

Abstract
It has been suggested that quorum sensing is an important signal transduction system regulating the expression of numerous virulence genes in bacterial pathogens. We previously revealed that SmcR, a LuxR homologue of Vibrio vulnificus, activates promoter S, an RpoS-dependent promoter of vvpE encoding a potential virulence factor elastase and binds in vitro to a binding site centered at −196.5. In this study, chromatin immunoprecipitation assays and promoter deletion analyses demonstrated that SmcR binds to the vvpE regulatory region in vivo and directly interacts with RNAP for activation of the vvpE expression. A search for regulatory genes involved in the regulation of elastase production singled out ihfA, which encodes for a subunit of integration host factor (IHF). Levels of both elastase activity and vvpE transcript decreased significantly as a result of inactivation of ihfA, and primer extension analyses demonstrated that IHF regulates the vvpE transcription by activating PS. Direct binding of IHF to the two distinct binding sites centered at −174 and −131, respectively, was determined using an electrophoretic mobility shift assay and a DNase I protection assay. Chromatin immunoprecipitation assays revealed that the interaction of SmcR with RNAP in vivo was mediated by IHF. Collectively, the results proposed a model whereby IHF positions SmcR to contact RNAP by looping the vvpE regulatory DNA, thus allowing precise control of the expression level of VvpE during the pathogenesis of V. vulnificus.
Keywords: Bacteria, Bacterial Genetics, Bacterial Signal Transduction, Bacterial Transcription, Gene Regulation, LuxR Homologues, Quorum Sensing, Vibrio vulnificus
Introduction
Many bacteria exchange diffusible signal molecules that accumulate extracellularly as a method to monitor their cell population densities (for recent reviews, see Refs. 1 and 2). This type of cell density-dependent regulation is termed quorum sensing and has been recognized as a global regulatory system controlling the expression of numerous genes in bacteria. The Vibrio harveyi regulation of bioluminescence is frequently used as a model for quorum sensing. LuxR, a transcriptional activator of the luminescence operon, is a quorum-sensing master regulator in V. harveyi, and its synthesis is controlled by the levels of three autoinducers: AI-1, AI-2, and CAI-1 (3). Thus far, homologues of LuxR, which are postulated to regulate virulence genes, have been identified in various pathogenic Vibrio spp. (4–9). However, until now, only a few studies have addressed the molecular mechanism by which the LuxR homologues modulate the expression of virulence genes (10–12).
As a LuxR homologue, SmcR has been identified from Vibrio vulnificus, a food-borne pathogenic bacterium (13). Upon analysis of the completed genome sequence, homologues of the genes required for sensing and responding to autoinducers, such as LuxO and LuxT, are also identified in V. vulnificus (14). It seemed logical to consider SmcR as a quorum-sensing regulator of V. vulnificus because of the similarities between the components of quorum-sensing systems in V. vulnificus and in V. harveyi. Recent works demonstrated that SmcR regulates numerous genes contributing to pathogenesis as well as survival of V. vulnificus (4, 10, 15–17). Among these SmcR target genes, vvpE encodes a potential virulence factor, elastase (elastolytic metalloprotease), and its regulation by SmcR is the best characterized in vitro (10, 18). We previously reported that the expression of vvpE is initiated by two different types of promoter, PL4 and PS (Fig. 1), in a growth phase-dependent manner. Although the basal level expression of vvpE is directed by PL, independent of SmcR, and remains low throughout the log and stationary growth phases, SmcR activates the RpoS-dependent promoter PS by directly binding to a SmcR-binding site in the stationary phase. The SmcR-binding site was determined using a DNase I protection assay in vitro and was centered at 196.5 bp upstream of the transcription start site of PS (Fig. 1) (10, 17).
FIGURE 1.

Sequence analysis of the vvpE upstream region. The transcription start sites of vvpE in the log (PL) and stationary (PS) phases are indicated by bent arrows. The positions of the putative −10 and −35 regions are underlined for the RpoS-dependent PS. The sequences proposed for the binding site of SmcR (SB) are presented in a dotted box. The IHF-binding sequences (IHFB1 and IHFB2) determined later in this study (Fig. 8) are presented in shaded boxes. The conserved nucleotide sequences for the binding of RNA polymerase α-subunit (α-CTD) (UP element) (33) are indicated above the V. vulnificus DNA sequence in capital letters. The ATG translation initiation codon and putative ribosome binding site (AGGA) are indicated in bold type. W, A or T; R, A or G; N, any base.
In general, activators binding this far upstream of the promoter cooperate and interact with additional transcriptional regulator(s) on the promoter DNA because the activators are not able to activate RNA polymerase (RNAP) directly (19, 20). Therefore, the additional regulatory proteins that convey the signal of the activator to RNAP and/or induce structural changes of the DNA (forming a DNA loop) to bring the activators closer to RNAP are required. Accordingly, in an effort to further elucidate the mechanism of SmcR for activation of vvpE expression at a molecular level, an additional regulator, integration host factor (IHF) (21), required for activation of SmcR-dependent PS, was identified in the present study. In addition, the two distinct IHF-binding sites located between the SmcR and RNAP binding were determined. Finally, a possible model whereby IHF positions SmcR to contact RNAP by looping the vvpE upstream DNA was experimentally suggested by promoter deletion analyses and confirmed by chromatin immunoprecipitation (ChIP) assays.
