel opening in the presence of ATP.
Keywords: Membrane Proteins, Purinergic Receptor, Receptor Structure-Function, Receptors, Signal Transduction, P2X, Gating, Ion Channel
Introduction
P2X receptors (P2X1–P2X7) are cell surface cation channels that transition from closed to open states upon binding ATP. They represent a physiologically and medically important (1), as well as a structurally distinct family of ligand-gated ion channels that are quite different to Cys-loop and ionotropic glutamate receptors (2–5). P2X receptors are found in many species from several phyla, implying important roles in diverse life forms (6, 7). A key goal is to understand how P2X receptors operate at the chemical level.
The first P2X receptor genes were cloned in 1994 (8, 9) leading to important progress over the last decade in understanding how P2X receptors function (10, 11) using biophysical and biochemical approaches. P2X subunits are thus known to possess intracellular N and C termini and two transmembrane (TM)2 segments separated by a large extracellular loop (12–14). The trimeric architecture of P2X receptors is also well established based on subunit concatemers, blue-native PAGE, and atomic force microscopy experiments (15–18). It is also well established that the P2X pore is lined by TM2 (19–22), with TM1 making little contribution to ion flow (23, 24). Moreover, careful studies of permeation and selectivity have suggested that the same area of TM2 is responsible for both ion selection and channel opening (25–31). Mutational approaches have shed light on the extracellular domain of P2X receptors, including how the ATP binding pocket may form (32), how the 10 conserved cysteines contribute to the fold of the protein (33, 34), and how cations such as Zn2+ profoundly affect receptor function (35–38). Finally, meticulous analysis of voltage-jump relaxations for ATP-evoked currents combined with mutagenesis has suggested that TM2 may display hinge-like flexibility (39, 40), and recently the P2X4 receptor open pore has been imaged with fast scanning atomic force microscopy (41). Most of these past studies were conducted on rat P2X2 (rP2X2) receptors and have provided a basis to explore P2X receptor pores and the gating process.
In 2009, the field was immeasurably advanced by the landmark achievement and report of a crystal structure of the zebrafish (zf) P2X4.1 receptor in the closed state (42). The P2X receptor topology was similar to that of acid-sensing ion channels (42, 43), a possibility that had been raised by earlier modeling studies (44). However, it is important to note that the extracellular domains of P2X and acid-sensing ion channels are distinct with little sequence or structural homology, whereas the pore domains share similar architectures (43). The availability of direct structural information for P2X receptors substantiated most of the aforementioned mutational studies and for the first time provided a framework to understand their atomic origins (5). Of most relevance here, it directly revealed that the P2X pore was indeed formed by TM2 α-helices arranged around a 3-fold molecular axis of symmetry. These were arranged ∼45° to the membrane normal and crossed about halfway along their length, where they presented a hydrophobic barrier (the “gate”) to ion flow (42). In this closed state, the pore resembles an hourglass shape with an extracellular vestibule that extended to Leu-340 (see Fig. 1A for P2X2 numbering). The gate is thought to be formed by an extended hydrophobic slab, with its center near Ala-344. The cytosolic end of TM2 displayed low resolution, but when viewed intracellularly the three TM2 helices were quite splayed with respect to the axis of the pore, forming an inverted cone-like intracellular vestibule starting from Ala-347. In view of the insights afforded by the closed state structure, one key question concerns the transitions in the pore during channel opening (5).
FIGURE 1.

P2X2 receptor TM2 SCAM with Cd2+. A, view of a trimeric rP2X2 receptor homology model based on zfP2X4.1. The TM2 domains are shown in gray. The amino acid sequence alignment to the right shows TM2 for zfP2X4.1 and rP2X2. In this alignment, gray boxes indicate residues that are identical; yellow box shows the only residue in TM2 that is absolutely conserved across all species, with one known exception (see under “Discussion”); and the red residues are those thought to be involved in calcium selectivity (27, 28). The solid line labeled as Gate indicates the region thought to be responsible for the gate in the zfP2X4.1 closed state structure. B, normalized traces show control (100 μm ATP alone) and test traces (100 μm ATP followed by 20 μm Cd2+) for WT rP2X2 and the indicated cysteine mutants. The arrows point to the location of the native amino acids in TM2 on a closed state homology model of the rP2X2 receptor. C, average data for percentage block of ATP-evoked currents by Cd2+ for the amino acids indicated in TM2. Number sign indicates a nonfunctional cysteine mutant. Clear block was observed at I332C, T336C, T339C, V343C, D349C, and L353C. Augmentation of the current was observed at G342C. All the recordings were performed at −60 mV. Asterisks indicate significant differences (see text).
Substituted cysteine accessibility mutagenesis (SCAM) (45, 46) has been used extensively in membrane protein research and has been valuable to demonstrate the role of TM2 in P2X receptors (10, 11, 24). However, a complete view of how TM2 transitions during channel opening is an ongoing goal for the field, not least because of inconsistencies between studies that have employed SCAM using different cysteine reactive reagents. Moreover, in one past study, data were not presented for many of the amino acids in TM2 (particularly in the cytosolic half), and of course, all these experiments were carried out prior to the availability of structural models (42). The most recent SCAM study places the narrowest part of the pore (the channel gate) in the outer half of TM2 but falls short of identifying it more precisely (24). Given that the pore is perhaps the most fundamental feature of any ion channel, we assessed transitions in TM2 during opening of the P2X2 receptor and interpreted our data within the context of a closed state homology model based on the zfP2X4.1 structure (42).
EXPERIMENTAL PROCEDURES
Homology Model of rP2X2 Receptor in the Closed State
A sequence alignment using ClustalW against zebrafish P2X4.1 was used to thread the rat P2X2 structure onto the zebra fish P2X4 structure (42) (PDB code 3H9V) using default parameters of the program MODELLER (47). The threaded structure was then manually inspected using Coot (48) and energy-minimized using Refmac (49). The model was validated in Coot showing that 98.8% of the residues reside in the most favorable region of the Ramachandran plot. The coordinates are submitted as supplemental material.
Molecular Biology
Some of the cysteine mutants were provided to us by Drs. T. Egan and M. Voigt. Others were generated using rat WT P2X2 in pcDNA3.1 as a template, which was available from previous work (50). Point mutations were introduced by QuikChange mutagenesis (Stratagene), and all constructs were sequenced before electrophysiological testing. cDNAs were propagated in DH5α Escherichia coli, and plasmids were purified using standard techniques.
HEK-293 Cell Transfection
HEK-293 cells (obtained from the ATCC) were maintained in 75-cm2 cell culture flasks (Corning Glass) in Dulbecco's modified Eagle's medium/F-12 media with Glutamax (Invitrogen) supplemented with 10% fetal bovine serum and penicillin/streptomycin. Cells were grown in a humidified atmosphere of 95% air, 5% CO2 at 37 °C in a cell culture incubator. The cells were split 1 in 10 when confluence reached 60–90%, which was generally every 4 days. Cells were prepared for transfection by plating onto 6-well plates at the time of splitting about 3 days before transfection. They were transfected at ∼60% confluence. For transient expression in HEK-293 cells, we used 0.5–1 μg of plasmid cDNA and the Effectene transfection reagent (Qiagen) for each well of a 6-well plate. The manufacturer's instructions were followed, with 4 μl of enhancer and 10 μl of Effectene used for each transfection. The transfection efficiency was 40–60%. We also used 200 ng of plasmids encoding yellow fluorescent protein as a transfection reporter.
