

Abstract
To find a suitable ligand from a complex antigen system is still a mission to be accomplished. Here we have explored a novel “library against proteome” panning strategy for ligand screening and antigen purification from a complex system using phage-displayed antibody technology. Human plasma proteome was targeted for phage library panning. During the process, the panning was carried out in solution, using a biotin/streptavidin beads separation system, for three rounds. Nine monoclonal phages, bound tightly to a number of unknown plasma proteins, were selected from the last round, six of which were directly employed as cross-linked matrices to purify their corresponding antigens from the plasma. The proteins isolated by G5 and E1 matrices were identified as amyloid protein and apolipoprotein A-I precursor, respectively. The results demonstrated that it was feasible to simultaneously obtain a number of ligand phages for various antigens, including low abundant proteins in a non-comparative proteome-wide system.
Keywords: Methods/Mass Spectrometry, Protein/Purification, Proteomics, Cross-linking, Phage Display, Proteome-wide, Screening
Introduction
Human plasma proteome is the most important field for proteomic research, clinical diagnosis, and treatments. Since the Human Proteome Organization Plasma Proteome Project started, thousands of human plasma proteins and peptides have been characterized at a high or low concentration in various methods such as two-dimensional liquid chromatography-tandem mass spectrometry. However, large quantities of plasma proteins still remain to be purified and characterized. In the present study, we aimed at a rapid purification and characterization method for unknown proteins in a whole proteome using a “library against proteome” panning strategy and cross-linked antibody phages.
Affinity technologies, which offer a one-step increase in purity and concentration of target molecules, remain as the most attractive method available for protein purification (1, 2). However, the traditional methods to generate affinity ligands such as monoclonal antibodies and synthetic compounds are expensive and time-consuming, which limit the application of affinity technologies (3, 4). Fortunately, the advances in phage display technology are providing a highly efficient and inexpensive way to acquire affinity ligands (5–7). In a phage-display library, ligands are displayed on the virion surface. By panning a library, phages with specific ligands against any given targets can be isolated from a large pool of candidates. Moreover, ligands with desired affinity can be obtained by either optimization of panning strategy or affinity maturation after the initial selection (8, 9).
Conventionally, the targets of phage panning were purified proteins, whereas simultaneous screening on complex antigens with a phage library was a relatively new concept. One approach was to use the subtractive selection among a few comparative systems such as cells or cellular extracts (10–11). Another approach was to pre-isolate complex proteins with two-dimensional electrophoresis before the panning (12). Recently a “library against library” strategy was developed combining one phage library with another library (such as the yeast-displayed system), as reported previously (13). However, screening through a proteome-wide system was rarely reported. In the present study, we have introduced a rapid procedure for phage-based panning through a non-comparative natural proteome-wide system. Different from the traditional panning strategies described above, we targeted human plasma proteome for the ligand screening strategy (also called the “library against proteome” strategy).
To overcome the difficulty in antigen characterization for each positive binder, we have adopted a new technique of phage cross-linking for our panning process. Usually, after a specific phage clone is isolated from the library, its ligand fragment is separately produced and immobilized to a support matrix for further use. However, in 1998, a new technique was developed to make soluble phages into an insoluble aggregate by directly cross-linking pVIII capsid proteins of filamentous phages (14) with cross-linkers. As the specific ligand is genetically fused on the phage surface, the aggregate serves as an effective affinity absorbent to purify the target antigen. Because of the high efficiency of library screening and the convenience of phage amplification, the direct availability of phage particles as affinity materials will represent a considerable reduction in cost, time, and efforts for proteins purification, and therefore facilitate high-throughput purification. We have modified the cross-linking approach to further reduce the cost.
In our strategy (Fig. 1), panning was carried out in solution using biotin/streptavidin beads. In each round of panning, phage particles with specific ligands bound to plasma proteins were enriched. Multiple ligands to various targets could be obtained simultaneously during the same round of phage library screening. In contrast to the difficulty in isolating antigens, monoclonal phage antibodies could be easily achieved by plating on dishes. Individual clones were picked up from the repertoire and cultured. The specificity of mono-phages for antigen proteins was examined by ELISA,2 and then positive mono-phages were prepared as cross-linked matrices to purify the corresponding antigen from plasma proteome. Purified antigens were identified with peptide sequence tag technology by matrix-assisted laser desorption/ionization time-of-flight & time-of-flight mass spectrometry (MALDI-TOF/TOF).
