

Abstract
We have studied oxygenation of fatty acids by cell extract of Pseudomonas aeruginosa 42A2. Oleic acid ((9Z)-18:1) was transformed to (10S)-hydroperoxy-(8E)-octadecenoic acid ((10S)-HPOME) and to (7S,10S)-dihydroxy-(8E)-octadecenoic acid (7,10-DiHOME). Experiments under oxygen-18 showed that 7,10-DiHOME contained oxygen from air and was formed sequentially from (10S)-HPOME by isomerization. (10R)-HPOME was not isomerized. The (10S)-dioxygenase and hydroperoxide isomerase activities co-eluted on ion exchange chromatography and on gel filtration with an apparent molecular size of ∼50 kDa. 16:1n-7, 18:2n-6, and 20:1n-11 were also oxygenated to 7,10-dihydroxy fatty acids, and (8Z)-18:1 was oxygenated to 6,9-dihydroxy-(7E)-octadecenoic acid. A series of fatty acids with the double bond positioned closer to ((6Z)-18:1, (5Z,9Z)-18:2) or more distant from the carboxyl group ((11Z)-, (13Z)-, and (15Z)-18:1) were poor substrates. The oxygenation mechanism was studied with [7S-2H]18:1n-9, [7R-2H]18:2n-6, and [8R-2H]18:2n-6 as substrates. The pro-R hydrogen at C-8 was lost in the biosynthesis of (10S)-HPODE, whereas the pro-S hydrogen was lost and the pro-R hydrogen was retained at C-7 during biosynthesis of the 7,10-dihydroxy metabolites. Analysis of the fatty acid composition of P. aeruginosa revealed relatively large amounts of (9E/Z)-16:1 and (11E/Z)-18:1 and only traces of 18:1n-9. We found that (11Z)-18:1 (vaccenic acid) was transformed to (11S,14S)-dihydroxy-(12E)-octadecenoic acid and to a mixture of 11- and 12-HPOME, possibly due to reverse orientation of (11Z)-18:1 at the active site compared with oleic acid. The reaction mechanism of the hydroperoxide isomerase suggests catalytic similarities to cytochrome P450.
Keywords: Cytochromes/Cytochrome P450, Enzymes/Lipid, Enzymes/Oxidase, Lipid/Fatty Acid, Methods/HPLC, Methods/Mass Spectrometry
Introduction
Unsaturated fatty acids can be oxygenated to a vast array of lipid mediators by lipoxygenases, heme-containing fatty acid dioxygenases, and P450.5 Lipoxygenases occur in plants and mammals and transform polyunsaturated fatty acids to hydroperoxides, which can be further transformed into leukotrienes, jasmonic acid, and other products of biological significance (1–3). Cyclooxygenases and fungal diol synthases also form hydroperoxides, which are isomerized to prostaglandin endoperoxides and to dihydroxy fatty acids, respectively (4, 5). P450 catalyzes hydroxylation of the ω-end of fatty acids, epoxidation of double bonds, and hydroxylation with double bond migration (6). Oxygenation of fatty acids has mainly been studied in eukaryotes, but many studies now show that Pseudomonas aeruginosa can oxidize unsaturated fatty acids to hydroperoxy and dihydroxy metabolites (7–15).
P. aeruginosa is an important opportunistic pathogen of immunocompromised patients, burn victims, and patients with cystic fibrosis (16, 17). Infections are difficult to treat with antibiotics because this Gram-negative bacterium also forms toxins and other virulence factors. In 1988, P. aeruginosa 42A2 was demonstrated to oxidize oleic acid to a new surfactant, a dihydroxy fatty acid metabolite (7). This product was later identified as (7S,10S)-dihydroxy-(8E)-octadecenoic acid ((7S,10S)-DiHOME) by Hou and co-workers using the P. aeruginosa strain PR3 (8, 10, 15). We now know that production of (7S,10S)-DiHOME is a characteristic of P. aeruginosa (13). Strains 42A2 and PR3 also formed 10-hydro(per)oxy-(8E)-octadecenoic acid (10-H(P)OME) (18), and the chirality was determined to be 10S (11). Palmitoleic and ricinoleic acids were also found to be oxygenated in the same way as oleic acid (9, 12–14).
The mechanisms of biosynthesis of (10S)-HPOME and (7S,10S)-DiHOME are not well understood. The genome of P. aeruginosa was sequenced in 2000, and this provides important information. One sequence (PA1169 of the Pseudomonas genome data base (19)) was designated as a putative lipoxygenase. A probable lipoxygenase was also detected in the genome of a second bacterium, Sorangium cellulosom (20). Vance et al. (21) found that P. aeruginosa secreted an arachidonate 15-lipoxygenase and expressed this protein by aid of the PA1169 sequence. As far as is known, lipoxygenases only oxidize oleic acid slowly (22).