Whole-cell Patch Clamp Recordings
HEK-293 cells were used for recordings 24–48 h post-transfection, gently mechanically dispersed, and plated onto glass coverslips 2–12 h before use. We included the dispersion step (20) to ensure adequate voltage clamp from single spherical cells. The extracellular recording solution included (mm) the following: NaCl 150, MgCl2 1, CaCl2 1, HEPES 10, and glucose 10 (pH 7.4); and the pipette solution included (mm) the following: KCl 154, EGTA 11 and HEPES 10. For some experiments, where stated, we added fresh 10 mm DTT to the intracellular solution within 3 h of its use. Whole-cell voltage clamp recordings were made with ∼3 megohms borosilicate glass electrodes (World Precision Instruments), using an Axopatch 200 or 200B amplifier controlled by a computer running pCLAMP10 software via a Digidata 1366 interface (Axon Instruments). Data were filtered at 2 kHz and digitized at 5 kHz. Drugs were applied to single cells using the Warner Instruments Perfusion Fast Step; solution exchange occurred in ∼4 ms (51). The timing of solution exchange was controlled by custom protocols in pCLAMP 10 and standardized so that ATP and/or Cd2+ was applied at known times to single cells.
Data Analysis
For each of the cysteine mutants we measured ATP-evoked responses (IATP) and also ATP responses during which we switched to Cd2+ (IATP/Cd). We then normalized IATP and IATP/Cd traces and superimposed them (see Fig. 1B). Next, to calculate the component of the current that was blocked by Cd2+, we subtracted IATP/Cd from IATP (see Fig. 4). This resulted in a flat trace in cases where Cd2+ produced no effect, whereas in cases where Cd2+ did block the current this resulted in a waveform that provided a measure Cd2+ association (k+1, in m−1 s−1), steady state block (in %), and dissociation (k−1, in s−1). The modification rate (k+1) was calculated from single exponential fits to the data (e.g. Fig. 4) to measure the time constant (τ, in seconds) for Cd2+ block. k+1 was then estimated as shown in Equation 1,
in units of m−1 s−1 as shown previously (24). In a specific set of experiments, the apparent equilibrium dissociation constant (Kapp) for the block of the ATP-evoked currents by Cd2+ was determined by considering the interaction of Cd2+ with mutant P2X2 receptors (R) with Reaction 1 (52),
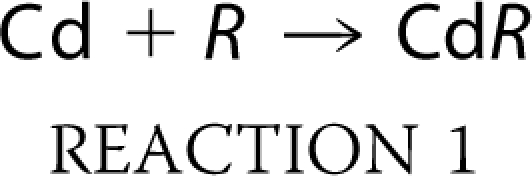 |
where the forward and reverse modification rate constants are given by k+1 and k−1, respectively, as shown in Equations 2 and 3,
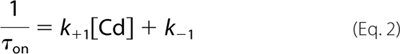 |
 |
and in Equation 4,
 |
where [Cd] is the concentration of Cd2+ (20 μm). All analysis was performed with Clampfit 10.1 (Molecular Devices), Origin 6.1 or 7.5 (OriginLab Corp.), or GraphPad Instat 3.0 (GraphPad Software). Homology models were viewed with PyMOL (Delano Scientific). The figures were assembled in CorelDraw version 12 (Corel Corp.). Data are the mean ± S.E. from at least five experiments. Significances were calculated using Student's t test.
FIGURE 4.

Traces used to assess reversibility and kinetics of Cd2+ block in TM2 of rP2X2. A shows average traces for current waveforms generated by subtracting control (ATP only) and test (ATP followed by ATP/Cd2+) records (as shown in Fig. 1). In the case of WT P2X2, the line was essentially flat as Cd2+ produced no block of ATP-evoked currents. In the other traces that are shown, clear current block is seen upon Cd2+ application that was reversible in all cases except D349C. The red lines are single exponential fits used to measure the time constant for Cd2+ block. The green traces are single exponential fits used to measure the time constant for Cd2+ dissociation. The time constants (τ) were used to measure the apparent affinity of Cd2+ (Kapp). B shows the Kapp values for each of the cysteine mutants that were significantly blocked by Cd2+ in the open state. The Cd2+ affinity was greatest for D349C as the block was irreversible (see text). This affinity was reduced in the P2X2-3T background (see text). All the recordings were performed at −60 mV.
Chemicals
All chemicals used were from Sigma or VWR Scientific.
RESULTS
Initial Studies and Rationale
To understand the rP2X2 pore, we began by generating a homology model (53) of the rP2X2 receptor in the closed state, based on the high resolution crystal structure of zfP2X4.1 (41) that has a sequence identity of 49% for the modeled region. The original report provided two structural models (42) as follows: a low resolution form used for phasing (PDB code 3i5d) and a higher resolution form used to build the final structure (PDB code 3h9v). Using the program MODELLER (47), we generated a model of rP2X2 consisting of residues 30–353, corresponding to zfP2X4.1 residues 32–361 of the higher resolution structure. Because the N and C termini were not present in the crystal structure, these regions were not modeled. Because the focus of this study is TM2, we only refer to the pore segment of the model and do not describe or investigate the extracellular aspects (the PDB file for the rP2X2 homology model is submitted as supplemental material). Fig. 1A shows a view of the rP2X2 receptor based on the model, with TM2 highlighted in gray, as well as an alignment of the residues in TM2 for zfP2X4.1 and rP2X2. In relation to this, Keceli and Kubo (54) have recently published a model of the rP2X2 receptor based on the zfP2X4.1 structure. In that model, the lower resolution form was used as the template. We obtained the PDB file for this homology model and compared it with ours. As expected, the two models were very similar with a root mean square deviation of 1.01 Å over 317 residues. Here, we use rP2X2 numbering when referring to individual amino acids, unless otherwise stated.
For our functional experiments, we did not attempt to delete the N and C termini of rP2X2 receptors to more closely resemble the form used to generate the structure by Kawate et al. (42). This is because past work shows that the N and C termini are vital for the function of P2X2 receptors (10). For example, deletion of the N terminus completely abolishes functional responses (55), and the C terminus of the P2X2 receptors contains a membrane stabilization motif, the absence of which results in negligible ATP-evoked responses (56). Thus, truncated P2X2 receptors would be of little use for the present electrophysiological experiments as they would not be functional. Consistent with these considerations, we note that the truncated form of the zfP2X4.1 receptor also displayed significantly impaired function (42).
We used Cd2+ as the cysteine-reactive probe in our work with P2X2 receptors. This is because MTS reagents and silver have been used extensively in the past but have generated results that are inconsistent between groups (10, 11, 24), and in one study the data were not reported for almost half of the residues in the lower half of TM2 (24). Past work on potassium (57) and amiloride-sensitive sodium channels (58–60) shows that the use of Cd2+ can provide information in addition to the use of MTS reagents and silver (57). This is because the ionic radii of Cd2+ and Na+ are similar at 0.95–1.02 Å. This is relevant as Na+ is the main permeant ion through P2X receptors, and thus Cd2+ is expected to provide data on the parts of the pore that interact with physiological ions. Finally, Cd2+ has not been used in any systematic way in previous studies on P2X receptors. We thus used 20 μm Cd2+ as a probe and interpreted our data within the context of the closed state model of rP2X2. We used 100 μm ATP to activate P2X2 receptors as this is a maximal dose evoking the highest open channel probability (61, 62).
Cd2+ Block at Substituted Cysteines in TM2 of Open P2X2 Receptors
We quantified the ability of 20 μm Cd2+ to block ATP-evoked currents carried by 150 mm Na+ in WT, as well as mutant rP2X2 receptors where every amino acid in TM2 had been independently mutated to a cysteine in the WT P2X2 receptor (Fig. 1A; from Ile-328 to Leu-353; see later for data on the Cys-less like P2X2-3T receptor).