FIGURE 1.

Schema of the library against proteome panning strategy. A, biotinylation of protein antigens in the plasma proteome. B, the panning process in the liquid phase started. The biotinylated antigens were mixed with the phage library. C, phages bound to different biotinylated antigens were separated by streptavidin beads from dissociative ones. D, elution and enrichment of antigen-bound phage particles. E, eluted phages were amplified to a secondary library for the next round of panning. F, monoclonalization of eluted phages from the final round of panning by plating on dishes. G, hundreds of mono-phages were picked and cultured. Their specificity to the proteome was examined by ELISA. H, positive mono-phages were selected and amplified. Different filamentous mono-phages were cross-linked to insoluble aggregates with the cross-linker glutaraldehyde. I, different cross-linked phages were used as affinity matrices to isolate the corresponding unknown protein antigens from the proteome. J, identification of the isolated proteins from different phage matrices with MALDI-TOF/TOF.
Our work has explored a new strategy of phage library panning on a whole proteome to obtain various specific ligands for their targeted antigens. To reduce the inherent bias of plasma proteome in the panning process, human serum albumin (HSA) and immunoglobulin G (IgG), which make up over 70% of the proteome altogether in mass, were mostly removed from the system before the biopanning.
EXPERIMENTAL PROCEDURES
Removal of HSA and IgG from Human Plasma by Affinity Chromatography
The human plasma sample was from the Jiangsu Province Blood Bank (Nanjing, China). Abundant HSA and IgG in the human plasma sample were removed by affinity chromatography with Cibacron Blue 3GA (6% cross-link bead agarose, Sigma) and Protein A-Sepharose 4B (prepared by ourselves). Briefly, 1 ml of the human plasma sample was diluted with 1 ml of the equilibration buffer (10 mm Tris-HCl, pH 7.6) and then loaded onto the dye-ligand Cibracron-Blue 3GA column equilibrated by 10 column volumes of the equilibration buffer at room temperature as reported previously (15). The flow-through part was collected and then loaded onto the Protein A column, meanwhile the part of eluate from the dye-ligand column was collected with the elution buffer (10 mm Tris-HCl, 1.5 m NaCl, pH 7.6). A similar separation procedure was applied to the Protein A column as described above. Respectively, the flow-through part was collected, and the eluate was obtained with 100 mm glycine, pH 2.5, and then neutralized by 1 m Tris, pH 7.4. The final flow-through part was named HPP/HSA&IgGminus.
Biotinylation of HPP/HSA&IgGminus
HPP/HSA&IgGminus should be biotinylated for the panning in liquid phase. 20 μl of 10 mm EZ-Link® Sulfo-NHS-LC-Biotin (Pierce) was added to 1 ml of HPP/HSA&IgGminus in phosphate-buffered saline (PBS), pH 7.4, and mixed well. The biotinylation was accomplished when the mixture stood at 4 °C for 2 h. The free biotin reagent was then removed by dialysis in PBS, pH 7.4, at 4 °C. The efficiency of the biotinylation was determined as follows. The biotinylated sample was filtered sufficiently by the ImmunoPure® immobilized streptavidin settled gel (Pierce). The efficiency was calculated by measuring the protein concentration in the sample before and after the filtration.