EXPERIMENTAL PROCEDURES
Strains, Plasmids, and Culture Conditions
The strains and plasmids used in this study are listed in Table 1. Unless noted otherwise, the V. vulnificus strains were grown in Luria-Bertani medium supplemented with 2.0% (w/v) NaCl (LBS). All of the media components were purchased from Difco, and the chemicals were purchased from Sigma.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristicsa | Reference or source |
|---|---|---|
| Strains | ||
| V. vulnificus | ||
| ATCC29307 | Clinical isolate | Laboratory collection |
| HS03 | ATCC29307, smcR::nptI | 10 |
| HS05 | ATCC29307, ΔihfA | This study |
| E. coli | ||
| SM10 λpir | Thi thr leu tonA lacY supE recA::RP4-2-Tc::Mu λpir, oriT of RP4, Kmr; conjugational donor | 26 |
| BL21(DE3) | F−, ompT, hsdS (rB−, mB−), gal(DE3) | Laboratory collection |
| Plasmids | ||
| pRK415 | Broad host range vector, IncP ori, oriT of RK2; Tcr | 34 |
| pDM4 | R6Kγ ori (requires π), oriT of RP4; Cmr | 43 |
| pHK0011 | pRK415 with promoterless luxAB; Tcr | 18 |
| pKC980 | pUC18 with vvpE; Apr | 32 |
| pHS201 | pHK0011 with 748-bp fragment of vvpE upstream region; Tcr | 10 |
| pHS206 | pH S201 with −206/−197substitution of vvpE upstream region; Tcr | This study |
| pHS176 | pH S201 with −176/−167substitution of vvpE upstream region; Tcr | This study |
| pHS136 | pH S201 with −136/−127substitution of vvpE upstream region; Tcr | This study |
| pHS106 | pH S201 with −106/−97substitution of vvpE upstream region; Tcr | This study |
| pHS56 | pH S201 with −56/−47substitution of vvpE upstream region; Tcr | This study |
| pHS221 | pH S201 with −101/−97deletion of vvpE upstream region; Tcr | This study |
| pHS222 | pH S201 with −106/−97deletion of vvpE upstream region; Tcr | This study |
| pHS301 | pGEM T with ihfA; Apr | This study |
| pHS302 | pGEM T with ΔihfA; Apr | This study |
| pHS3021 | pDM4 with ΔihfA; Cmr | This study |
| pHS303 | pRK415 with ihfA; Tcr | This study |
| pHS304 | pRSET C with ihfA; Apr | This study |
| pHS305 | pRSET C with ihfB; Apr | This study |
a Apr, ampicillin-resistant; Cmr, chloramphenicol-resistant; Kmr, kanamycin-resistant; Tcr, tetracycline-resistant.
ChIP Assay
The ChIP experiments were performed using formaldehyde cross-linking as described by Rhee et al. (22). Briefly, the cross-linked chromatin in the wild type, smcR, or ihfA mutant cells (Table 1) was fragmented by sonication to result in sheared chromatins with an average length of 300 bp. When required, the average lengths of the sheared chromatins were reduced by further treatment with DNase I (at a final concentration of 5 μg/ml) (Sigma). One-half of the clarified supernatant was saved as the total input sheared chromatin (positive control) prior to the reaction with the anti-SmcR antibody (10), whereas the sheared chromatin (100 μl) from the other half of the supernatant was reacted with 10 μl of the anti-SmcR antibody overnight at 4 °C. The resulting chromatin-antibody complex was specifically precipitated with protein A-Sepharose and washed, and the sheared chromatins were eluted using the method described elsewhere (22). The cross-linkings were reversed, and DNAs were purified and analyzed by PCR using a pair of the primers specific to the vvpE promoter region as listed in Table 2.
TABLE 2.
Oligonucleotides used in this study
| Oligonucleotidea | Oligonucleotide sequence (5′–3′)b | Locationc | Use(s) |
|---|---|---|---|
| Hybridized to the vvpE regulatory region | |||
| VVPE031 | TTATTTCTGAGATGACTGTCCCA | −236 to −214 | ChIP assay, electrophoretic mobility shift assay, DNaseI footprinting |
| VVPE032 | AGCCAACTTCACCAAAAAAAT | −36 to −56 | |
| VVPE033 | TATTTCTGAGATGACTGTCC | −235 to −212 | Amplification of DNA S and Fd |
| VVPE034 | ACTAGTAATAAAATTGTGC | −158 to −177 | DNA S |
| VVPE035 | CTATACAGGGCGTAAAAAC | −75 to −57 | DNA R |
| VVPE036 | GACACTGCGAGCTCAACA | +7 to −11 | DNA R and F |
| Hybridized to the vvpE coding region | |||
| VVPE-RTF | GCGGTTTCGTTCTCGGTGTTG | VV2_0974 | Real time PCR |
| VVPE-RTR | TGCCTTCTGTGTCGGTTAATCTTG | VV2_0974 | Real time PCR |
| VVPE9905 | GACGTTGATTGAGTTTCATTATCG | VV2_0974 | Primer extension |
| Hybridized to the ihfA coding region | |||
| IhfA01 | GATACTGTGCTTGTGGGTGGATTG | VV1_2375 | Cloning of ihfA |
| IhfA01-1 | GGGTGGATCCGAAGGTTCTCTGCCAAATAG | VV1_2375 | Construction of the ihfA mutant |
| IhfA02 | GTAAAGAGCACCGCACTGTTGACG | VV1_2375 | Cloning of ihfA |
| IhfA02-1 | CTTCGGATCCACCCGGTGAAGACATTCC | VV1_2375 | Construction of the ihfA mutant |
| IhfA05 | GAATCTAGAGAGGAAAGTTTATGGCGCTC | VV1_2375 | Complementation of the ihfA mutant |
| IhfA06 | GTTGAATTCTGTATTCATAGTTCCATTAC | VV1_2375 | Complementation of the ihfA mutant Purification of IhfA |
| IhfA07 | ATACTGCAGATGGCGCTCACAAAAGCCG | VV1_2375 | Purification of IhfA |
| Hybridized to the ihfB coding region | |||
| IhfB01 | ATACTGCAGATGACTAAGTCTGAACTGATTG | VV1_2980 | Purification of IhfB |
| IhfB02 | GTTGAATTCTCACGATTTTATGTAAATCAG | VV1_2980 | Purification of IhfB |
a The oligonucleotides were designed using the genomic sequence of V. vulnificus CMCP6 (GenBankTM accession numbers AE016795 and AE016796).
b Regions of oligonucleotides not complementary to corresponding genes are underlined.
c Shown are the oligonucleotide positions, where +1 is the transcription start site of PS. Locus tag numbers are based on the database of the V. vulnificus CMCP genome.
d The amplified vvpE upstream DNA fragments are shown in Fig. 9B.
Generation of Mutation in the vvpE Upstream Region
A set of mutant vvpE upstream sequences were developed by substituting a 10-bp wild type sequence with the mutant sequence (5′-GTGGATCCTC-3′) using the PCR-mediated linker-scanning mutation method as described previously (17, 23). Briefly, VVPE001 that contained an XbaI restriction site followed by bases corresponding to the 5′-end of the vvpE promoter region was used in conjunction with one of the primers of Bn to amplify the 5′ amplicons. The Bn antisense primers contained the mutant sequence (Table 3). Similarly, 3′ amplicons were amplified using primers An and VVPE006 that carries a KpnI restriction site. The An sense primers contained a sequence complementary to the Bn primers. Similar experimental conditions were used for deletion of internal 5- and 10-bp sequences of the vvpE regulatory region, except that primers AΔ and BΔ were used in place of An and Bn, respectively, as indicated in Table 3. Second stage PCR was performed using VVPE001 and VVPE006 as a pair of primers and the mixture of the two amplicons as the template to result in the vvpE regulatory region with the substitution or deletion mutant sequences.