On the other hand, heme-containing fatty acid dioxygenases can oxidize oleic acid to hydroperoxy fatty acids, but there are no apparent homologues to cyclooxygenases, (10R)-DOX, linoleate diol synthase, or α-DOX in the genome of P. aeruginosa. Cytochrome P450 can oxygenate fatty acids and isomerize hydroperoxy fatty acids (6, 23). Interestingly, the genome of P. aeruginosa contains two genes with homology to P450 (PA2475 and PA3331). (10S)-HOME has been suggested to be transformed to (7S,10S)-DiHOME by hydroxylation (11, 15), but isomerization of 10S-HPOME to the 7,10-diol is also a possibility as judged from the hydroperoxide isomerase activity of linoleate diol synthase (5, 23, 24).
The goal of the present study was to determine the mechanism of biosynthesis of 7,10-DiHOME. Our first objective was to partially purify the diol synthase activity and to determine the apparent molecular weight. Second, the mechanism of biosynthesis of 7,10-dihydroxy metabolites was investigated with (i) stereospecifically deuterated fatty acids or (ii) (10S)- and (10R)-HPOME as substrates and with (iii) experiments under oxygen-18 gas. We also performed a systematic investigation of the substrate specificity and products with a series of regioisomeric 18:1 fatty acids. Finally, we found that P. aeruginosa was rich in vaccenic acid, and we therefore investigated its oxygenation by P. aeruginosa 42A2 by semipurified cell extracts.
EXPERIMENTAL PROCEDURES
Materials
Fatty acids were dissolved in ethanol and stored in stock solutions (50–100 mm) at −20 °C. 18:1n-9 (99%), 18:1n-7, 18:2n-6 (99%), 18:3n-6 (99%), and 18:3n-3 (99%) were from Sigma; all other fatty acids (98–99%) were from Larodan (Malmö, Sweden) or Lipidox (Stockholm, Sweden). Oleic acid (90%) from Fluka was used to induce enzyme expression. Piperonyl butoxide (Fluka) and 1-aminobenzotriazole (Fluka) were dissolved in ethanol (1 m stock solutions). [(8R)-2H]18:2n-6 (64% 2H) and [(7R)-2H]18:2n-6 (41% 2H) were prepared as described (5, 25)6; [(7S)-2H]18:1 (∼70% 2H) was obtained by the same method. Oxygen-18 (99%) was from Icon (Summit, NJ). P. aeruginosa (42A2) was grown as described (27). Precoated TLC plates (0.25-mm Silica Gel 60A, 20 × 20 or 5 × 20 cm), molybdatophosphoric acid, and peptone from casein and soy meal were from Merck. SepPak/C18 cartridges were from Waters (Milford, MA).
Preparation and Purification of Diol Synthase Cell Fraction
P. aeruginosa 42A2 was grown overnight on agar plates (30 g/liter TSA: 15 g of tryptone (digest of casein), 5 g of NaCl, 5 g of soytone, and 15 g of agar per liter) and collected. The cells were centrifuged and suspended in Ringer's solution (OD ∼2), and 1 ml was transferred to 50 ml of TSB: 17 g of peptone, 3 g of peptone, 2.5 g of glucose, 5 g of NaCl, and 2.5 g of KH2PO4. Enzyme activity was induced by growing with 1% oleic acid (18 h, 30 °C; 250 rpm). The cells were washed twice with Ringer, resuspended in 0.05 m Tris-HCl (pH 7.0; +4 °C), and sonicated (three times for 1 min each) on ice (Branson Sonifier B-15). After centrifugation (8000 × g, 5 min, +4 °C), the supernatant was filtered and then concentrated and washed by diafiltration (Amicon Ultra-15, Millipore; final volume, 1 ml). An aliquot of this preparation (10–20 μl) was used for most studies of the enzyme activity and is referred to as the semipurified cell extract. The protein was purified by gel filtration (Tricorn Superdex 200 10/300 GL (GE Healthcare)), eluted with 0.05 m Tris-HCl (pH 7.0) at 1 ml/min (0.5 megapascals) in a chromatography system (ÄKTA FPLC, GE Healthcare). Molecular mass standards were BSA (66 kDa), ovalbumin (45 kDa), trypsinogen (24 kDa), and lysozyme (14.3 kDa) from Sigma. The fractions with enzyme activity were combined and purified by chromatography on Mono-Q 5/50 with a gradient in 0.05 m Tris-HCl (pH 7.0) from 0 to 1 m NaCl in 20 min (flow 2 ml/min). An aliquot (10 μl) of each fraction (0.5–0.3 ml) was assayed for diol synthase activity using oleic acid as substrate, and the released products were analyzed by TLC (hexane/diethyl ether/acetic acid; 75:15:10). The plates were sprayed with molybdatophosphoric acid and heated to visualize the products.