Panning of the Phage Library
The human single fold single-chain antibody variable fragment (scFv) library (Tomlinson I+J) was a semi-synthetic single-chain human VH+Vκ library constructed with random CDR2 and CDR3 primers, which was a gift from Medical Research Council, UK. Using this scFv library and KM13 helper phage, a modified biopanning protocol was employed as previously described (16–18). Briefly, 0.5 ml of the biotinylated HPP/HSA&IgGminus and 0.5 ml of the original library (0.25 ml of I library plus 0.25 ml of J library, 1013 pfu/ml) were mixed well and rotated for 4 h at 4 °C. The mixture was then added to 200 μl of ImmunoPure® immobilized streptavidin settled gel and incubated with mixing for 2 h at room temperature. The gel was washed with PBS, pH 7.4, with centrifugation for 5 times. After dissociative proteins and nonspecific phages were washed away, phage particles bound to the protein beads complex were digested by 500 μl of 1 mg/ml trypsin with shaking for 10 min at room temperature. 250 μl of the digested phages was used to infect 1.75 ml of freshly prepared Escherichia coli TG1 (A600 = 0.6) cultured in the 2×TY medium (0.5% NaCl, 1.6% Tryptone, 1% yeast extract, w/v). After incubation at 37 °C for 30 min, the infected TG1 was plated on the TYE dish (0.8% NaCl, 1% Tryptone, 0.5% yeast extract, 1.5% agar, w/v, with 100 μg/ml ampicillin) and incubated at 37 °C overnight. One the next day all the clones were collected with 2 ml of 2×TY, 50 μl of which were cultivated in 25 ml of 2×TY medium with 100 μg/ml ampicillin. After incubation for 1 h at 37 °C, the culture was rescued by 40 μl of KM13 helper phage (1012 pfu/ml), and then incubation was continued at 37 °C for 30 min and was spun down at 3,000 × g for 10 min. The pellets were cultivated in 100 ml of 2×TY medium with 100 μg/ml ampicillin and 50 μg/ml kanamycin and incubated at 30 °C overnight. On the next day the culture was centrifuged at 11,000 × g for 10 min, and the supernatant was mixed with one-fourth volume of polyethylene glycol/NaCl (20% polyethylene glycol 6,000, w/v, 2.5 m NaCl) and stood on ice for 2 h. The mixture was centrifuged at 11,000 × g for 10 min, and the pellets were dissolved in 2 ml of PBS to make a secondary library for the next round of biopanning. The panning was done after three rounds.
Monoclonal Phage ELISA
Selected phage clones from the third round of digestion were cultivated and amplified separately. Each mono-phage culture supernatant was added into micro-wells that were coated with HPP/HSA&IgGminus and the negative control, respectively, and then blocked with 3% bovine serum albumin, w/v. After incubation for 1 h at room temperature and washing with PBS containing 0.2% Tween 20 three times, the positive binders were detected by using horseradish peroxidase-conjugated anti-M13 antibody (anti-M13/HRP, Amersham Biosciences) and 3,3′,5,5′-tetramethylbenzidine substrate (KPL, Gaithersburg, MD). Phage clones were considered to be positive if their ELISA signals on HPP/HSA&IgGminus were 3-fold higher than that on the negative control.
Preparation of Cross-linked Phages and Optimization of Conditions for Cross-linking
Phage particles were cross-linked according to the previous method with a revision (14, 19). Briefly, 1 ml of mono-phages (3 × 1013 pfu/ml) in coupling buffer (pH 3–10, the standard coupling buffer used for further study was 100 mm MOPS-HCl, pH 7) were mixed with 150 μl of glutaraldehyde (up to 23.4 μm). To this mixture, 150 μl of 50% polyethylene glycol 6000 (w/v) was added and mixed. The mixture was rotated overnight at room temperature. Unreacted aldehydic groups of glutaraldehyde were quenched by adding 8 ml of 1 m ethanolamine-HCl (pH 9) and 890 μl of 5 m NaCl, and the mixture was shaken for 2 h at room temperature. Cross-linked phages were then spun down and washed with Tris-buffered saline at least five times by centrifugation. The final pellets were suspended in Tris-buffered saline containing 0.02% NaN3 and stored at 4 °C.
To achieve effective cross-linking, the conditions for phage cross-linking were optimized. Instead of NHS-dextran (14, glutaraldehyde was used as the cross-linker in this study, and the solution containing amino groups should not be used. A broad range of cross-linking conditions, including buffer pH (3.5–9), glutaraldehyde (23.4 nm to 2.34 mm), and phage particles (>3.6 × 1011pfu/ml) was tested. The amount of cross-linked phages was determined spectrophotometrically, and the cross-linking ratio was calculated under each condition.