TABLE 3.
Oligonucleotides used in PCR-mediated mutation of the vvpE promoter
| Oligonucleotidea | Oligonucleotide sequence (5′–3′)b | Locationc | Use |
|---|---|---|---|
| VVPE001 | GTAGGTACCACTCAAGCTGACGAACTTGATC | −335 to −314 | Construction of all pHS-lux reportersd |
| VVPE006 | GAATCTAGACCGATACAGAAGGCAGATCGGC | 392 to 413 | |
| A206/197 | TCTGTGGATCCTCTCTGCGTAAAAAAGA | −196 to −180 | pHS206 |
| B206/197 | AGAGAGGATCCACAGATAAATGGGACAG | −207 to −223 | pHS206 |
| A176/167 | AAAGTGGATCCTCTTACTAGTGTTTATC | −166 to −150 | pHS176 |
| B176/167 | GTGGAGGATCCACCTTTTTCTTTTTTAC | −177 to −193 | pHS176 |
| A136/127 | AATGTGGATCCTCAAACGAAATCGCAGT | −126 to −110 | pHS136 |
| B136/127 | AATGAGGATCCACATAGCGATAAACACT | −137 to −153 | pHS136 |
| A106/97 | ATGTGGATCCTCTATAACATTGCGTCAG | −96 to −81 | pHS106 |
| B106/97 | TAGAGGATCCACATCTCACTGCGATTTC | −107 to −122 | pHS106 |
| A56/47 | AACGTGGATCCTCTGAAGTTGGCTGGTG | −46 to −30 | pHS56 |
| B56/47 | TCAGAGGATCCACGTTTTTACGCCCTGT | −57 to −73 | pHS56 |
| AΔ | TATAACATTGCGTCAGAGTTTCTATAC | −96 to −70 | pHS221 and pHS222 |
| BΔ−101/−97 | TGACGCAATGTTATAAATCCATCTCACTGC | −82 to −116 (Δ −101 to −97) | pHS221 |
| BΔ−106/−97 | TGACGCAATGTTATAATCTCACTGCGATTTC | −82 to −127 (Δ −106 to −97) | pHS222 |
a The oligonucleotides were designed using the genomic sequence of V. vulnificus CMCP6 (GenBankTM accession numbers AE016795 and AE016796).
b Regions of oligonucleotides not complementary to corresponding genes are underlined.
c Shown are the oligonucleotide positions, where +1 is the transcription start site of PS.
Construction of vvpE-luxAB Transcriptional Fusions and Measurement of Cellular Luminescence
The PCR products of the vvpE regulatory region with the mutant sequences were digested with XbaI and KpnI and inserted into pHK0011 that had been digested with the same enzymes. The latter plasmid carries promoterless luxAB luciferase genes (18). The resulting vvpE-luxAB fusion pHS reporters (Table 1; see also Figs. 3A and 4A), as confirmed by DNA sequencing, were then transferred into V. vulnificus ATCC29307 by conjugation. The cellular luminescence of the cultures was measured with a luminometer (Lumat model 9501, Berthold, Germany) and expressed in arbitrary relative light units (RLUs), as described previously (10).
FIGURE 3.

Mapping of cis-acting regulatory elements required for the vvpE expression. A, construction of vvpE-luxAB fusion pHS reporters. PCR fragments carrying the vvpE regulatory region with 10-bp substitutions (black boxes) were subcloned into pHK0011 (18) to create each pHS reporter. Solid lines, the vvpE upstream region; shaded blocks, the vvpE coding region; open blocks, the luxAB. The wild type vvpE regulatory region is shown on top with the proposed −10 and −35 regions, and the binding site for SmcR (SB). The reporter pHS201 that carries the wild type vvpE regulatory region was used as a positive control. B, cellular luminescences were determined from the stationary phase culture (A600 = 2.0) of V. vulnificus containing each pHS reporter as indicated. The error bars represent the S.E.
FIGURE 4.

Phase stringency of the SmcR binding for the vvpE expression. A, construction of vvpE-luxAB fusion pHS reporters. PCR fragments carrying the vvpE regulatory region with 5- or 10-bp deletions (Δ) were subcloned into pHK0011 (18) to create each pHS reporter. Solid lines, the vvpE upstream region; shaded blocks, the vvpE coding region; open blocks, the luxAB. The wild type vvpE regulatory region is shown on top with the proposed −10 and −35 regions and the binding site for SmcR (SB). The reporter pHS201 and pHS106 were used as positive controls. B, cellular luminescences were determined from the stationary phase culture (A600 = 2.0) of V. vulnificus containing each pHS reporter as indicated. The error bars represent the S.E.
Identification and Cloning of V. vulnificus ihfA
A mutant that exhibited decreased proteolytic activity on the LBS plate supplemented with skim milk (1.5%, w/v) was screened from a library of V. vulnificus mutants generated by random transposon mutagenesis using a mini-Tn5 lacZ1 (24). A DNA segment flanking the transposon insertion was amplified by PCR as described previously (25). Because a data base search for homology to the amino acid sequence deduced from the resulting PCR product singled out the V. vulnificus IhfA, a subunit of IHF, a DNA fragment containing the whole ihfA open reading frame was amplified by PCR using the primers, IhfA01 and IhfA02 (Table 2). The amplified 1,520-bp DNA fragment was ligated into pGEM-T Easy (Promega, Madison, WI) to result in pHS301 (Table 1).
Generation of the ihfA Deletion Mutant
The ihfA gene on pHS301 was inactivated in vitro by deletion of about two-thirds (164 of 288 bp) of the ihfA open reading frame using the PCR-mediated linker-scanning mutation method as described above. Pairs of primers IhfA01 and IhfA01-1 (for amplification of the 5′ amplicon) or IhfA02 and IhfA02-1 (for amplification of the 3′ amplicon) were designed and used as listed in Table 2. The 164-bp deleted ihfA was amplified by PCR using the mixture of both amplicons as the template and IhfA01 and IhfA02 as primers. The resulting 1,356-bp DNA fragment containing ΔihfA was ligated with SphI-SalI-digested pDM4 (43) forming pHS3021. To generate the ΔihfA mutant by homologous recombination, Escherichia coli SM10 λ pir, tra (containing pHS3021) (26) was used as a conjugal donor to V. vulnificus ATCC29307. The conjugation and isolation of the transconjugants were conducted using the method previously described (18).