Control recordings were made for all mutants to monitor the degree of desensitization (100 μm ATP was applied for 20–30 s; see black traces in Fig. 1B). Using triple barrel tubing and fast stepping between barrels, recordings were also made for each mutant in TM2 in the presence of ATP and Cd2+. For these experiments, we applied 100 μm ATP for ∼5 s and then switched to a solution containing 100 μm ATP plus 20 μm Cd2+ for 10–20 s. Following this, we returned the cell back to a solution containing 100 μm ATP alone for ∼5 s (Fig. 1B). In other words, Cd2+ was applied when most of the channels were expected to be open (62).
As predicted (11), most of the cysteine mutants in TM2 were functional (with the exception of L334C, A335C, and F346C). In addition, Cd2+ had no effect on WT P2X2, changing the steady state ATP-evoked current insignificantly by <1% (Table 1; n = 5; Fig. 1, B and C). These results are consistent with past work (19, 20). The WT and mutant P2X receptors all showed some degree of desensitization that was variable between mutants; however, by normalizing and superimposing the control and test traces for every mutant we were easily able to identify mutants that were blocked by Cd2+ (Fig. 1B; see under “Data Analysis”).
TABLE 1.
Summary of open state hits for TM2 cysteine mutants in relation to WT rP2X2 receptors
ND indicates that these values could not be calculated because there was no significant block of the ATP-evoked currents. Kapp is the apparent affinity, as described in the text. IATP was measured with 100 mm ATP, and Cd2+ block was assessed with 20 mm of the cation. The traces for WT P2X2 and the mutants listed in this table are provided in the figures. The data are shown as mean ± S.E. from 5 to 6 experiments as indicated in the text. The Kapp for D349C mutants is shown as an estimate because binding at this site was irreversible over the time course of the experiments reported in this study.
| IATP | Desensitization | Block with Cd2+ | Cd2+k+1 (×105) | Cd2+Kapp | |
|---|---|---|---|---|---|
| −nA | tau; s | % | m−1s−1 | μm | |
| WT P2X2 | 3.8 ± 0.5 | 18.5 ± 3.9 | 0.8 ± 2.0 | ND | ND |
| I332C | 3.3 ± 0.7 | 5.9 ± 1.1 | 46 ± 2.4 | 3.9 ± 0.3 | 4.0 ± 0.5 |
| T336C | 2.5 ± 0.5 | 10.2 ± 2.2 | 54 ± 4 | 5.8 ± 0.7 | 2.7 ± 1.0 |
| T339C | 3.8 ± 0.8 | 10.7 ± 4.0 | 48 ± 8 | 2.1 ± 0.5 | 5.7 ± 2.1 |
| G342C | 4.6 ± 0.7 | 0.8 ± 0.1 | −25 ± 4.8 | 1.7 ± 0.2 | 14 ± 0.2 |
| V343C | 2.6 ± 0.7 | 6.9 ± 1.9 | 33 ± 9 | 2.2 ± 1.1 | 126 ± 60 |
| D349C | 0.3 ± 0.07 | 10.8 ± 1.4 | 37 ± 3 | 0.2 ± 0.01 | ∼0.4 |
| L353C | 3.4 ± 0.5 | 1.0 ± 0.2 | 9.3 ± 3.3 | 0.8 ± 0.07 | 32 ± 0.6 |
To account for the contribution of desensitization, we calculated the percentage of Cd2+ block by subtracting the normalized control and test traces for every mutant (e.g. Fig. 4). With this approach, we found that Cd2+ significantly reduced the ATP-evoked currents for six mutants between 10 and 50% when compared with control (I332C, p < 0.01; T336C, p < 0.01; T339C, p < 0.01; V343C, p < 0.01; D349C, p < 0.01; and L353C, p < 0.05, see Fig. 1C and Table 1).
It was notable that the peak 100 μm ATP-evoked currents at D349C (∼300 pA) were smaller than those for the other accessible residues in TM2, which in comparison were all greater than 2 nA per cell (Table 1). The smaller currents at D349C could conceivably be due to a lower open channel probability, lower single channel conductance, lower expression levels, or any combination of these possibilities. It is notable, however, that past work shows that D349C mutants express at levels equivalent to WT P2X2 (19). Irrespectively, D349C ATP-evoked responses were readily and reproducibly recorded and analyzed (e.g. Fig. 1B and Fig. 6) thus posing no problems. The smaller currents for the D349C mutants may explain why the data for this residue were not reported in previous work (20, 24).
FIGURE 6.

Interactions between native Cys-348 and D349C. A, extracellular view from above the membrane showing TM2 helices in the P2X2 pore and the positions of the native cysteine at Cys-348 in relation to the Asp-349 from three subunits. B, representative normalized control and test traces for WT P2X2. 100 μm ATP-evoked inward currents were not blocked by 20 μm Cd2+ (also see Fig. 1). C, as in B but for D349C mutants in the P2X2-3T background. D, as in B but with D349C mutants in the WT P2X2 background. Note the significant block by Cd2+ that exceeds that expected by desensitization alone (broken line). E, as in D but for cells that had been dialyzed with 10 mm DTT for 2–5 min. In this case the kinetics for Cd2+ block were considerably slower, and the degree of block was reduced. F, summary of experiments such as those shown in E for seven cells. The presence of DTT in the intracellular solution markedly reduced the modification rate and the percentage current block by 20 μm Cd2+. All the recordings were performed at −60 mV.
Based on past work with SCAM (46), we interpret the mutants (Fig. 1) in the following manner. If a cysteine mutant was not affected by Cd2+, we concluded that the introduced sulfhydryl either did not project into the water-filled pore or that Cd2+ binding to it had no effect on the Na+ current (i.e. it is in a wide vestibule). Conversely, if a mutant was affected by Cd2+, we concluded that the sulfhydryl did project into the water-filled pore and that Cd2+ binding to it significantly affected Na+ flow through the pore. We made no interpretation of pore orientation of the three nonfunctional mutants in TM2 (Fig. 1C). Therefore, our data indicate that I332C, T336C, T339C, V343C, D349C, and L353C define a water-accessible face of TM2 during the open state of P2X2 receptors. Reassuringly, all our “hits” lined one helical face of TM2 (Fig. 1B), which would be consistent with the observations reported here.
Observations on G342C
Cd2+ significantly augmented ATP-evoked currents for G342C mutants by ∼20% (p < 0.01; Fig. 1B; Table 1). Overall, it was notable that the only residue in our screen to be augmented upon Cd2+ application was the well conserved glycine at position 342 (Fig. 1A). Although Cd2+ did not block ATP-evoked currents at G342C, the modification rate (k+1) for augmentation for this mutant was fast (1.7 ± 0.2 × 105 m−1 s−1; n = 5), and the augmentation was completely reversible (τ = 0.49 ± 0.08 s), implying an apparent affinity kapp of 14.0 ± 2.1 μm (n = 5). The k+1 for Cd2+ at Gly-342 was thus similar to that for Cd2+ block at I332C, T336C, and T339C in the open state (Table 1 and below) indicating that Gly-342 is accessible in the open state. An important role for Gly-342 in accessibility experiments and in channel gating to distinct states has been reported previously (20, 25). Glycine residues are also known to be involved with endowing TM2 with flexibility (39). Our data suggest that Cd2+ binding to G342C increases Na+ current through P2X2 receptors.