Bioconversion Assays
Fatty acids (1–5 mm) in 0.05 m Tris-HCl (pH 7.0) were prepared from stock solutions in ethanol, and incubated with 0.02 volume of the semipurified enzyme preparation described above (usually 0.5 mg of protein/ml, Bradford assay (28)) for up to 1 h at 37 °C with occasional vortexing. The reaction was terminated by acidification to pH 2–3 with 0.5 m HCl and then extracted with ethyl acetate or on a C18 cartridge (SepPak/C18). Products were usually analyzed by preparative TLC, followed by LC-MS/MS analysis. The Km of the semipurified fraction was determined in triplicate by measuring the amount of 7,10-diol (LC-MS/MS analysis) formed from 0.7, 1.1, 1.5, and 1.9 mm oleic acid (10 min, +37 °C). The effect of CO on the oxygenation of oleic acid was assessed in buffer with dissolved CO and under partial CO atmosphere. The effects of P450 inhibitors (piperonyl butoxide and 1-aminobenzotriazole) at 1 and 5 mm concentrations with oleic acid as a substrate were analyzed by TLC.
LC- and GC-MS Analysis
Reversed phase-HPLC with MS/MS analysis was performed with a Surveyor MS pump (ThermoFisher) and an analytical octadecyl silica column (5 μm; 2.0 × 150 mm; Phenomenex), which was usually eluted with methanol/water/acetic acid, 800:200:0.05, at 0.3 ml/min. The effluent was subject to electrospray ionization in an ion trap mass spectrometer (LTQ, ThermoFisher). The heated transfer capillary was set at 315 °C, the ion isolation width was set at 1.5 atomic mass units, and the collision energy was set at 25–35 (arbitrary scale). Prostaglandin F1α (100 ng/min) was infused for tuning. Products formed from stereospecifically deuterated 18:2n-6 were analyzed as described (5).6 The deuterium content of the [2H]18:2n-6 was determined by LC-MS/MS analysis (m/z 276–282 → full scan) or by GC-MS analysis.
Normal phase-HPLC with MS/MS analysis was performed with a silicic acid column (5 μm; Kromasil 100SI, 250 × 2 mm, Dalco Chromtech) using 1–3% isopropyl alcohol in hexane for separation of hydroxy fatty acids and 5–7% isopropyl alcohol in hexane for separation of DiHOME (0.3–0.5 ml/min; Constrametric 3200 pump, LDC/MiltonRoy). The effluent was combined with isopropyl alcohol/water (3:2; 0.2–0.3 ml/min) from a second pump (Surveyor MS pump) (5). The combined effluents were introduced by electrospray ionization into the ion trap mass spectrometer (LTQ, ThermoFisher). Steric analysis of 8-HODE was performed by CP-HPLC-MS/MS (Chiralcel-OBH) (29), whereas hydroperoxides formed from (9Z)-18:1 and (11Z)-18:1 by photooxidation were separated by CP-HPLC (Reprosil Chiral NR or NR-R) (30).
GC-MS analysis and synthesis of fatty acid methyl ester and trimethylsilyl ether derivatives were performed as described (31). Carbon values were estimated from the retention times of saturated fatty acids methyl esters (31). Steric analysis of 11,14-DiHOME was performed after ozonolysis of (−)-menthoxycarbonyl derivatives as described (32).
RESULTS
Oleate Diol Synthase Activity
Crude or semipurified cell extracts of P. aeruginosa rapidly transformed 18:1n-9 to (7S,10S)-DiHOME and (10S)-HPOME (Fig. 1). (10S)-HOME was also detected along with small amounts of 8-HOME by LC-MS/MS analysis. Preparation and purification of 1-mg amounts of the main enzymatic products in cultures of P. aeruginosa are described in the supplemental material. (10S)-HPOME was a poor substrate in comparison with oleic acid, whereas transformation of (10S)-HOME to the 7,10-diol could not be detected. A comparison of the oxidation of oleic acid and equal amounts of oleic acid plus (10S)-HPOME showed that the latter rather inhibited than augmented the biosynthesis of the 7,10-diol (TLC analysis). This diol synthase activity showed a pH optimum around pH 7.0, and the reaction rate was optimal at 37 °C (supplemental Fig. S1) and was linear for at least 10 min (data not shown). The Km was estimated to be 1.7 mm (supplemental material). The formation of DiHOME appeared to be reduced ∼30 and ∼50% by 1 and 5 mm 1-aminobenzotriazole (a nonspecific and irreversible P450 inhibitor) (33), respectively, whereas neither CO nor the P450 inhibitor piperonyl butoxide (5 mm) (34) appeared to inhibit the diol synthase activities.