RNA Purification and Transcript Analysis
Total cellular RNAs from the V. vulnificus strains were isolated using an RNeasy minikit (Qiagen). For quantitative reverse transcription-PCR, cDNA was synthesized with an iScriptTM cDNA synthesis kit (Bio-Rad) according to the manufacturer's procedure. Real time PCR amplification of the cDNA was performed with a pair of primers, VVPE-RTF and VVPE-RTR (Table 2), using the Chromo 4 real time PCR detection system (Bio-Rad) as described previously (27). Relative expression levels of the vvpE transcript were calculated using the 16 S rRNA expression level as the internal reference for normalization.
For the primer extension experiments, an end-labeled 24-base primer VVPE9905 (Table 2) complementary to the coding region of vvpE was added to the RNA and then extended with SuperScript II RNase H− reverse transcriptase (Invitrogen) as previously described (10). The cDNA products were purified and resolved on a sequencing gel alongside sequencing ladders generated from pKC980 (Table 1) with the same primer used for the primer extension. The gels were visualized using a phosphorimaging analyzer (model BAS1500; Fuji Photo Film Co. Ltd., Tokyo, Japan).
Overexpression and Purification of V. vulnificus IHF
The ihfA coding region was amplified by PCR using the primers IhfA06 and IhfA07 (Table 2) and then subcloned into a His6 tagging expression vector, pRSET C (Invitrogen), to result in pHS304. The His-tagged IhfA protein was then expressed in E. coli BL21(DE3), and the protein was purified by affinity chromatography according to the manufacturer's procedure (Qiagen). In a similar way, the ihfB coding region was amplified by PCR using the primers IhfB01 and IhfB02 (Table 2). The expression and purification of the His-tagged IhfB were carried out using pHS305, carrying the ihfB coding region. The purified proteins were dialyzed against storage buffer (28), and equal concentrations of IhfA and IhfB were mixed and then used as a V. vulnificus IHF protein. The protein concentrations were determined by the method of Bradford (29), with bovine serum albumin as the standard.
Gel Mobility Shift Assay and DNase I Footprinting
The 201-bp upstream region of vvpE, extending from −236 to −36 (hereafter, +1 is the transcription start site of PS), was amplified by PCR using 32P-labeled VVPE031 and unlabeled VVPE032 as the primers (Table 2). Binding of IHF to the labeled DNA (7 nm) and electrophoretic analysis of the DNA-IHF complexes were carried out by the procedure described previously (17, 30). The same labeled 201-bp DNA was used for the DNase I protection assays. The binding of IHF to the labeled DNA, and DNase I digestion of the DNA-IHF complexes followed the procedure previously described by Choi et al. (31). After precipitation with ethanol, the digested DNA products were resolved on a sequencing gel alongside sequencing ladders of pKC980 generated using VVPE031 as a primer. The gels were visualized as described above for the primer extension analysis.
Measurement of Elastase Activity and Data Analysis
Cultures of the V. vulnificus strains were grown at 30 °C under aeration, and the growth was monitored by measuring the A600 of the cultures. The cultures were harvested at an A600 value of 2.0, and the elastase activities in the stationary phase were determined according to the procedure previously described (32). Averages ± S.E. were calculated from at least three independent experiments.
RESULTS
SmcR Directly Binds to the vvpE Regulatory Region in Vivo
Because SmcR binding at −196.5 is unusually distant for the protein to interact directly with RNAP, we examined whether the SmcR binding to the vvpE regulatory region occurs indeed in V. vulnificus. For this purpose, the cross-linked chromatin from the wild type and smcR mutant HS03 cells was immunoprecipitated using the antibody against SmcR (Fig. 2). As positive controls, the input chromatin from both the wild type and HS03 appeared to carry the vvpE regulatory region when determined based on PCR using the primers VVPE031 and VVPE032 (Fig. 2). The primers were designed to specifically amplify the vvpE regulatory region that contains the SmcR-binding site and is 201 bp in length (Table 2). After reversing the cross-links, the vvpE regulatory fragment was detected in the chromatin precipitate of the wild type, induced in the presence of the anti-SmcR antibody. The presence of the vvpE regulatory region in the precipitated chromatin was caused by the specific binding of the SmcR protein to the DNA, because no vvpE regulatory DNA was detected in the precipitate induced in the absence of the anti-SmcR antibody. Consistent with this, no detectable level of the vvpE regulatory fragment was present in the anti-SmcR immunoprecipitate of the smcR mutant (Fig. 2), verifying that the SmcR protein directly binds to the vvpE regulatory region in V. vulnificus as well as in vitro.
FIGURE 2.
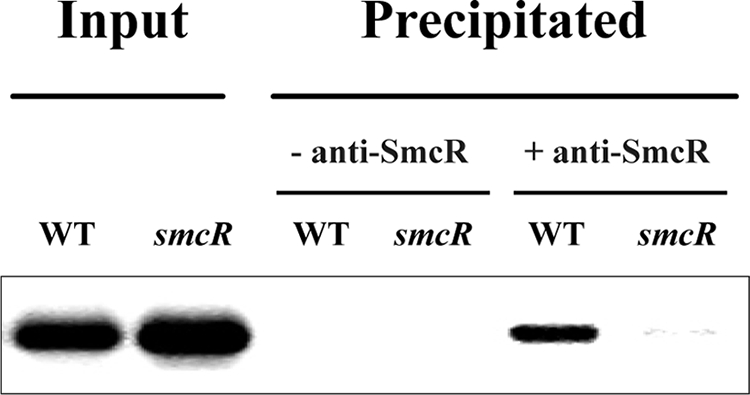
SmcR binding to the vvpE regulatory region in vivo. The cells were cross-linked, washed, and then sonicated to produce sheared chromatin as described under “Experimental Procedures.” The DNA was purified from the sheared chromatins before precipitation (input, positive control) and after precipitation in the presence (+) or absence (−) of the anti-SmcR antibody. The DNA was then amplified by PCR using primers VVPE031 and VVPE032 (Table 2). WT, wild type; smcR, smcR mutant.