Purification of Human Plasma Proteins by Cross-linked Phages
10 ml of HPP/HSA&IgGminus was mixed with the insoluble cross-linked phages prepared from 20 ml of soluble phages (1013 pfu/ml), and the mixture was rotated at 4 °C overnight. After washing with Tris-buffered saline containing 0.2% Tween 20 five times at least, the adsorbed proteins were eluted from cross-linked phages by 100 mm triethylamine or 100 mm HCl then neutralized by 1 m Tris, pH 7.4, and examined in SDS-PAGE.
Identification of Unknown Protein Antigens by MALDI-TOF/ TOF
The unique unknown protein bands of different eluates in SDS-PAGE gel were cut off and then identified by MALDI-TOF/TOF (Beijing Proteome Research Center). Briefly, the procedures of tryptic in-gel digestion and MALDI-TOF/TOF were done according to the previous protocol (20–22). The results of MALDI-TOF/TOF were searched with the Mascot engine (Matrix Science, available on-line). The search results with scores > 55 were considered to be significant. Unless otherwise stated all chemicals mentioned above used in this work were purchased from Sigma and were of the highest purity available.
RESULTS
Removal of HSA and IgG from Human Plasma
Fresh human plasma passed through the Cibacron Blue 3GA column and then Protein A column. Both the flow-through and eluate were collected and examined by SDS-PAGE (Fig. 2). The results showed that the amount of HSA or IgG was reduced significantly. The human plasma sample with HSA and IgG removed was named as HPP/HSA&IgGminus.
FIGURE 2.
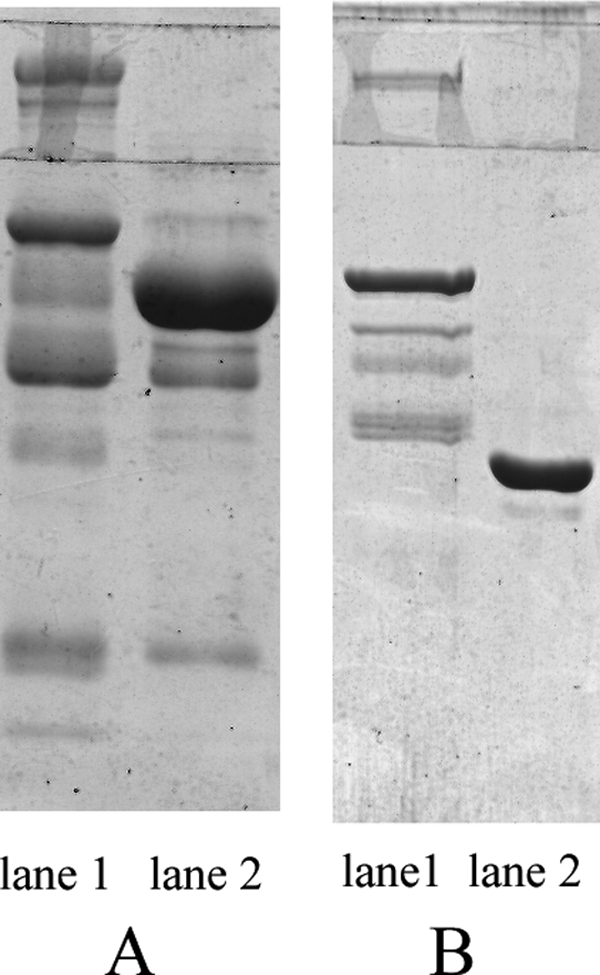
Significant reduction of HSA and IgG in human plasma. A, human plasma was loaded onto the Cibacron Blue 3GA column. The flow-through and eluate were visualized by Coomassie Blue staining, respectively. Lane 1 shows the flow-through part and lane 2 the eluate part, mostly HSA. B, the flow-through part of Cibacron Blue 3GA column was loaded onto the Protein A column. The final flow-through and the new eluate were visualized by Coomassie Blue staining, respectively. Lane 1 shows the final flow-through part and lane 2 the new eluate part, mostly IgG heavy chain. The figure shows that the amount of HSA and IgG in the final flow-through was reduced significantly.
Biotinylation of HPP/HSA&IgGminus
The sample of HPP/HSA&IgGminus was biotinylated with biotin reagents. The efficiency of biotinylation was 91.6% as determined using the protein quantification in the sample before and after the streptavidin bead filtration (Table 1).