FIGURE 1.

LC-MS/MS analysis of products formed from oleic acid by the diol synthase preparation. Top, chromatogram showing the elution of (10S)-HOME (m/z 297 → full scan). Bottom, elution of 7,10-DiHOME and (10S)-HPOME (m/z 313 → full scan). The C18 column was eluted with 80% methanol.
Partial Enzyme Purification
The diol synthase activity was subject to sequential purification by gel filtration and by ion exchange chromatography (Mono-Q), as shown in Fig. 2. SDS-PAGE analysis showed multiple protein bands at this stage; TLC analysis showed that the dioxygenase activity described below was not separated from the hydroperoxide isomerase activity. The elution of the enzyme activity on gel filtration in relation to protein standards (ovalbumin, 45 kDa; bovine serum albumin, 66 kDa) suggested an apparent molecular mass of ∼50 kDa.
FIGURE 2.

Partial purification of the diol synthase activity of P. aeruginosa. A, gel filtration with TLC analysis of enzyme activity. The enzyme eluted between 15 and 17 min, suggesting a molecular size of ∼50 kDa based on the elution time of protein standards. B, ion exchange chromatography. The NaCl gradient started at 5 min, and the diol synthase activity eluted after 7.5 min (TLC analysis) at ∼0.3 m NaCl in the elution buffer.
Substrate Specificity
We next examined the substrate specificity of the diol synthase of the semipurified cell extracts. We first studied a series of regioisomeric 18:1 fatty acids. The results are summarized in Table 1 along with previous data on oleic, palmitoleic, and ricinoleic acids. With the exception of vaccenic acid, the common denominator of substrates was an unsaturated carbon chain from the carboxyl group to the cis double bond at position 9,10, whereas a double bond at 8,9 resulted in slow but significant conversion. Additional double bonds between the carboxyl group and the 9Z double bond inhibited transformation. 7,10-DiHOME showed an unexpected fragmentation during MS/MS analysis due to fragments with charge migration to the ω-end; a systematic study of its MS/MS spectrum is reported elsewhere.7
TABLE 1.
Summary of the oxygenation of fatty acids to diols by the diol synthase activity of the extract of P. aeruginosa
| Fatty acids | Diols |
|---|---|
| 16:1 | |
| (9Z)-16:1 | (7S,10S)-Dihydroxy-(8E)-hexadecenoic acida |
| 18:1 | |
| (8Z)-18:1 | (6S,9S)-Dihydroxy-(7E)-octadecenoic acidb |
| (9Z)-18:1 | (7S,10S)-Dihydroxy-(8E)-octadecenoic acid |
| (9E)-18:1 | NDc |
| (11Z)-81:1 | (11S,14S)-Dihydroxy-(12E)-octadecenoic acid |
| (13Z)-18:1 | ND |
| Ricinoleic acid | (7S,10S,12R)-trihydroxy-(8E)-octadecenoic acidd |
| 18:2 | |
| (9Z,12Z)-18:2 | (7S,10S)-Dihydroxy-(8E,12Z)-octadecadienoic acide |
| (5Z,9Z)-18:2 | ND |
| C20-C24 | |
| (9Z)-20:1 | (7S,10S)-Dihydroxy-(8E)-eicosenoic acid |
| (13Z)-22:1 | ND |
| (15Z)-24:1 | ND |
We also investigated 18:2n-6, 20:1n-9, 20:1n-11, and 20:2n-6 (Table 1) and found that 18:2n-6 (supplemental Fig. S4) and 20:1n-11 were oxidized, whereas the methyl ester of oleic acid was not a substrate. The results suggested that substrate might bind the active site ‘head‘ first because the distance from the carboxyl group to the first double bond appeared to be critical and not the distance from the ω-end to the double bond. We found one exception. The oxygenation of vaccenic acid might be due to reverse orientation at the active site (see below).
Oxygenation Mechanism
The Hydroperoxide Isomerase Activity
Experiments performed under oxygen-18 atmosphere with oleic acid as a substrate revealed that both hydroxyl groups of 7,10-DiHOME contained either oxygen-18 or oxygen-16, but we could not detect significant formation of the diol with both isotopes present (Fig. 3). In a separate experiment under oxygen-18, we were able to isolate [18O2](10S)-HPOME (>95% 18O2). The latter was transformed by the enzyme to [18O2]7,10-DiHOME (supplemental material). We conclude that 7,10-DiHOME is formed from (10S)-HPODE in a hydroperoxide isomerase reaction.
FIGURE 3.