Mapping of cis-Acting Regulatory Elements in the vvpE Regulatory Region
To delineate the cis-DNA sequences required for activation of vvpE, transcriptional fusions of the mutant vvpE regulatory regions were made to the luxAB reporter gene (Fig. 3A). Culture luminescence was used to quantify the capacity of each vvpE upstream fragment to activate vvpE (Fig. 3B). For the V. vulnificus containing pHS201, a plasmid carrying an intact vvpE regulatory region, the luminescence activity was about 3.8 × 106 RLU. The luminescence was reduced in the strains that carried either pHS206, pHS176, pHS136, or pHS56, indicating that the important cis-acting regulatory element(s) for the activation of vvpE was mutated in the vvpE regulatory region of the transcriptional fusions tested.
Because the sequence for SmcR binding extended from −207 to −186 (10, 17), the reduced luminescence from the cell containing pHS206 appeared to be due to the lack of part of the SmcR binding sequence on the transcriptional fusion. The sequences from −56 to −47 scored a reasonable homology to the UP element consensus sequences of the promoters recognized by the α-CTD of E. coli RNAP (Fig. 1) (33), implying that the vvpE regulatory region of pHS56 did not harbor UP element sufficient for the activation of vvpE by the V. vulnificus RNAP. The light produced by the cells carrying either pHS176 or pHS136 was significantly reduced, suggesting that the upstream region extending from −176 to −127 is also required for activation of vvpE by an as yet unknown mechanism. Interestingly, luminescence of pHS106 did not significantly decrease and was comparable with that of pHS201, indicating that the sequences from −106 to −97 did not harbor any cis-acting regulatory elements essential for the activation of vvpE.
The Correct Phasing of SmcR Binding Is Required for the Activation of vvpE
One possible mechanism for the activation of vvpE by SmcR bound at −196.5 is that a transcription factor(s) bring SmcR to RNAP for their direct interaction. To examine this possibility, the transcriptional fusions of the vvpE regulatory region with either 5- or 10-bp deletions within the sequences from −106 to −97 were constructed to the luxAB reporter genes, and their luminescence was compared with that of pHS201 (Fig. 4). Luminescence of pHS221 carrying the 5-bp deletion from −101 to −97 significantly decreased. Because the sequences from −106 to −97 were not essential for the vvpE expression and the 5-bp deletion results in a half-integral turn of DNA, the decreased luminescence of pHS221 presumably resulted from the altered phasing of SmcR on the transcriptional fusion. In contrast, luminescence of pHS222 carrying the 10-bp deletion from −106 to −97 was greater than that of pHS201, indicating that reducing the distance between SmcR and RNAP increased the vvpE expression only when a specific phasing of SmcR was maintained. These results suggested that SmcR contacts directly with RNAP for the activation of vvpE. However, because SmcR did not appear to bend the vvpE upstream DNA (data not shown), the results suggested that the direct contact between SmcR and RNAP was presumably mediated by yet unidentified protein(s).
Effect of ihfA Mutation on Production of Elastase
To examine the role of IHF, the V. vulnificus ihfA isogenic mutant was constructed by allelic exchanges. Double cross-overs, in which the wild type ihfA on the V. vulnificus chromosome was replaced with the ΔihfA allele, were confirmed using PCR as previously described (data not shown) (10). The ihfA mutant chosen for further analysis was named HS05 (Table 1). For the wild type, elastase was produced and reached a maximum at 29 units during stationary growth (Fig. 5A). When compared with parental wild type, HS05 produced much less elastase that was almost 4-fold lower than that of the wild type, indicating that the elastase expression is positively regulated by IHF. Real time PCR was used to reconfirm positive regulation of the elastase expression by IHF and revealed that the vvpE transcript level also significantly decreased as a result of ihfA inactivation. These results demonstrated that IHF activates vvpE at the transcription level.
FIGURE 5.

Dependence of elastase production and vvpE transcription on IHF. Samples from the stationary phase cultures (A600 = 2.0) of the wild type, smcR, and ihfA mutant were removed and analyzed for the elastase activity (A) and vvpE transcript (B). For a complementation test, when the cultures of the ihfA mutant containing pHS303 reached an A600 of 0.6, IPTG was added to a final concentration of 0.1 mm to induce the expression of recombinant ihfA. WT, wild type; smcR, smcR mutant; ihfA, ihfA mutant. The error bars represent S.E.
We examined whether the introduction of pHS303 carrying a recombinant ihfA could complement the decrease of the vvpE expression in HS05. For this purpose, pHS303 was constructed by subcloning ihfA amplified by PCR using the primers IhfA05 and IhfA06 into the broad host range vector pRK415 (Table 1) (34). The elastase activity and vvpE transcript level of HS05 (pHS303) was restored to the wild type levels (Fig. 5). Therefore, the decreased vvpE expression in HS05 was confirmed to result from the inactivation of functional ihfA rather than any polar effects on genes downstream of ihfA. Because SmcR has been previously demonstrated as a key activator for the vvpE expression (10), it is perhaps not surprising that the residual levels of elastase and vvpE transcript in HS05 were still significantly higher than those observed in the smcR mutant HS03 (Fig. 5).
Effect of IHF on vvpE Expression Is Mediated through PS
The presence of two promoters for vvpE expression raises the question of whether the activation by IHF is through the stationary phase-induced promoter (PS) or the constitutive promoter (PL). To answer this question, the activities of PL and PS in the wild type and ihfA mutant were determined by primer extension analyses (Fig. 6). As such, RNAs were prepared from cultures grown to the log phase or stationary phase. PL activities were observed in the cells grown to both the log phase and the stationary phase. Plus, when determined based on the intensity of the bands of the reverse transcripts, the PL activities were not significantly changed by the inactivation of ihfA. These results were consistent with previous observations by the current authors in which the activity of PL was found to be constitutive regardless of the growth phase. In contrast to PL, no band corresponding to PS activity was detected in the stationary phase RNA from the ihfA mutant, indicating that the PS activity is positively regulated by IHF. When recombinant ihfA was reintroduced to complement the ihfA mutant, the decreased PS activity was restored to the wild type level. Accordingly, these results indicate that IHF is involved in the regulation of vvpE transcription by activating the SmcR-dependent promoter PS rather than PL.
FIGURE 6.

Activities of PL and PS in V. vulnificus with different genetic backgrounds. The vvpE promoter activities were determined separately by primer extension of the RNA derived from the wild type, ihfA mutant, and complemented strain, as indicated. Total RNA was prepared from the log phase (L; A600 = 0.6) and stationary phase (S; A600 = 2.0) of each culture. Lanes G, A, T, and C represent the nucleotide sequencing ladders of pKC980. The asterisks indicate the sites of the transcription starts for PS and PL, respectively. WT, wild type; ihfA, ihfA mutant.