Modification Rates for Cd2+ Block at Substituted Cysteines in TM2 of Closed and Open P2X2 Receptors
We considered it important to measure modification rates for Cd2+ block in addition to the steady state percentage block values reported in Fig. 1. First, we measured the time constants for the Cd2+ block of ATP-evoked currents for open I332C, T336C, T339C, V343C, D349C, and L353C mutants and calculated their modification rates as described under “Data Analysis.” Second, we also assessed I332C, T336C, T339C, V343C, D349C, and L353C mutants for block by Cd2+ when applied to closed P2X2 receptors at various time points prior to a test pulse of ATP (Fig. 2). We found that only I332C and T336C were significantly blocked by Cd2+ in the closed state with kinetics on the subsecond time scale (Fig. 2, A and B).
FIGURE 2.

Closed state accessibility for I332C and T336C to Cd2+. A, traces for I332C mutants. The left-hand panel shows two responses evoked by 100 μm ATP applied ∼100 s apart. Note the responses were similar. The middle panel shows one control response and then a second test response to ATP that was preceded in this case by the application of 20 μm Cd2+ for 100 ms. Note that Cd2+ caused a slight outward current as it blocked the standing current for this mutant but also reduced the size of the subsequent ATP-evoked response. It is important to note that Cd2+ was removed from the bath 10 ms before ATP, i.e. was applied only to closed channels. The right-hand graph summarizes data from such experiments whereby Cd2+ was applied for differing durations before a test pulse of ATP. The line is a single exponential fit to the data. Such experiments were used to calculate the closed state modification rate for Cd2+ block (see text for further details). B, as in A but for T336C mutants. Note that in this case that Cd2+ did not cause any outward current because T336C mutants did not display any standing currents. All the recordings were performed at −60 mV.
The modification rates for I332C and T336C in the closed state were not significantly different from those observed for these mutations in the open state (Fig. 3A). This suggests that the sulfhydryls at these positions project into a water-filled cavity that is equally accessible from the outside of the cell in both the closed and open states (Fig. 3B), i.e. in an outer vestibule. Our data with I332C were consistent with the expectations derived from the closed state P2X2 model based on the zfP2X4.1 structure (42). Moreover, the modification rates we report for the outer vestibule are similar to those reported for a potassium channel pore (57).
FIGURE 3.

Modification rates for Cd2+ block for cysteine mutants in TM2 of P2X2. A shows a plot for the rate of modification for cysteine mutants that were accessible in the open state (Fig. 1), which are indicated as open. The closed state modification rates are also shown for I332C and T336C (as shown in Fig. 2). For the other mutants, closed state reactivity could not be measured, as there was no detectable block of the ATP-evoked currents. In these cases, we estimate the closed state rates to be less than 10 m−1 s−1 (these values are shown as estimates with half-filled circles). As reported in the text, we measured significant standing currents for the I332C mutant that were blocked by the bath application of 20 μm Cd2+ (supplemental Fig. 1). The modification rate for block of the standing currents is also shown in this plot with a red symbol. In some cases the symbols are larger than the error bars in A and note that the x axis is logarithmic. B, schematic summarizing the data for mutants that were accessible in the closed and open states for P2X2. A switch in accessibility between closed and open states occurs between T336C and T339C. All the recordings were performed at −60 mV.
With mutant I332C, we detected significant standing currents. In the most straightforward interpretation, this suggests that a fraction of these mutant channels were open in the absence of ATP. Similar standing currents have been well documented in nicotinic receptors with mutations at or near the gate and are usually interpreted to represent stabilization of the open state (63). For I332C, the standing currents (in the absence of ATP) were −82 ± 12 pA/picofarads (n = 6). They were reversibly blocked by 35 ± 4% by 20 μm Cd2+, with a modification rate of ∼3 × 106 m−1 s−1 (supplemental Fig. 1; Fig. 3A). Standing currents were not observed for the other mutants that were accessible in the open state.
The analysis of the experiments described in the preceding paragraphs are summarized in Fig. 3A and show that the fastest open state modification rates were for I332C and T336C (∼6 × 105 m−1 s−1), 3-fold slower for T339C and V343C (∼2 × 105 m−1 s−1), and 30-fold slower for D349C and L353C (2 × 104 m−1 s−1) deeper into the pore. Thus, the modification rate for the water-accessible surface of the pore falls slowly with distance along the length of TM2.
In contrast with the observations at I332C and T336C (Fig. 2), for the T339C, V343C, D349C, and L353C mutants, we did not measure any closed state block of ATP-evoked currents. This was the case even when 20 μm was applied for up to 60 s before a test pulse of ATP (less than 5% change in current for greater than five cells for each mutation). At the most conservative estimate, based on these data, we calculated modification rates to be slower than 10 m−1 s−1 for the closed state. These data thus imply a dramatic increase in modification rate for T339C, V343C, D349C, and L353C upon channel opening (Fig. 3, A and B), presumably as Cd2+ is allowed access to deeper parts of the pore. Because the switch in accessibility between closed and open states occurs near Thr-336 and Thr-339, this implies that the closed channel gate is likely to be between these residues. Based on the closed state model, these data are also consistent with the suggestion that the gate is formed near Thr-336 and Thr-339 (42).
Cd2+ Is Not Coordinated at the Gate in the Open State of P2X2 Receptors
We were cognizant of the possibility that Cd2+ may be irreversibly bound if it interacted with multiple sulfhydryl groups along the axis of the pore. We tested for this possibility and found that Cd2+ block was fully reversible at all accessible sites in the P2X2 pore with the exception of D349C (Fig. 1B and Fig. 4). To analyze the data unambiguously, we arithmetically calculated the waveform of the blocked current by digital subtraction of traces such as those shown in Fig. 1. Thus, for WT P2X2, we observed essentially a flat line (Fig. 4A), which indicated Cd2+ had no effect. In contrast, for I332C, T336C, T339C, V343C, D349C, and L353C mutants, we observed clear waveforms that provided a measure of Cd2+ binding and unbinding to the introduced sulfhydryl moieties (Fig. 4A).
We calculated apparent affinities (Kapp) by analyzing the association and dissociation kinetics for Cd2+ block at I332C, T336C, T339C, V343C, and L353C mutants and assumed a simple bimolecular reaction (see under “Data Analysis”). The apparent affinities were all low at 4.0 ± 0.5, 2.7 ± 1.0, 5.7 ± 2.1, 126 ± 60, and 32.1 ± 5.1 μm for I332C, T336C, T339C, V343C, and L353C (n = 5–11), respectively. These data are consistent with fully reversible Cd2+ binding that was evident from the traces of the mutants that span the gate region of the pore (i.e. I332C to T339C; Fig. 1 and Fig. 4).
Our results at T336C differ from a single previous report using Cd2+ that concluded this cation was irreversibly coordinated at this site (24). However, our data were very clear and consistent between cells (supplemental Fig. 2) and show fully reversible Cd2+ block at T336C. We have no completely satisfying explanation for this difference between our work and the single past observation. We also could not compare our data precisely to the previous report because average data were not included in the aforementioned studies (24). In relation to this, a systematic analysis of Cd2+ coordination number in proteins of known structure shows that three or four cysteines are strongly preferred, with the mean distance to the cation being 2.51 Å (64). However, the distance between Thr-336 residues of open channels, if they are at the gate as our data and the model suggests (see below and Fig. 7), is expected to be greater than 8 Å, which is the minimal open pore diameter based on ion replacement studies (11, 61, 65–69). Thus, we believe Cd2+ is not tightly bound at T336C and most other TM2 hits because the number of available cysteines and the distance between them is not optimal for coordination.
FIGURE 7.