LC-MS analysis of the carboxylate anions of 7,10-DiHOME formed under an atmosphere of oxygen-18. The MS spectrum of [7,10-16O2]7,10-DiHOME shows a carboxylate anion at m/z 313, whereas [7,10-18O2]7,10-DiHOME has the corresponding ion at m/z 317. We could not detect [7,10-16O18O]7,10-DiHOME at m/z 315, suggesting that both hydroxyl oxygens were derived from the same molecule of O2. Spectra were recorded with zoom scan resolution.
We next prepared (10R)-HPOME by photooxidation and purified this stereoisomer by CP-HPLC. (10R)-HPOME was not a substrate (TLC analysis; supplemental Fig. S2), whereas 10S-HPOME was isomerized to 7,10-DiHOME under identical conditions.
The reaction mechanism was studied with [(7S)-2H]18:1n-9 as a substrate. As shown in Fig. 4, the deuterium label was retained in the 10-hydroperoxide but lost in the 7,10-diol. These results suggest suprafacial oxygenation at C-7. This was confirmed by bioconversion of [(7R)-2H]18:2n-6, which resulted in the [(7R)-2H]7,10-DiHOME (supplemental material). This label was also retained in 13-HODE, formed by the lipoxygenase activity of this preparation (21).
FIGURE 4.

LC-MS analysis of products formed during oxidation of [(7S)-2H]18:1 (∼70% 2H) to (10S)-HPODE and to (7S,10S)-DiHOME. A, partial MS spectrum at the retention time of (10S)-HPODE. The major signal of the carboxylate anion at m/z 314 shows that [7S-2H] was retained. B, partial MS spectrum at the retention time of 7S,10S-DiHOME. The signal intensities at m/z 313 and m/z 314 show that hydrogen was eliminated, suggesting abstraction of the pro-S hydrogen at C-7. Spectra were recorded with zoom scan resolution.
The (10S)-Dioxygenase Activity
Linoleic acid was oxidized slowly compared with oleic acid. We found that the deuterium label of [(8R)-2H]18:2n-6 was lost in 10-HODE and also in 7,10-dihydroxy-(8E,12Z)-octadecadienoic acid, but we confirmed that the label was retained in the lipoxygenase product 13-HODE of the very same preparation (supplemental Fig. S5). Finally, small amounts of 8-HODE were also detected. Steric analysis by CP-HPLC suggested that 8-HODE was a mixture of the S and R stereoisomers in a ∼3:7 ratio and could in part be produced enzymatically.
Oxygenation of Vaccenic Acid
Fatty acid analysis showed that P. aeruginosa cell membranes contain palmitoleic ((9Z)-16:1), palmitelaidic ((9E)-16:1), and cis and trans vaccenic acids (18:1n-7), as shown in Fig. 5. Only traces of oleic acid could be detected (data not shown).
FIGURE 5.
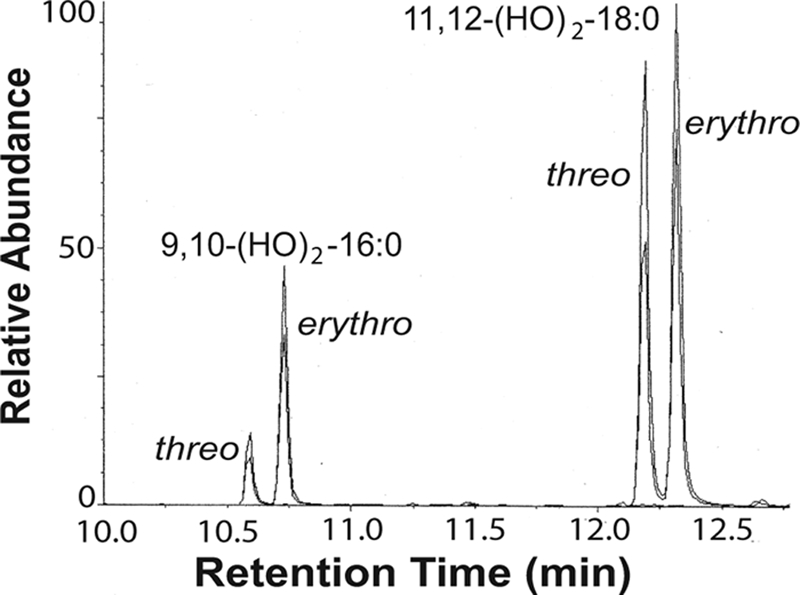
Analysis of the cis and trans configuration of monounsaturated fatty acids in P. aeruginosa after trans hydroxylation to the corresponding vicinal dihydroxy fatty acids. Palmitoleic and vaccenic acids were identified as the main products, and only traces of oleic acid could be detected. Fatty acids with cis configuration form threo diols.