IHF Binds Specifically to the vvpE Regulatory Region
The 201-bp DNA fragment encompassing the vvpE regulatory region was incubated with increasing amounts of IHF (IhfA and IhfB) and then subjected to electrophoresis. As seen in Fig. 7, the addition of IHF at a concentration of 200 nm resulted in a shift of the 201-bp DNA fragment to a single band with a slower mobility. The binding of IHF was also specific, because assays were performed in the presence of 1 μg of poly(dI-dC) as a nonspecific competitor. In a second gel mobility shift assay, the same, but unlabeled, 201-bp DNA fragment was used as a self-competitor to confirm the specific binding of IHF to the vvpE regulatory region (Fig. 7). The unlabeled 201-bp DNA competed for the binding of IHF in a dose-dependent manner (Fig. 7), confirming that IHF binds specifically to the DNA. These results suggested that IHF activates PS by directly binding to the vvpE regulatory region.
FIGURE 7.

Gel mobility shift assay for binding of IHF to the vvpE regulatory region. The 201-bp DNA fragment of the vvpE upstream region was radioactively labeled and then used as a probe DNA. Lanes 1–5, the radiolabeled fragments (7 nm) were mixed with increasing amounts of IHF (0, 100, 200, 300, and 400 nm in lanes 1–5, respectively) and then resolved on a 5% polyacrylamide gel. Lanes 6–9, for a competition analysis, the same yet unlabeled 201-bp DNA fragment was used as a self-competitor DNA. Various amounts of the self-competitor DNA were added to the reaction mixture containing 7 nm of the labeled DNA prior to the addition of 400 nm of IHF. Lanes 6–9, probe DNA incubated with 35, 70, 175, and 350 nm of competitor DNA, respectively. B, bound DNA; F, free DNA.
Identification of the IHF-binding Sites
As shown in Fig. 8A, the DNase I footprinting performed with IHF revealed two clear protection patterns in the upstream region of PS extending from −183 to −165 (centered at −174, IHFB1) and from −137 to −125 (centered at −131, IHFB2), respectively (Figs. 1 and 8A). Several nucleotides also showed enhanced cleavages, which have been frequently observed in DNase I protection analyses of the binding sites of transcriptional regulatory proteins with DNA bending activities, such as cAMP receptor protein (10). Both sequences were almost equally protected by the same level of IHF, indicating that IHF bound to the two sites with a comparable affinity. The pattern of protection was consistent with the result of gel mobility shift assays where only a single DNA-IHF complex was produced (Fig. 7). These IHF-binding sites were further supported by the finding that the sequences spanning from −176 to −167 and −136 and −127 mapped by mutational analysis of the vvpE upstream region are essential cis-regulatory elements for the vvpE expression (Fig. 3).
FIGURE 8.

IHF-binding sites in the vvpE regulatory region. A, DNase I protection analysis of IHF binding to the vvpE regulatory region. Lane 1, no IHF added; lanes 2–4, IHF at 200, 400, and 600 nm, respectively. Lanes G, A, T, and C represent the nucleotide sequencing ladders of pKC980. The nucleotides showing an enhanced cleavage in the presence of IHF are indicated by the black boxes, whereas the regions protected by IHF are indicated by the shaded boxes. B, schematic representation of the vvpE regulatory region with the proposed binding sites for SmcR (SB), IHF (IHFB1 and IHFB2), and RNAP, where +1 is the transcription start site of PS. The arrow represents the transcriptional direction and coding region of vvpE, and the solid line represents the upstream region of vvpE, respectively. The sequences for binding of SmcR (SB) and RNAP were determined previously (10, 18). The sequences for the binding of IHF, proposed by the DNase I protection assay, are represented by shaded boxes. The consensus sequences for the binding of IHF from E. coli (37) were shown below the V. vulnificus DNA sequence. W, A or T; R, A or G; N, any base.
IHF Induces a Protein-Protein Interaction between SmcR and RNAP
Although IHF associated with the activation of the vvpE expression was identified, its mechanism of the activation had yet to be determined. Accordingly, the cross-linked chromatins from the wild type, smcR mutant HS03, and ihfA mutant HS05 cells were immunoprecipitated using the antibody against SmcR (Fig. 9A). As positive controls, the input chromatins from the wild type, HS03, and HS05 appeared to carry the DNAs for SmcR (S) and RNAP (R) binding (Fig. 9), based on PCR using the primers VVPE033 and VVPE034 (for S) and VVPE035 and VVPE036 (for R) (Table 2). PCR amplification of the DNA covering SmcR- and RNAP-binding sites together (F) from the input chromatin and chromatin precipitate was not successful using primers VVPE033 and VVPE036, indicating that fragmentation of chromatin using sonication and DNase I treatment was sufficient to separate the SmcR- and RNAP-binding sites on the resulting chromatins. Neither S nor R fragment was detected in the precipitate induced in the absence of the anti-SmcR antibody (negative controls).
FIGURE 9.

IHF induces direct interaction between SmcR and RNAP. A, direct interaction between SmcR and RNAP on the vvpE regulatory region was analyzed using a ChIP assay. The cells were cross-linked, washed, and then sonicated to produce sheared chromatin as described under “Experimental Procedures.” DNase I was added to the sheared chromatins to reduce their average sizes. The DNA was purified from the DNase I-treated chromatin fragments before precipitation (input, positive control) and after precipitation in the presence (+) or absence (−) of the anti-SmcR antibody. The DNA was then amplified by a PCR using the following primers: VVPE033 and VVPE034 (for DNA S), VVPE035 and VVPE036 (for DNA R), and VVPE033 and VVPE036 (for DNA F) (Table 2). WT, wild type; smcR, smcR mutant; ihfA, ihfA mutant. B, a proposed role of IHF in the activation of PS. IHF is able to increase the activity of PS by inducing DNA looping and thereby enhancing the interaction between SmcR and RNAP in the wild type (left panel) but not in the ihfA mutant (right panel).