Summary of SCAM data for open and closed P2X2 receptors in relation to the closed state rP2X2 homology model. A, views of the membrane-spanning segment of rP2X2 based on the closed state homology model. The upper panel is a view from the extracellular side looking into the pore, and the middle panel is a view in the plane of the membrane. The residues highlighted in dark blue are Ile-332 and Thr-336, which displayed equally fast modification rates with Cd2+ in the closed and open states. The lower panel shows a schematic summary of the SCAM data for the closed state. For clarity, only two of three subunits are represented. B shows the open state hits highlighted on the closed state model of rP2X2 (upper and middle panels). The residues marked in cyan are T339C, V343C, D349C, and L353C, which displayed Cd2+ modification only in the open state. These were located deeper into the pore after the gate, which we propose is between Thr-336 and Thr-339. The lower panel is a schematic summary of the SCAM hits for the open state. Our data suggest that the open state of the rP2X2 receptor is associated with opening of the gate at Thr-336/Thr-339 and movements near the cytosolic end of the pore that render the TM2 helices significantly less splayed. For simplicity, G342C is not shown in the schematic but is represented in closed state homology model in red. C, surface representations of the extracellular vestibule for rP2X2 receptors. The region of approach to the gate is shown, as viewed from the extracellular side looking into the pore and in the plane of the membrane. The point of most occlusion is at Thr-336, which is consistent with the SCAM data (see B). The approximate distance between the top of TM2 and the point of maximum occlusion (*) at Thr-336 is 10 Å, as shown by the arrow. D, as in C but for the zfP2X4.1 structure. The point of most occlusion is at Leu-340 (*) and the approximate distance from the top of TM2 to this position is 6.5 Å.
Cd2+ Is Tightly Bound at D349C in the Open State of P2X2 Receptors
In contrast to the reversible binding at most sites in TM2, Cd2+ block at D349C was slow (τon = 2.7 ± 0.2 s; n = 10) but also irreversible over 120 s (supplemental Fig. 2). A conservative estimate of the time constant for Cd2+ dissociation is thus >120 s, which would mean the estimated affinity would be higher than ∼400 nm. Based on these empirical observations, we suggest that Cd2+ is more tightly bound at D349C than any other accessible site in TM2 of P2X2, even though the modification rate was slowest here (∼2 × 104 m−1 s−1). D349C, like many residues in the cytosolic half of TM2, escaped similar evaluation by Li et al. (24). However, our finding that D349C and L353C contribute to the inner part of the permeation pathway is consistent with single channel analysis (29) and experiments using MTS reagents (19, 20) on P2X2 receptors. Also, Asp-349 is the only extremely well conserved negative charge in TM2, and our data are consistent with the structural interpretation that this residue helps to attract cations to the inner pore vestibule of P2X receptors (42).
Interactions between Native Cys-348 and D349C Revealed by Studying Modification Rates for Wild Type and “Cys-less Like” P2X2 Receptors
Our data show that the block of ATP-evoked currents by Cd2+ at introduced cysteines was site- and channel state-specific, revealed one face of a TM2 helix, and showed that Cd2+ had no effect on wild type P2X2 receptors (Figs. 1 and 2). Based on previous experiences with the interpretation of SCAM experiments (46), we take these data to indicate a water-accessible surface of TM2 associated with the open state, whereby Cd2+ binding impairs Na+ flow through the pore (Fig. 2B). However, in the most ideal cases, SCAM experiments are performed in channels that have been mutated to remove all native cysteine residues, to generate “Cys-less” proteins (46). This approach is not feasible for P2X receptors as several conserved cysteines form disulfide bonds and are known to be vital for the fold of the protein (42) and for receptor function (33, 34). Instead, we repeated our experiments for I332C, T336C, T339C, V343C, D349C, and L353C mutants in P2X2-3T receptors (24), which lack two “free” cysteines in the P2X2 receptor cytosolic domain (Cys-9 and Cys-430), as well as one in TM2 (Cys-348).
Fig. 5 shows the relationship between the modification rates for I332C, T336C, T339C, V343C, and D349C mutants in WT P2X2 and P2X2-3T backgrounds. For I332C, T336C, T339C, V343C, and L353C mutants, the modification rates were essentially identical between these two constructs, providing strong evidence that native cysteines do not impact the Cd2+ block. However, for D349C, we found that the ability of 20 μm Cd2+ to block the 100 μm ATP-evoked currents was attenuated by more than four orders of magnitude from the expected value in the P2X2-3T background, if the free cysteines had no effect (cyan symbols in Fig. 5).
FIGURE 5.

Modification rates for Cd2+ block at single cysteine mutations in WT P2X2 and P2X2-3T receptors. The diagonal line is the line of identity between the x and y axis. The data for D349C deviate from this line because the modification rate for this residue in the P2X2-3T receptor background was minimal. The other indicated mutants sit close to the line of identity, suggesting that they behave similarly in WT P2X2 and P2X2-3T backgrounds. All the recordings were performed at −60 mV.
The data presented in Fig. 5 suggest that irreversible Cd2+ binding to D349C mutants in the WT P2X2 background was due to an interaction between the native cysteine at position Cys-348 and D349C during the open state of P2X2 receptors. We were intrigued by this empirical observation because the inner aspects of each of the three TM2 helices within the receptor are considerably distanced from each other, relative to the axis of the pore in the closed state structure. The estimated distance between Cys-348 and Asp-349 residues of neighboring subunits is 28.6 and 25.2 Å, respectively (Fig. 6A). Cd2+ coordination prefers four cysteine residues with a mean distance between the cation and the electron donor being 2.51 Å (64). The distance between the α-carbons of Cys-348 and D349C on the same TM2 helix is 3.8 Å. Because tight Cd2+ binding is thought to require at least three or four cysteines (57, 64), these considerations imply that two cysteines from the same TM2 helix are unlikely to result in the tight Cd2+ binding that was observed experimentally. Moreover, the modification rate at D349C was slow in the WT P2X2 background (Fig. 6D) and nonexistent for WT P2X2 alone as well as for D349C in P2X2-3T (Fig. 6, B and C), implying a Kapp of >100 μm. We interpret these data to indicate that Cd2+ is tightly bound at this position, having the possibility to be fully coordinated over extended time periods.
In studies of amiloride-sensitive sodium channels, tight Cd2+ binding by cysteines can be significantly reduced by the application of the reducing agent DTT (60). In light of this, we repeated the experiments shown in Fig. 4D with 10 mm DTT in the intracellular solution (cells were dialyzed for 2–5 min before commencing with the recordings). We found that Cd2+ binding at D349C was significantly reduced in the WT P2X2 background, both in terms of modification rate and the percentage of current block (Fig. 6F). Based on the association and dissociation kinetics for Cd2+ block in cells dialyzed with DTT, we estimated Kapp to be 27.8 ± 4.4 μm (n = 7), close to values for the other single cysteine mutants in TM2 (Fig. 4B). Thus, tight Cd2+ binding at D349C requires the native Cys-348 and is reduced by DTT. We interpret these data to indicate that TM2 helices are significantly less divergent in the open state and present Cys-348 and D349C from neighboring subunits to the permeation pathway in the intracellular vestibule.
DISCUSSION
There are three main findings of this study. First, by using Cd2+ as a cysteine-reactive probe, we have identified the water-accessible face of TM2 lining the extra- and intracellular vestibules in the closed and open state of P2X2 receptors. Second, our data suggest that the closed channel gate is between Thr-336 and Thr-339. Third, our data suggest the intracellular vestibule is more restrictive in the open state, as compared with its dimensions in the closed state homology model. These features are summarized in schematics shown in Fig. 7, A–C.
When the first P2X receptor cDNAs were cloned (8, 9), sequence analysis suggested that the region preceding TM2 may form a re-entrant loop (70). This hypothesis meant that the early SCAM and mutagenesis experiments aimed at understanding the pore largely focused on TM2 of rP2X2 receptors. These experiments showed that this domain directly contributed to the ion permeation pathway (19, 20) and that P2X channels could be locked into distinct permeability states by state-selective mutants in TM2 (25, 26). However, these experiments did not support the idea of a re-entrant loop as a key feature of the P2X receptor pore, a view confirmed by the recent crystal structure (42).