It is known that (9Z)-16:1 is oxidized by the diol synthase of P. aeruginosa (14), but cis vaccenic acid has not been investigated. We found that this fatty acid was oxygenated but to an unexpected metabolite, (11S,14S)-DiHOME. The latter was identified by GC-MS analysis (Fig. 6A) after purification by TLC. The C value was 20.8. The position of the double bond was determined by ozonolysis, and the configuration of the alcohols by separation of (−)-menthoxycarbonyl derivatives of the biological product and authentic standards on GC-MS (Fig. 6B).
FIGURE 6.

GC-MS analysis of 11,14-DiHOME and steric analysis after ozonolysis. A, electron impact mass spectrum of the trimethylsilyl ether methyl ester derivative of 11,14-DiHOME. The C value was 20.8. B, steric analysis of the hydroxyl groups of 11,14-DiHOME. 11,14-DiHOME was derivatized with (−)-menthoxycarbonyl chloride and subject to ozonolysis, which yielded 2-hydroxyhexanoic and 2-hydroxydodecane-1,12-dioic acid derivatives. Top, the reconstructed ion chromatogram shows that the derivative of 2-hydroxyhexanoic acid had the same retention time as the 2S stereoisomer (co-injection with this derivative of (2R,2S)-hydroxyhexanoic acid). Bottom, the reconstructed ion chromatogram shows that the derivative of 2-hydroxydodecane-1,12-dioc acid had the same retention time as the 2S stereoisomer (co-injection with (2R,2S)-hydroxydodecane-1,12-dioic acid). Analysis of the biological products alone yielded single peaks (data not shown). The results suggest that (11S,14S)-dihydroxy-(12E)-octadecenoic acid was formed. Conditions were as follows: GC oven at 260 °C, isothermal.
LC-MS/MS analysis revealed that biosynthesis of (11S,14S)-DiHOME was accompanied by formation of a mixture of 11-HPOME and 12-HPOME (supplemental Fig. S6). The 11- and 12-hydroperoxides of (11Z)-18:1 were prepared by photooxidation with methylene blue in methanol (35), separated, and analyzed by CP-HPLC-MS. Their triple mass spectra (supplemental Fig. S7) confirmed that a mixture of 11-HPOME and 12-HPOME were formed from (11Z)-18:1. It seems likely that (11S)-HPOME is isomerized to (11S,14S)-DiHOME, but this was not further investigated.
DISCUSSION
We report that P. aeruginosa expresses a fatty acid diol synthase responsible for the sequential transformation of oleic acid to (10S)-HPOME and (7S,10S)-DiHOME. This enzyme also oxygenates, albeit slowly, linoleic acid and some other fatty acids. Transformation of stereospecifically deuterated oleic and linoleic acids suggested that 10S-hydroperoxides were formed by abstraction of the pro-R hydrogen at C-8 and antarafacial oxygenation, whereas the 7,10-diols were formed by abstraction of the pro-S hydrogen at C-7 and suprafacial oxygenation. The reaction mechanism is outlined in Fig. 7.
FIGURE 7.

Mechanism of biosynthesis of the oleic acid diol synthase of P. aeruginosa. The nature of the (10S)-DOX enzyme is unknown, whereas the reaction mechanism of the hydroperoxide isomerase is consistent with P450 catalysis.
A systematic investigation of the substrate specificity suggested that the enzyme required a saturated carbon chain from the carboxyl group to the first cis double bond between C-8 and C-9 or between C-9 and C-10, whereas fatty acids with a longer carbon chain ((13Z)-18:1 and (15Z)-18:1) were not oxidized (Table 1). These observations and studies of (9Z)-16:1, (9Z)-20:1, and (11Z)-20:1 suggested that the oxygenated fatty acids bound to the enzyme with their carboxyl groups at a fixed position relative to the catalytic site. We found one exception; (11Z)-18:1 was transformed to (11S,14S)-DiHOME and 11-HPOME, possibly by reverse orientation at the active site (Fig. 8 and supplemental Fig. S6).
FIGURE 8.

Schematic view of the oxidation of vaccenic acid ((11Z)-18:1) by P. aeruginosa. The 11,14-diol can be visualized as formed from (11S)-HPOME by reverse orientation of the fatty acid in the two active sites compared with the orientation of oleic acid (cf. Fig. 7). 12-HPOME was also formed (supplemental Fig. S6), but this compound was apparently not isomerized to a diol.