After reversing the cross-links, both the S and R fragments were detected in the chromatin precipitate of the wild type, induced in the presence of the anti-SmcR antibody. The presence of the DNA fragment for RNAP binding (R) in the chromatin precipitate indicated that the SmcR protein directly contacts RNAP to enhance the activity of PS. Consistent with this, the R fragment was not detected in the anti-SmcR immunoprecipitate of the smcR mutant. It was noteworthy that the direct interaction of SmcR with RNAP was dependent on the presence of functional IHF, because no R fragment was detected in the chromatin precipitate induced in the ihfA mutant. This dependence of SmcR on IHF for direct interaction with RNAP, along with the two IHF-binding sites observed between SmcR- and RNAP-binding sites, suggests a possible model whereby IHF introduces a bend to the vvpE regulatory region and thus brings SmcR to RNAP as proposed in Fig. 9B.
In summary, SmcR binding to the vvpE regulatory region in vivo was demonstrated by a ChIP assay. Mutational analyses of the vvpE upstream region suggested that direct interaction of SmcR with RNAP is essential for activation of the RpoS-dependent promoter PS. An additional regulator, IHF, was identified and was required for the activation of PS by directly binding two distinct sites between SmcR- and RNAP-binding sites. Finally, a working model in which IHF brings SmcR to contact RNAP by looping the vvpE regulatory DNA was proposed and confirmed by ChIP assays.
DISCUSSION
Among global regulators controlling numerous genes contributing to pathogenesis as well as survival of the pathogenic Vibrio spp., Vibrio cholerae HapR, Vibrio parahemeolyticus OpaR, Vibrio anguillarum VanT, and V. vulnificus SmcR have been demonstrated as LuxR homologues (4–7, 10, 15–17). Although a number of the promoters regulated by LuxR homologues have been reported (3, 11, 12, 17, 35, 36), the molecular mechanism by which the proteins modulate the expression of the promoters has yet to be extensively studied (10–12, 35). Therefore, the question of whether the LuxR homologues collaborate with any other regulator proteins and whether (and how) the LuxR homologues interact directly with RNAP for regulation of the target promoters has not yet been addressed.
IHF is a member of the nucleoid-associated proteins (for a recent review, see Ref. 21) and a heterodimeric protein composed of the α and β subunits, encoded by the ihfA (himA) and ihfB (himD) genes, respectively. Among the nucleoid-associated proteins, IHF has long been an exception because it recognizes and binds to a specific asymmetric binding site on DNA. The consensus IHF-binding sequence from E. coli is WATCAANNNNTTR, where W stands for A or T and R stands for A or G (37). The V. vulnificus IHF-binding sequences in the vvpE regulatory region, IHFB1 and IHFB2, scored 85 and 69% homologies to the consensus sequence, respectively (Fig. 8B). IHF has been known as a global regulator to control the transcriptions of over 100 genes of various functions in E. coli (38). The amino acid sequences of the V. vulnificus IhfA and IhfB were 87 and 81% identical to those of IhfA and IhfB from E. coli, respectively, and their identity appeared evenly throughout the whole proteins (data not shown). It is not yet clear whether IHF also acts as a global regulator in V. vulnificus. Nonetheless, the similarity in the binding sequences and the high level of identity in the amino acid sequences of IHF proteins from V. vulnificus and E. coli indicated that they might perform a similar function in their global gene regulation.
Although IHF can control bacterial transcription by different mechanisms, its ability to bend DNA as much as 180° and thus facilitate a protein-protein interaction between an upstream activator and RNAP represents a key feature as a transcriptional regulator (39). In addition to the SmcR-binding site within the vvpE regulatory region mentioned above, many of the known binding sites of SmcR and LuxR are also unusually distant from the promoter (17, 35). For example, LuxR binding at region A (centered at −250.5) and at region B (centered at −117) for activation of luxCDABEGH operon in V. harveyi is also exceptionally distant (35). It is apparent from this study that the ability of SmcR to function as a transcriptional activator depends on the presence of IHF that brings the protein at −165.5 to RNAP for direct contacts (Figs. 5, 6, and 9). This finding, along with the previously observed LuxR-binding sites well removed from the target promoter, suggests that the involvement of other transcriptional factors that promote direct contacts between the LuxR homologues and RNAP may be a common feature inherited in the regulation of genes by quorum sensing.
Additional levels of control for the precise coordinate expression of the virulence factors may be obtainable through the participation of multiple global regulators. Elastolytic protease is a putative virulence factor that has been proposed to account for the destructive nature of V. vulnificus infections. Elastase has been hypothesized as an important virulence factor for V. vulnificus by several studies (for a recent review, see Refs. 40). Nonetheless, it is essential to understand the mechanism whereby the expression pattern and level of elastase are modified during infection to further understand the role of elastase in pathogenesis. We previously demonstrated that vvpE expression is activated by the RpoS-dependent promoter, PS, in the stationary phase, which is under the control of SmcR (10, 18). RpoS (σS or σ38) is a stationary phase-specific sigma factor and controls the expression of numerous genes responsible for increased resistance of bacteria to a range of environmental stresses (41, 42). What seems likely is that RpoS would make expression of vvpE temporally coordinated along with expression of the stress resistance genes when V. vulnificus cells encounter increased stresses imposed by the host immune defense system during infection. LuxR homologues, including SmcR, of Vibrio spp. are proposed to sense the place where their cell densities reach higher than critical levels (1, 2). It is still difficult to define the additional signal(s) integrated by IHF into the regulation of vvpE; however, it is likely that IHF allows more precise tuning of the elastase expression by optimizing SmcR activation of the RpoS-dependent promoter PS. Whereas the collaboration between RpoS, SmcR, and IHF and its implications in pathogenesis of V. vulnificus has yet to be explored further, the overall success of the organism during pathogenesis would be enhanced through this fine tuning of the temporally and spatially coordinated expression of elastase.
Supplementary Material
This work was supported by World Class University Program Grant R32-2008-000-10183-0, a 21C Frontier Microbial Genomics and Applications Center Program grant, National Research Laboratory Grant R0A-2007-000-20039-0, and funds from the Ministry of Education, Science and Technology of the Republic of Korea (to S. H. C.).
- PL
- promoter L
- PS
- promoter S
- ChIP
- chromatin immunoprecipitation
- IHF
- integration host factor
- RNAP
- RNA polymerase
- RLU
- relative light unit.