Subsequently, attention shifted to TM1, in part by the realization that an ion-selective pore may require greater than three α-helical TM2 domains (71). In contrast to the experiences with TM2, SCAM experiments aimed at elucidating a role for TM1 were less clear, leading to the conclusion that this domain affected channel gating (21, 22, 72). Later, the region responsible for calcium ion selection was identified in TM2 using mutagenesis coupled with relative ion permeability and fractional calcium current measurements (27, 28). When identical methods were applied to TM1, they revealed no evidence that this domain contributed to ion selection or flow (23). Thus, by early 2008, it was well established that the major pore-lining segment in P2X receptors was TM2. This realization gained further support from Swartz and co-workers (24) who systematically showed that the main pore-lining segment was indeed TM2. Furthermore, this study also suggested that the gate was located in the outer half of TM2 (24).
The publication of the zfP2X4.1 structure in the closed state laid to rest any lingering concerns about which TM domains contribute to the P2X pore (42). The structure showed that three α-helical TM2 domains form the pore and that the TM1 domains are located more peripherally. The TM2 domains cross about halfway along their length and thus support the view that the closed channel gate is located in the outer half of TM2. The structure also suggested that the gate may be formed by an extended region of TM2 stretching from Leu-340 to Ala-347 of zfP2X4.1 (i.e. Ile-332 to Thr-339 of P2X2).
The experiments reported in this study began with the published mutagenesis and the recent structure as a framework. We aimed to functionally evaluate the location of the closed channel gate and also to assess accessibility to deeper parts of the pore during channel opening. Our interpretations are based on the best available structural data from Gouaux and co-workers (42). It is important to note that zfP2X4.1 and rP2X2 receptors are distinct receptor isoforms and that the structure is of a mutated form of zfP2X4.1 (42). Thus, our interpretations should be appraised with these considerations in mind. However, our functional measurements for rP2X2 are consistent with expectations based on the structure of zfP2X4.1, providing confirmatory validation of the P2X2 homology model.
For the closed state, our data are consistent with the zfP2X4.1 structure. We find that Ile-332 and Thr-336 are freely accessible in the closed state with fast modification rates consistent with exposure of the introduced sulfhydryl groups to a water-filled extracellular vestibule (Fig. 7A). These amino acids correspond to Leu-340 and Ala-344 of the zfP2X4.1 protein, thought to be part of an extended gate, described as a “hydrophobic slab,” stretching to Ala-347 (Thr-339 of P2X2). In this structure, the center of the gate was located at Ala-344, which corresponds to the last residue that was accessible in the closed state of P2X2 receptors (Thr-336). The structure showed that the pore was most occluded by the side chain of Leu-340 (Ile-332 of rP2X2). In our homology model of rP2X2 receptors the maximal occlusion is predicted to be at Thr-336, i.e. one helical turn away (Fig. 7C). Consistent with this, our experimental data also show that the closed channel gate for Cd2+ ions is at Thr-336, i.e. at the center of the hydrophobic slab (Ala-344 of zfP2X4.1) proposed by Kawate et al. (42) to form an extended gate. From this perspective, the agreement between our functional measurements and the closed state structure is strong.
Why is the point of maximum occlusion in the structure at position Leu-340 (Ile-332 of rP2X2), although our empirical data supported by the homology model suggest it is at Thr-336? If the gate is formed by an extended hydrophobic slab, one possibility is that the precise occlusion point may vary in a receptor sequence and state-dependent manner. Thus, there is an Ala (small side chain) at the equivalent of Thr-336 in zfP2X4.1, but in most P2X isoforms and orthologues this is often a bulkier residue (usually Ser or Thr). Presumably, the pore would be more occluded at this position for subunits with more voluminous side chains (e.g. rP2X2). In addition to this, the side chain of Ile at 332 of rP2X2 is smaller than that of Leu at 340 in zfP2X4.1, thereby leading to a less occluded pore at this position in rP2X2. Another possibility is that the closed state measured physiologically may be different from the closed state of the receptor in the crystal without a lipid environment. If this slightly changed the Leu-340 rotamer position, this could allow access of cations to Ala-344 in the closed state. Further work with molecular dynamics simulations may shed light on this issue, but our experiments for a functional protein suggest the gate for Cd2+ is at Thr-336, i.e. one helical turn lower than Ile-332 (Leu-340 of zfP2X4.1; see Fig. 7, C and D), but nonetheless clearly within the area identified as the gate region by Kawate et al. (42).
As expected, upon channel opening we observed increased accessibility to deeper parts of the TM2 domain (Fig. 7, A and B). Specifically, the first residue (from the outside in) that became accessible in the open state was Thr-339, corresponding to Ala-347 of the zfP2X4.1 structure and defining the lower limit of the hydrophobic slab (42). Based on these functional data, interpreted within the framework of the closed state model, we propose that the closed gate for Cd2+ is between Thr-336 and Thr-339. This suggestion would be consistent with past work by us (28) and by others (27, 29) demonstrating that polar residues in this region contribute to ion selection and that the gate and selectivity filter are both cast from the same region of the protein. This view is also supported by interpretations of the closed state structure (42).
We also detected significant Cd2+ block at deeper sites in the pore with a pattern highlighting one helical face of TM2 (Fig. 3; Fig. 7, A and B). In addition, we observed augmentation of ATP-evoked currents at G342C mutants by Cd2+. In the simplest interpretation, this suggests that Cd2+ binding to G342C stabilizes the open state. Indeed, previous work has shown that mutants at Gly-342 can lock the channel into distinct open and closed states (25). However, further work is warranted to fully explore the role of Gly-342, as another interpretation is that Cd2+ binding to G342C reverses channel desensitization thus leading to the augmented current that was observed. This view is not negated by the past mutagenesis work as many mutants at Gly-342 also displayed altered desensitization kinetics (25).
Further into the pore, we found that V343C also lines the permeation pathway with a modification rate somewhat slower than at I332C and T336C implying this region corresponds to a wider cavity internal to the gate. Accordingly, in the closed zfP2X4.1 structure, this corresponds to Leu-351 at the apex of the inverted cone that defines the start of a wider intracellular vestibule (42).