TABLE 1.
Biotinylation efficiency was determined by protein quantification before and after the streptavidin beads filtration
| Mean protein concentration | Volume | Biotinylation efficiency | |
|---|---|---|---|
| μg/ml | ml | % | |
| Before filtration | 1066.2 | 0.2 | 91.6 |
| After filtration | 23.8 | 0.75 | 91.6 |
Library Panning and Ligand Identification by ELISA
The panning of scFv library Tomlinson I+J that targeted HPP/HSA&IgGminus was conducted in liquid phase based on the biotin/streptavidin system. Phage clones bound to dissociative plasma proteins were steadily enriched and titered after three rounds of screening (two orders of magnitude increase in recovery), which suggested no need for further panning (Table 2). Nine phage clones from the last round were selected by ELISA for their affinity to HPP/HSA&IgGminus (Fig. 3), including clone F4, which also showed specific binding to the negative control as the nonspecific control.
TABLE 2.
Enrichment of antigen-specific phages in the panning
| Round | 1 | 2 | 3 |
|---|---|---|---|
| Phage applied (pfu) | 1.6 × 1013 | 1.5 × 1012 | 6.9 × 1013 |
| Phage eluted (pfu) | 7.8 × 108 | 1.4 × 109 | 7.3 × 1010 |
| Recovery (eluted/applied) | 4.9 × 10−5 | 9.3 × 10−4 | 1.0 × 10−3 |
FIGURE 3.
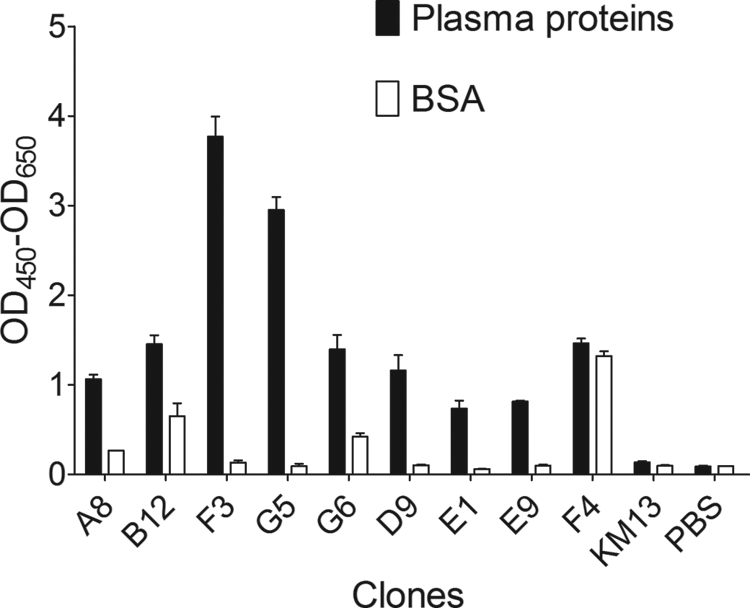
Identification of plasma proteins-specific mono-phages by ELISA. Different monoclonal phage culture supernatants were used as the first antibody against the plasma proteins and the negative control. The binding of each mono-phage was detected by anti-M13/HRP and 3,3′,5,5′-tetramethylbenzidine. KM13 helper phage was used as a negative binding control. The figure shows eight significantly positive mono-phage clones among hundreds of tests and one nonspecific clone F4.
Optimization of Conditions for Cross-linking
Various cross-linking conditions were explored, in which the optimal condition for cross-linking was neutral pH, the molar ratio of glutaraldehyde to phage pVIII subunits (3000 pVIII subunits per phage particle) should be higher than 0.27. The concentration of phage particles was less important. When the concentration decreased by 125-fold (from 4.5 × 1013 pfu/ml to 3.6 × 1011 pfu/ml), the cross-linking efficiency decreased only by 43%, from 95% to 52% (Fig. 4). For the rest of the study, a large batch of phage matrix was made by using 23.4 μm glutaraldehyde as the cross-linker, MOPS as the coupling buffer, and the phage concentration of 3 × 1013 pfu/ml at pH 7, which led to a less intensive cross-linking with molar ratio 0.27.
FIGURE 4.