An obvious question is the nature of the (10S)-DOX and hydroperoxide isomerase enzyme activities. The hydroperoxide isomerase showed an unexpected feature; (10S)-HPOME was slowly transformed to the 7,10-diol compared with oleic acid and also reduced the rate of oxygenation of oleic acid. As far as is known, hydroperoxide isomerases belong to the family of P450, and suprafacial hydrogen abstraction and oxygenation are typical for this enzyme family (23). The genome of P. aeruginosa PAO1, which is closely related to 42A2 and often used for comparison, contains only two P450 homologues. Whether these two P450s possess hydroperoxide isomerase activity clearly merits further investigation.
The (10S)-DOX activities are of interest in their own right. The genome of P. aeruginosa contains no obvious homologues to heme-containing fatty acid dioxygenases (cyclooxygenases, linoleate diol synthases, 10-DOX, and α-DOX), and monounsaturated fatty acids are poor substrates for lipoxygenases (22). Furthermore, partial purification of the (10S)-DOX and hydroperoxide isomerase activities suggested that they were linked. The molecular mass was estimated by gel filtration to be ∼50 kDa. The observation that (10S)-HPODE was transformed to the 7,10-diol less efficiently than its precursor, oleic acid, suggested that (10S)-DOX and hydroperoxide isomerase activities are functionally coupled. The (10S)-DOX activity probably involves hydrogen abstraction at C-8 by a catalytic metal so that the double bond migrates (9Z → 8E), and the carbon radical centered at C-10 reacts with molecular oxygen and forms a hydroperoxide in analogy with (10R)-DOX of aspergilli (26). Determination of whether the 7,10-diol synthase consists of one enzyme with both dioxygenase and hydroperoxide isomerase activities or two separate enzymes awaits further studies.
What is the biological function of the diol synthase of P. aeruginosa? Oleic acid is the preferred substrate, and enzyme expression is induced by growing P. aeruginosa in the presence of oleic acid. This bacterium contains little oleic acid compared with palmitoleic and vaccenic acids. This rare prokaryote enzyme may be associated with the modification of the membrane lipids and/or associated with the lipid modification. Alternatively, the combined (10S)-DOX and diol synthase activities could contribute to detoxification of fatty acids in the bacterial environment, promoting colonization and growth. This bioconversion might also contribute to production of oxylipins during growth on plant tissues rich in oleic acid. In this context, the antifungal activity of (7S,10S)-DiHOME is of interest (see Ref. 15 for a review). We conclude that P. aeruginosa expresses a rare diol synthase activity never described before among bacteria.
Supplementary Material

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S7.
Jernerén, F., Sesma, A., Francheschetti, M., Hamberg, M., and Oliw, E. H., (2010) J. Biol. Chem. 285, 5308–5316.
Nilsson, T., Martinez, E., Manresa, A., and Oliw, E. H. (2010) Rapid Commun. Mass Spectrom. 24, 777–783.
- P450
- cytochrome P450
- CP
- chiral phase
- 7,10-DiHOME
- (7S,10S)-dihydroxy-(8E)-eicosenoic acid
- (7S,10S)-DiHOME
- (7S,10S)-dihydroxy-(8E)-octadecenoic acid
- DiHOME
- dihydroxyeicosenoic acid
- (7S,10S)-DiHOME
- (7S,10S)-dihydroxy-(8E)-octadecenoic acid
- (11S,14S)-DiHOME
- (11S,14S)-dihydroxy-(12E)-octadecenoic acid
- DOX
- dioxygenase
- GC
- gas chromatography
- MS
- mass spectrometry
- MS/MS
- tandem MS
- (10S)-HOME
- (10S)-hydroxy-(8E)-octadecenoic acid
- 8-HODE
- 8-hydroxylinoleic acid
- HPLC
- high performance liquid chromatography
- (10S)-HPOME
- (10S)-hydroperoxy-(8E)-octadecenoic acid
- 11-HPOME
- 11-hydroperoxy-(12E)-octadecenoic acid
- 12-HPOME
- 12-hydroperoxy-(10E)-octadecenoic acid
- LC
- liquid chromatography.