One of the interesting features of the closed zfP2X4.1 receptor is that the lower aspect of TM2 is somewhat splayed and forms an intracellular vestibule (radius ∼6 Å) that includes the well conserved negative charge of Asp-357 (Asp-349 of rP2X2). This residue was also accessible in the open state of rP2X2 receptors, but only if the D349C mutation was introduced in the WT P2X2 background. Our data suggest that Cd2+ is tightly bound at this site in the open state, reminiscent of past work (19, 20). Perhaps the most relevant finding for D349C is that Cd2+ binding was tight and irreversible over the time course of our experiments. The possibility that the irreversible nature of the block, and therefore the underlying change, was due to Cd2+ itself is worth considering, especially if the interaction between cysteines from distinct subunits is a low probability event that is stabilized by Cd2+. Such an interpretation would not detract from our empirical observations or conclusions. We note, however, that the irreversible nature of the block at D349C is not specific to Cd2+, as it has been observed with MTS reagents (20). Overall, we interpret our findings to suggest that the intracellular vestibule formed by the lower half of TM2 in P2X2 receptors may slowly change shape during the activated state (explaining the slow modification rate) and thus tightly bind Cd2+ leading to substantial block of Na+ currents through the pore. This would also explain smaller Cd2+ block at L353C mutants (Figs. 1 and 7) (20). It is interesting to note that mutations at Asp-349 alter desensitization of P2X2 receptors, implying that this residue may regulate the equilibrium between open and desensitized states (73). Past work shows that aspartate at the position equivalent to Asp-349 in hP2X5 receptors is vital for receptor assembly, perhaps via helix-helix interactions (74). However, these experiments do not exclude additional functional roles for this aspartate for the majority of receptors in which this residue is absolutely conserved. In fact, we are aware of only one P2X receptor orthologue lacking aspartate at this location (from the green algae Osteococcus tauri), and this displays unusually low calcium permeability, which can be increased by re-introducing aspartate (65). Moreover, recent single channel measurements directly reveal that changing the side chain at Asp-349 can affect conductance (29). These studies support our suggestion that this region of the pore contributes in a meaningful way to ion flow. Furthermore, the slow rate of block by Cd2+ at D349C is consistent with seconds time scale conformational changes that have been measured for the N and C termini of P2X2 receptors using spectroscopic methods (50). These termini start where the TM domains end. We could not incorporate the N and C termini into the closed state rP2X2 model, as both were not observed in the zfP2X4.1 structure (42) upon which our model is based. Once we have a structure of a full-length P2X receptor or of a receptor in the open state, it will be interesting to examine if the intracellular vestibule formed by the lower limits of TM2 is similar to that of the closed state. Another useful approach for the future has been provided by Shinozaki et al. (41) who used fast-scanning atomic force microscopy to image changes in the pore of single rat P2X4 receptors. In those studies, the pore was shown to dilate considerably in the absence of Ca2+ ions but undergo smaller changes in the presence of Ca2+ (41). Our data on TM2 most likely reflect this latter state as we performed our experiments under physiological levels of Ca2+.
In past work using Ca2+-free conditions, we and others have shown that rP2X2 receptors undergo “pore dilation” (25, 26, 61), but we did not investigate this property in this study. However, in future work it would be interesting to return to the issue of whether larger conformational changes occur in the P2X2 receptor pore in the absence of Ca2+ using systematic analysis of both TM1 and TM2. Such experiments may be best approached by capitalizing on the data and mutants reported in this study, in combination with atomic force microscopy (41) or electron paramagnetic resonance (75) on isolated P2X2 receptors carrying mutants that lock the channels into either small or large pore states (10). These experiments will be feasible when methods to purify functional P2X2 receptors in sufficient quantities have been developed and would negate the complications of “host cell type” on pore properties (10). As far as we know, such methods do not currently exist.
In summary, our experiments are consistent with the closed state structure of the zfP2X4.1 receptor and also reveal an accessible surface of the rP2X2 pore during the open state. Our studies thus provide the basis for more detailed evaluations of the P2X receptor pore in the closed, open, and desensitized states in the post structure era (5, 42). This would contribute to a more complete understanding of channel gating and to a better appreciation of the diversity of mechanisms employed by ion channels. It will be particularly interesting to functionally determine whether common gating principles (76) exist among structurally diverse ligand-gated P2X, nicotinic, and glutamate receptors.
Supplementary Material
Acknowledgments
We thank Dr. Y. Kubo for sharing details of the rP2X2 homology model. We are grateful to Drs. G. Buell, T. Egan, M. Voigt, and K. Swartz for sharing plasmids from their laboratories. We thank Dr. H. Singh for contributions in the early stages of this work. We also thank Drs. K. Philipson, D. Papazian, T. Egan, E. Shigetomi, and E. Toulme for comments and discussions.
This work was supported, in whole or in part, by a National Institutes of Health Grant GM070925-04. This work was also supported by unrestricted funds from UCLA (to B. S. K.), by a S&R Foundation Ryuji Ueno Award for Ion Channels or Barrier Function Research from the American Physiological Society and by a Stein-Oppenheimer Foundation Endowment Award (to B. S. K.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2 and File 1.
- TM
- transmembrane domain
- DTT
- dithiothreitol
- MTS
- methanethiosulfonate
- SCAM
- substituted cysteine accessibility mutagenesis
- WT
- wild type
- zf
- zebrafish
- r
- rat
- PDB
- Protein Data Bank.
REFERENCES
- 1.Burnstock G. (2007) Physiol. Rev. 87, 659–797 [DOI] [PubMed] [Google Scholar]
- 2.North R. A. (1996) CIBA Found. Symp. 198, 91–109 [DOI] [PubMed] [Google Scholar]
- 3.Khakh B. S. (2001) Nat. Rev. Neurosci. 2, 165–174 [DOI] [PubMed] [Google Scholar]
- 4.Khakh B. S., North R. A. (2006) Nature 442, 527–532 [DOI] [PubMed] [Google Scholar]
- 5.Young M. T. (2010) Trends Biochem. Sci. 35, 83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fountain S. J., Burnstock G. (2009) Purinergic Signal. 5, 269–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burnstock G., Verkhratsky A. (2009) Acta Physiol. 195, 415–447 [DOI] [PubMed] [Google Scholar]
- 8.Brake A. J., Wagenbach M. J., Julius D. (1994) Nature 371, 519–523 [DOI] [PubMed] [Google Scholar]
- 9.Valera S., Hussy N., Evans R. J., Adami N., North R. A., Surprenant A., Buell G. (1994) Nature 371, 516–519 [DOI] [PubMed] [Google Scholar]
- 10.North R. A. (2002) Physiol. Rev. 82, 1013–1067 [DOI] [PubMed] [Google Scholar]
- 11.Egan T. M., Samways D. S., Li Z. (2006) Pflugers Arch. 452, 501–512 [DOI] [PubMed] [Google Scholar]
- 12.Newbolt A., Stoop R., Virginio C., Surprenant A., North R. A., Buell G., Rassendren F. (1998) J. Biol. Chem. 273, 15177–15182 [DOI] [PubMed] [Google Scholar]
- 13.Torres G. E., Egan T. M., Voigt M. M. (1998) FEBS Lett. 425, 19–23 [DOI] [PubMed] [Google Scholar]
- 14.Torres G. E., Egan T. M., Voigt M. M. (1998) Biochemistry 37, 14845–14851 [DOI] [PubMed] [Google Scholar]