Optimization of conditions for cross-linking. A broad range of conditions was considered to effect the cross-linking. A, the effect of pH on cross-linking was tested. The cross-linking was conducted in different coupling buffers: 1 m citric acid, pH adjusted to 3–5.5 with NaOH; 1 m MOPS, pH adjusted to 7–8 with NaOH; 1 m NaHCO3, pH adjusted to 8.5–10 with HCl or NaOH. The linker and phage concentration used were 23.4 μm and 3 × 1013 pfu/ml, respectively. In the figure, neutral pH showed an optimal cross-linking. B, the effect of molar ratio on cross-linking was tested. This experiment was conducted in MOPS, pH 7, and with fixed phage concentration (3 × 1013 pfu/ml). In the figure, the concentration of glutaraldehyde varied from 23.4 nm to 2.34 mm, corresponding to the molar ratio of glutaraldehyde to phage pVIII subunits from 2.7 × 10−4 to 27. The molar ratio 0.27 showed the best cross-linking. C, the effect of phage concentration on cross-linking was tested. The experiment was conducted in MOPS, pH 7, and with the linker concentration fixed (23.4 μm) while the phage concentration varied from 3.6 × 1011 to 4.5 × 1013. In the figure, the cross-linking result shows a less significant variation.
Identification of Plasma Antigens Purified by Cross-linked Phages
Six selected positive clones were amplified and made into cross-linked phage matrices to purify proteins from human plasma as affinity chromatography beads. The eluates from phage matrices were visualized for protein bands by Coomassie Blue staining on SDS-PAGE (Fig. 5). As shown in Fig. 5, two unique protein bands in the eluates of G5 and E1, respectively, were picked and cut off from the gels for the identification by MALDI-TOF/TOF (Beijing Proteome Research Center). They were finally identified to be Amyloid protein (Mr 29,767) and Apolipoprotein A-I precursor (Mr 30,758) (score > 61, p < 0.05). Additionally, they were confirmed with Western blotting using their corresponding antibodies (data not shown).
FIGURE 5.

Protein isolation from plasma proteins by cross-linked phages. Positive mono-phages were made into cross-linked matrices to isolate corresponding plasma proteins. The results were visualized by Coomassie Blue staining. A, two clones were eluted with 100 mm triethylamine. The figure shows, from left to right, the flow-through (lane 1 is F3 and lane 2 is G5), the eluate (lane 3 is F3 and lane 4 is G5), and as a negative control, the Tris-buffered saline flow-through (lane 5 is F3 and lane 6 is G5). In the G5 eluate, one unique band appeared with an approximate molecular mass of 25–30 kDa (as indicated by the arrow). B, four clones were eluted with 100 mm HCl. The figure shows the part of the eluate of the four clones (lanes 1 and 2, D9; lanes 3 and 4, E1; lanes 5 and 6, E9; and lanes 7 and 8, F4). In the E1 eluate, one unique band appeared with an approximate molecular mass of 30 kDa (as indicated by the arrow).
DISCUSSION
Our data demonstrated that monoclonal phage-displayed scFv ligands specific for human plasma proteins could be quickly screened from a phage display library using the library against proteome strategy. The selected plasma protein-specific phages could be directly used as affinity matrices by cross-linking for the isolation of corresponding proteins from the complex. This approach represents an efficient way to prepare affinity ligands for multiple antigens in a proteome, even without any actual purification of antigens.
Compared with the conventional procedures for ligand preparation such as monoclonal or polyclonal antibody and chemical synthesis/screening, phage library display is much more powerful. Therefore, many efforts are aimed at applying phage library for ligand identification (23, 24). However, strategies of panning on a complex antigen system were always under studies in contrast to ligand screening against one individual antigen. As reported here, we applied a phage-displayed scFv library for panning on a whole proteome. Our results showed the validity of the library against proteome strategy and the potential in high-throughput ligand screening. In this study, panning was carried out in solution to maintain the natural conformations of plasma proteins to the greatest extent. As a result, the clones bound to proteins with artificial conformations when antigens were immobilized on a solid matrix were avoided (16).