REFERENCES
- 1.Fuqua C., Greenberg E. P. (2002) Nat. Rev. Mol. Cell Biol. 3, 685–695 [DOI] [PubMed] [Google Scholar]
- 2.Ng W. L., Bassler B. L. (2009) Annu. Rev. Genet. 43, 197–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waters C. M., Bassler B. L. (2006) Genes Dev. 20, 2754–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDougald D., Rice S. A., Kjelleberg S. (2001) J. Bacteriol. 183, 758–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jobling M. G., Holmes R. K. (1997) Mol. Microbiol. 26, 1023–1034 [DOI] [PubMed] [Google Scholar]
- 6.McCarter L. L. (1998) J. Bacteriol. 180, 3166–3173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Croxatto A., Chalker V. J., Lauritz J., Jass J., Hardman A., Williams P., Cámara M., Milton D. L. (2002) J. Bacteriol. 184, 1617–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu J., Miller M. B., Vance R. E., Dziejman M., Bassler B. L., Mekalanos J. J. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 3129–3134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beyhan S., Bilecen K., Salama S. R., Casper-Lindley C., Yildiz F. H. (2007) J. Bacteriol. 189, 388–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeong H. S., Lee M. H., Lee K. H., Park S. J., Choi S. H. (2003) J. Biol. Chem. 278, 45072–45081 [DOI] [PubMed] [Google Scholar]
- 11.Kovacikova G., Skorupski K. (2002) Mol. Microbiol. 46, 1135–1147 [DOI] [PubMed] [Google Scholar]
- 12.Lin W., Kovacikova G., Skorupski K. (2007) Mol. Microbiol. 64, 953–967 [DOI] [PubMed] [Google Scholar]
- 13.McDougald D., Rice S. A., Kjelleberg S. (2000) Gene 248, 213–221 [DOI] [PubMed] [Google Scholar]
- 14.Roh J. B., Lee M. A., Lee H. J., Kim S. M., Cho Y., Kim Y. J., Seok Y. J., Park S. J., Lee K. H. (2006) J. Biol. Chem. 281, 34775–34784 [DOI] [PubMed] [Google Scholar]
- 15.Shao C. P., Hor L. I. (2001) J. Bacteriol. 183, 1369–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee J. H., Rhee J. E., Park U., Ju H. M., Lee B. C., Kim T. S., Jeong H. S., Choi S. H. (2007) J. Microbiol. Biotechnol. 17, 325–334 [PubMed] [Google Scholar]
- 17.Lee D. H., Jeong H. S., Jeong H. G., Kim K. M., Kim H., Choi S. H. (2008) J. Biol. Chem. 283, 23610–23618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeong H. S., Jeong K. C., Choi H. K., Park K. J., Lee K. H., Rhee J. H., Choi S. H. (2001) J. Biol. Chem. 276, 13875–13880 [DOI] [PubMed] [Google Scholar]
- 19.Barnard A., Wolfe A., Busby S. (2004) Curr. Opin. Microbiol. 7, 102–108 [DOI] [PubMed] [Google Scholar]
- 20.Browning D. F., Busby S. J. (2004) Nat. Rev. Microbiol. 2, 57–65 [DOI] [PubMed] [Google Scholar]
- 21.Dorman C. J. (2009) Adv. Appl. Microbiol. 67, 47–64 [DOI] [PubMed] [Google Scholar]
- 22.Rhee J. E., Kim K. S., Choi S. H. (2005) J. Bacteriol. 187, 7870–7875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J. H., Kim M. W., Kim B. S., Kim S. M., Lee B. C., Kim T. S., Choi S. H. (2007) J. Microbiol. 45, 146–152 [PubMed] [Google Scholar]
- 24.de Lorenzo V., Herrero M., Jakubzik U., Timmis K. N. (1990) J. Bacteriol. 172, 6568–6572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim H. J., Lee J. H., Rhee J. E., Jeong H. S., Choi H. K., Chung H. J., Ryu S., Choi S. H. (2002) J. Microbiol. Biotechnol. 12, 318–326 [Google Scholar]
- 26.Miller V. L., Mekalanos J. J. (1988) J. Bacteriol. 170, 2575–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee B. C., Lee J. H., Kim M. W., Kim B. S., Oh M. H., Kim K. S., Kim T. S., Choi S. H. (2008) Infect. Immun. 76, 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sambrook J., Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, 3rd Ed., pp. A1.7–A1.18, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 29.Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 30.Lee J. H., Choi S. H. (2006) Mol. Microbiol. 60, 513–524 [DOI] [PubMed] [Google Scholar]
- 31.Choi H. K., Park N. Y., Kim D. I., Chung H. J., Ryu S., Choi S. H. (2002) J. Biol. Chem. 277, 47292–47299 [DOI] [PubMed] [Google Scholar]
- 32.Jeong K. C., Jeong H. S., Rhee J. H., Lee S. E., Chung S. S., Starks A. M., Escudero G. M., Gulig P. A., Choi S. H. (2000) Infect. Immun. 68, 5096–5106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Estrem S. T., Gaal T., Ross W., Gourse R. L. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 9761–9766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keen N. T., Tamaki S., Kobayashi D., Trollinger D. (1988) Gene 70, 191–197 [DOI] [PubMed] [Google Scholar]
- 35.Miyamoto C. M., Smith E. E., Swartzman E., Cao J. G., Graham A. F., Meighen E. A. (1994) Mol. Microbiol. 14, 255–262 [DOI] [PubMed] [Google Scholar]
- 36.Pompeani A. J., Irgon J. J., Berger M. F., Bulyk M. L., Wingreen N. S., Bassler B. L. (2008) Mol. Microbiol. 70, 76–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Craig N. L., Nash H. A. (1984) Cell 39, 707–716 [DOI] [PubMed] [Google Scholar]
- 38.Arfin S. M., Long A. D., Ito E. T., Tolleri L., Riehle M. M., Paegle E. S., Hatfield G. W. (2000) J. Biol. Chem. 275, 29672–29684 [DOI] [PubMed] [Google Scholar]
- 39.Rice P. A., Yang S., Mizuuchi K., Nash H. A. (1996) Cell 87, 1295–1306 [DOI] [PubMed] [Google Scholar]
- 40.Miyoshi S., Shinoda S. (2000) Microbes Infect. 2, 91–98 [DOI] [PubMed] [Google Scholar]
- 41.Lange R., Hengge-Aronis R. (1991) Mol. Microbiol. 5, 49–59 [DOI] [PubMed] [Google Scholar]
- 42.Tanaka K., Takayanagi Y., Fujita N., Ishihama A., Takahashi H. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 3511–3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Milton D. L., O'Toole R., Horstedt P., Wolf-Watz H. (1996) J. Bacteriol. 178, 1310–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.