- 15.Stoop R., Thomas S., Rassendren F., Kawashima E., Buell G., Surprenant A., North R. A. (1999) Mol. Pharmacol. 56, 973–981 [DOI] [PubMed] [Google Scholar]
- 16.Nicke A., Bäumert H. G., Rettinger J., Eichele A., Lambrecht G., Mutschler E., Schmalzing G. (1998) EMBO J. 17, 3016–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrera N. P., Ormond S. J., Henderson R. M., Murrell-Lagnado R. D., Edwardson J. M. (2005) J. Biol. Chem. 280, 10759–10765 [DOI] [PubMed] [Google Scholar]
- 18.Jiang L. H., Kim M., Spelta V., Bo X., Surprenant A., North R. A. (2003) J. Neurosci. 23, 8903–8910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rassendren F., Buell G., Newbolt A., North R. A., Surprenant A. (1997) EMBO J. 16, 3446–3454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Egan T. M., Haines W. R., Voigt M. M. (1998) J. Neurosci. 18, 2350–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang L. H., Rassendren F., Spelta V., Surprenant A., North R. A. (2001) J. Biol. Chem. 276, 14902–14908 [DOI] [PubMed] [Google Scholar]
- 22.Haines W. R., Migita K., Cox J. A., Egan T. M., Voigt M. M. (2001) J. Biol. Chem. 276, 32793–32798 [DOI] [PubMed] [Google Scholar]
- 23.Samways D. S., Migita K., Li Z., Egan T. M. (2008) J. Biol. Chem. 283, 5110–5117 [DOI] [PubMed] [Google Scholar]
- 24.Li M., Chang T. H., Silberberg S. D., Swartz K. J. (2008) Nat. Neurosci. 11, 883–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khakh B. S., Bao X. R., Labarca C., Lester H. A. (1999) Nat. Neurosci. 2, 322–330 [DOI] [PubMed] [Google Scholar]
- 26.Virginio C., MacKenzie A., Rassendren F. A., North R. A., Surprenant A. (1999) Nat. Neurosci. 2, 315–321 [DOI] [PubMed] [Google Scholar]
- 27.Migita K., Haines W. R., Voigt M. M., Egan T. M. (2001) J. Biol. Chem. 276, 30934–30941 [DOI] [PubMed] [Google Scholar]
- 28.Egan T. M., Khakh B. S. (2004) J. Neurosci. 24, 3413–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao L., Broomhead H. E., Young M. T., North R. A. (2009) J. Neurosci. 29, 14257–14264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao L., Young M. T., Broomhead H. E., Fountain S. J., North R. A. (2007) J. Neurosci. 27, 12916–12923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khakh B. S., Lester H. A. (1999) Neuron 23, 653–658 [DOI] [PubMed] [Google Scholar]
- 32.Evans R. J. (2009) Eur. Biophys. J. 38, 319–327 [DOI] [PubMed] [Google Scholar]
- 33.Ennion S. J., Evans R. J. (2002) Mol. Pharmacol. 61, 303–311 [DOI] [PubMed] [Google Scholar]
- 34.Clyne J. D., Wang L. F., Hume R. I. (2002) J. Neurosci. 22, 3873–3880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clyne J. D., LaPointe L. D., Hume R. I. (2002) J. Physiol. 539, 347–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagaya N., Tittle R. K., Saar N., Dellal S. S., Hume R. I. (2005) J. Biol. Chem. 280, 25982–25993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tittle R. K., Power J. M., Hume R. I. (2007) J. Biol. Chem. 282, 19526–19533 [DOI] [PubMed] [Google Scholar]
- 38.Tittle R. K., Hume R. I. (2008) J. Neurosci. 28, 11131–11140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fujiwara Y., Keceli B., Nakajo K., Kubo Y. (2009) J. Gen. Physiol. 133, 93–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kubo Y., Fujiwara Y., Keceli B., Nakajo K. (2009) J. Physiol. 587, 5317–5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shinozaki Y., Sumitomo K., Tsuda M., Koizumi S., Inoue K., Torimitsu K. (2009) PLoS Biol. 7, e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawate T., Michel J. C., Birdsong W. T., Gouaux E. (2009) Nature 460, 592–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gonzales E. B., Kawate T., Gouaux E. (2009) Nature 460, 599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guerlet G., Taly A., Prado de Carvalho L., Martz A., Jiang R., Specht A., Le Novère N., Grutter T. (2008) Biochem. Biophys. Res. Commun. 375, 405–409 [DOI] [PubMed] [Google Scholar]
- 45.Akabas M. H., Stauffer D. A., Xu M., Karlin A. (1992) Science 258, 307–310 [DOI] [PubMed] [Google Scholar]
- 46.Karlin A., Akabas M. H. (1998) Methods Enzymol. 293, 123–145 [DOI] [PubMed] [Google Scholar]
- 47.Sali A., Blundell T. L. (1993) J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 48.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 49.Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 50.Chaumont S., Khakh B. S. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 12063–12068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richler E., Chaumont S., Shigetomi E., Sagasti A., Khakh B. S. (2008) Nat. Methods 5, 87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khakh B. S., Proctor W. R., Dunwiddie T. V., Labarca C., Lester H. A. (1999) J. Neurosci. 19, 7289–7299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rhodes G. (1993) Crystallography Made Crystal Clear, 2nd Ed., pp. 237–246, Academic Press, San Diego, CA [Google Scholar]
- 54.Keceli B., Kubo Y. (2009) J. Physiol. 587, 5801–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Torres G. E., Egan T. M., Voigt M. M. (1999) J. Biol. Chem. 274, 22359–22365 [DOI] [PubMed] [Google Scholar]
- 56.Chaumont S., Jiang L. H., Penna A., North R. A., Rassendren F. (2004) J. Biol. Chem. 279, 29628–29638 [DOI] [PubMed] [Google Scholar]
- 57.Liu Y., Holmgren M., Jurman M. E., Yellen G. (1997) Neuron 19, 175–184 [DOI] [PubMed] [Google Scholar]
- 58.Takeda A. N., Gautschi I., van Bemmelen M. X., Schild L. (2007) J. Biol. Chem. 282, 31928–31936 [DOI] [PubMed] [Google Scholar]
- 59.Kellenberger S., Gautschi I., Pfister Y., Schild L. (2005) J. Biol. Chem. 280, 7739–7747 [DOI] [PubMed] [Google Scholar]
- 60.Sheng S., Perry C. J., Kashlan O. B., Kleyman T. R. (2005) J. Biol. Chem. 280, 8513–8522 [DOI] [PubMed] [Google Scholar]
- 61.Eickhorst A. N., Berson A., Cockayne D., Lester H. A., Khakh B. S. (2002) J. Gen. Physiol. 120, 119–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ding S., Sachs F. (1999) J. Gen. Physiol. 113, 695–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lester H. A., Dibas M. I., Dahan D. S., Leite J. F., Dougherty D. A. (2004) Trends Neurosci. 27, 329–336 [DOI] [PubMed] [Google Scholar]
- 64.Dokmanić I., Sikić M., Tomić S. (2008) Acta Crystallogr. D Biol. Crystallogr. 64, 257–263 [DOI] [PubMed] [Google Scholar]
- 65.Fountain S. J., Cao L., Young M. T., North R. A. (2008) J. Biol. Chem. 283, 15122–15126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fountain S. J., Parkinson K., Young M. T., Cao L., Thompson C. R., North R. A. (2007) Nature 448, 200–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Evans R. J., Lewis C., Virginio C., Lundstrom K., Buell G., Surprenant A., North R. A. (1996) J. Physiol. 497, 413–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Agboh K. C., Webb T. E., Evans R. J., Ennion S. J. (2004) J. Biol. Chem. 279, 41650–41657 [DOI] [PubMed] [Google Scholar]
- 69.Ding S., Sachs F. (1999) J. Membr. Biol. 172, 215–223 [DOI] [PubMed] [Google Scholar]
- 70.MacKinnon R. (1995) Neuron 14, 889–892 [DOI] [PubMed] [Google Scholar]
- 71.Spencer R. H., Rees D. C. (2002) Annu. Rev. Biophys. Biomol. Struct. 31, 207–233 [DOI] [PubMed] [Google Scholar]
- 72.Haines W. R., Voigt M. M., Migita K., Torres G. E., Egan T. M. (2001) J. Neurosci. 21, 5885–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou Z., Monsma L. R., Hume R. I. (1998) Biochem. Biophys. Res. Commun. 252, 541–545 [DOI] [PubMed] [Google Scholar]
- 74.Duckwitz W., Hausmann R., Aschrafi A., Schmalzing G. (2006) J. Biol. Chem. 281, 39561–39572 [DOI] [PubMed] [Google Scholar]
- 75.Perozo E., Cuello L. G., Cortes D. M., Liu Y. S., Sompornpisut P. (2002) Novartis Found. Symp. 245, 146–158 [DOI] [PubMed] [Google Scholar]
- 76.Unwin N. (2000) Philos. Trans. R. Soc. Lond. B Biol. Sci. 355, 1813–1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.