Although phage library-derived ligands were extensively studied (23, 25, 26), employing phage particles directly as affinity ligands was a relatively new concept. In the present work, phage-displayed scFv fragments were cross-linked to serve as affinity matrices for a favorable alternative, compared with phage-displayed peptides or wild-type phages as reported before (14, 19, 27). The matrices were strong enough to become support materials as well as appropriate ligands for affinity purification. Cross-linked phage matrix has great advantages over phage ligands immobilized on a support matrix. First, the outcome of cross-linking provided a phage density at least one order of magnitude higher than that of non-cross-linking (data not shown). Second, cross-linking would reduce nonspecific binding to target proteins, as the charged functions of phage capsid proteins were blocked by glutaraldehyde. Thirdly, cross-linked phages were much more stable than non-cross-linked phages; therefore, they had higher resistance to eluants (data not shown). Last but not the least, the use of cross-linked phages rather than commercial beads as affinity matrix reduced the cost of affinity purification evidently.
We applied glutaraldehyde rather than NHS-dextran as the cross-linker not only for the difference in their prices, but also for their molecular sizes. Although plasma samples are pre-treated to remove the abundant plasma proteins such as HSA and IgG, they usually remain in larger quantities in the samples. Their molecular masses are higher than 50 kDa. In this study, we were “fishing” ligands for plasma proteins with low abundance. To avoid the interference from the abundant proteins, we chose glutaraldehyde as the cross-linker simply to limit the inner space of the phage matrices, compared with NHS-dextran (∼139 NHS groups on each dextran chain). However, this also explained why two antigens obtained with this method had molecular masses of <30 kDa. The cross-linking reagents with different sizes should be used to obtain the ligands for all sizes of plasma proteins.
Our data confirmed another way to screen panels of phage ligands simultaneously on a proteome-wide scale, as anticipated (28–29), which made the phage-based affinity technology a valuable tool for the proteomics research. Compared with the proteomics technologies currently available such as the shotgun method, our approach could be used not only to characterize the target antigens but also to obtain the corresponding specific phage ligands both in phenotype and genotype. In the present work, two important plasma antigens, amyloid protein and apolipoprotein A-I precursor, were purified from human plasma proteome along with their specific ligands in both forms of DNA and protein (data not shown). The results implied that using our method any proteins known or unknown that were difficult to be isolated could be enriched, purified, and identified from a complicated proteome. It is expected that more ligands specific for plasma proteins will be mined when we continually improve the fishing conditions as using different sizes of cross-linkers. In fact, this approach should be also suitable for non-protein molecules. It could serve as a platform for high-throughput purification and ligand screening.
The work was supported by the Ministry of Education, China (Grants 20060284042 and 20060284045).
- ELISA
- enzyme-linked immunosorbent assay
- MALDI
- matrix-assisted laser desorption ionization time-of-flight
- TOF
- time of flight
- HSA
- human serum albumin
- PBS
- phosphate-buffered saline
- scFv
- single-chain antibody variable fragment
- pfu
- plaque- forming unit(s)
- MOPS
- 4-morpholinepropanesulfonic acid
- NHS
- N-hydroxysuccinimide.