REFERENCES
- 1.Kuhn H., Saam J., Eibach S., Holzhütter H. G., Ivanov I., Walther M. (2005) Biochem. Biophys. Res. Commun. 338, 93–101 [DOI] [PubMed] [Google Scholar]
- 2.Liavonchanka A., Feussner I. (2006) J. Plant Physiol. 163, 348–357 [DOI] [PubMed] [Google Scholar]
- 3.Schneider C., Pratt D. A., Porter N. A., Brash A. R. (2007) Chem. Biol. 14, 473–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith W. L. (2008) Trends Biochem. Sci. 33, 27–37 [DOI] [PubMed] [Google Scholar]
- 5.Garscha U., Jernerén F., Chung D., Keller N. P., Hamberg M., Oliw E. H. (2007) J. Biol. Chem. 282, 34707–34718 [DOI] [PubMed] [Google Scholar]
- 6.Oliw E. H. (1994) Prog. Lipid Res. 33, 329–354 [DOI] [PubMed] [Google Scholar]
- 7.Mercade E., Robert M., Espuny M. J., Bosch M. P., Manresa M. A., Parra J. L., Guinea J. (1988) J. Am. Oil Chem. Soc. 65, 1915–1916 [Google Scholar]
- 8.Hou C. T., Bagby M. O., Plattner R. D., Koritala S. (1991) J. Am. Oil Chem. Soc. 68, 99–101 [Google Scholar]
- 9.Kuo T. M., Manthey L. K., Hou C. T. (1998) J. Am. Oil Chem. Soc. 75, 875–879 [Google Scholar]
- 10.Gardner H. W., Hou C. T. (1999) J. Am. Oil Chem. Soc. 76, 1151–1156 [Google Scholar]
- 11.Kim H., Gardner H. W., Hou C. T. (2000) J. Am. Oil Chem. Soc. 77, 95–99 [Google Scholar]
- 12.Kuo T. M., Kim H., Hou C. T. (2001) Curr. Microbiol. 43, 198–203 [DOI] [PubMed] [Google Scholar]
- 13.Kuo T. M., Nakamura L. K. (2004) Curr. Microbiol. 49, 261–266 [DOI] [PubMed] [Google Scholar]
- 14.Bae J. H., Kim D. S., Suh M. J., Oh S. R., Lee I. J., Kang S. C., Hou C. T., Kim H. R. (2007) Appl. Microbiol. Biotechnol. 75, 435–440 [DOI] [PubMed] [Google Scholar]
- 15.Hou C. T. (2009) Nat. Biotechnol. 26, 2–10 [Google Scholar]
- 16.Yetkin G., Otlu B., Cicek A., Kuzucu C., Durmaz R. (2006) Am. J. Infect. Control 34, 188–192 [DOI] [PubMed] [Google Scholar]
- 17.Lyczak J. B., Cannon C. L., Pier G. B. (2002) Clin. Microbiol. Rev. 15, 194–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guerrero A., Casals I., Busquets M., Leon Y., Manresa A. (1997) Biochim. Biophys. Acta 1347, 75–81 [DOI] [PubMed] [Google Scholar]
- 19.Winsor G. L., Van Rossum T., Lo R., Khaira B., Whiteside M. D., Hancock R. E., Brinkman F. S. (2009) Nucleic Acids Res. 37, D483–D488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porta H., Rocha-Sosa M. (2001) Microbiology 147, 3199–3200 [DOI] [PubMed] [Google Scholar]
- 21.Vance R. E., Hong S., Gronert K., Serhan C. N., Mekalanos J. J. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 2135–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clapp C. H., Senchak S. E., Stover T. J., Potter T. C., Findeis P. M., Novak M. J. (2001) J. Am. Chem. Soc. 123, 747–748 [DOI] [PubMed] [Google Scholar]
- 23.Brash A. R. (2009) Phytochemistry 70, 1522–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brodhun F., Göbel C., Hornung E., Feussner I. (2009) J. Biol. Chem. 284, 11792–11805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamberg M., Zhang L. Y., Brodowsky I. D., Oliw E. H. (1994) Arch. Biochem. Biophys. 309, 77–80 [DOI] [PubMed] [Google Scholar]
- 26.Garscha U., Oliw E. H. (2009) J. Biol. Chem. 284, 13755–13765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vidal-Mas J., Busquets M., Manresa A. (2005) Antonie Van Leeuwenhoek 87, 245–251 [DOI] [PubMed] [Google Scholar]
- 28.Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 29.Garscha U., Oliw E. H. (2007) Anal. Biochem. 367, 238–246 [DOI] [PubMed] [Google Scholar]
- 30.Garscha U., Nilsson T., Oliw E. H. (2008) J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 872, 90–98 [DOI] [PubMed] [Google Scholar]
- 31.Oliw E. H., Stark K., Bylund J. (2001) Biochem. Pharmacol. 62, 407–415 [DOI] [PubMed] [Google Scholar]
- 32.Hamberg M. (1971) Anal. Biochem. 43, 515–526 [DOI] [PubMed] [Google Scholar]
- 33.El-Kattan A. F., Poe J., Buchholz L., Thomas H. V., Brodfuehrer J., Clark A. (2008) Drug Metab. Lett. 2, 120–124 [DOI] [PubMed] [Google Scholar]
- 34.Ning D., Wang H., Zhuang Y. (2009) Biodegradation, in press [DOI] [PubMed] [Google Scholar]
- 35.Chacón J. N., Gaggini P., Sinclair R. S., Smith F. J. (2000) Chem. Phys. Lipids 107, 107–120 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.