REFERENCES
- 1.Hage D. S. (1999) Clin. Chem. 45, 593–615 [PubMed] [Google Scholar]
- 2.Labrou N., Clonis Y. D. (1994) J. Biotechnol. 36, 95–119 [DOI] [PubMed] [Google Scholar]
- 3.Adamson S. R. (1998) Dev. Biol. Stand. 93, 89–96 [PubMed] [Google Scholar]
- 4.Girot P., Moroux Y., Duteil X. P., Nguyen C., Boschetti E. (1990) J. Chromatogr. 510, 213–223 [DOI] [PubMed] [Google Scholar]
- 5.Huang P. Y., Carbonell R. G. (1999) Biotechnol. Bioeng. 63, 633–641 [PubMed] [Google Scholar]
- 6.Amatschek K., Necina R., Hahn R., Schallaun E., Schwinn H., Josic D., Jungbauer A. (2000) J. High Resol. Chromatogr. 23, 47–58 [DOI] [PubMed] [Google Scholar]
- 7.Hammond D. J. (1998) Chromatographia 47, 475–476 [Google Scholar]
- 8.Kretzschmar T., Zimmermann C., Geiser M. (1995) Anal. Biochem. 224, 413–419 [DOI] [PubMed] [Google Scholar]
- 9.Hawkins R. E., Russell S. J., Winter G. (1992) J. Mol. Biol. 226, 889–896 [DOI] [PubMed] [Google Scholar]
- 10.Hoogenboom H. R., Lutgerink J. T., Pelsers M. M., Rousch M. J., Coote J., Van Neer N., De Bruïne A., Van Nieuwenhoven F. A., Glatz J. F., Arends J. W. (1999) Eur. J. Biochem. 260, 774–784 [DOI] [PubMed] [Google Scholar]
- 11.Stausbøl-Grøn B., Wind T., Kjaer S., Kahns L., Hansen N. J., Kristensen P., Clark B. F. (1996) FEBS Lett. 391, 71–75 [DOI] [PubMed] [Google Scholar]
- 12.Bowley D. R., Jones T. M., Burton D. R., Lerner R. A. (2009) Proc. Natl. Acad. Sci. 106, 1380–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furuta M., Ito T., Eguchi C., Tanaka T., Wakabayashi-Takai E., Kaneko K. (2002) J. Biochem. 132, 245–251 [DOI] [PubMed] [Google Scholar]
- 14.Smith G. P., Petrenko V. A., Matthews L. J. (1998) J. Immunol. Methods 215, 151–161 [DOI] [PubMed] [Google Scholar]
- 15.Su Z. G., Jiang W., Feng P. S., Gao K. Y. (1991) Chin. J. Biotechnol. 7, 161–168 [PubMed] [Google Scholar]
- 16.McConnell S. J., Dinh T., Le M. H., Spinella D. G. (1999) BioTechniques 26, 208–210, 214 [DOI] [PubMed] [Google Scholar]
- 17.Kay B. K., Winter J., McCafferty J. (1996) Phage display of peptides and proteins: A Laboratory Manual, Academic Press, San Diego, CA [Google Scholar]
- 18.Goletz A., Christensen P. A., Kristensen P., Blohm D., Tomlinson I., Winter G., Karsten U. (2002) J. Mol. Biol. 315, 1087–1097 [DOI] [PubMed] [Google Scholar]
- 19.Samoylova T. I., Cox N. R., Morrison N. E., Globa L. P., Romanov V., Baker H. J., Petrenko V. A. (2004) BioTechniques 37, 254–260 [DOI] [PubMed] [Google Scholar]
- 20.Hellman U., Wernsted C., Góñez J., Heldin C. H. (1995) Anal. Biochem. 224, 451–455 [DOI] [PubMed] [Google Scholar]
- 21.Shevchenko A., Wilm M., Vorm O., Mann M. (1996) Anal. Chem. 68, 850–858 [DOI] [PubMed] [Google Scholar]
- 22.Hortin G. L. (2006) Clin. Chem. 52, 1223–1237 [DOI] [PubMed] [Google Scholar]
- 23.Kelley B. D., Booth J., Tannatt M., Wub Q. L., Ladner R., Yuc J., Potter D., Ley A. (2004) J. Chromatogr A 1038, 121–130 [DOI] [PubMed] [Google Scholar]
- 24.Breccia J., Krook M., Ohlin M., Hatti-Kaul R. (2003) Enzyme Microb. Technol. 33, 244–249 [Google Scholar]
- 25.Labrou N. E. (2003) J. Chromatogr. B 790, 67–78 [DOI] [PubMed] [Google Scholar]
- 26.Nord K., Nord O., Uhlén M., Kelley B., Ljungqvist C., Nygren P. A. (2001) Eur. J. Biochem. 268, 4269–4277 [DOI] [PubMed] [Google Scholar]
- 27.Kouzmitcheva G. A., Petrenko V. A., Smith G. P. (2001) Clin. Diagn. Lab. Immunol. 8, 150–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu B., Huang L., Sihlbom C., Burlingame A., Marks J. D. (2002) J. Mol. Biol. 315, 1063–1073 [DOI] [PubMed] [Google Scholar]
- 29.Hust M., Dübel S. (2004) Trends Biotechnol. 22, 8–14 [DOI] [PubMed] [Google Scholar]