

Abstract
We show that Mycobacterium smegmatis has an enzyme catalyzing transfer of maltose from [14C]maltose 1-phosphate to glycogen. This enzyme was purified 90-fold from crude extracts and characterized. Maltose transfer required addition of an acceptor. Liver, oyster, or mycobacterial glycogens were the best acceptors, whereas amylopectin had good activity, but amylose was a poor acceptor. Maltosaccharides inhibited the transfer of maltose from [14C]maltose-1-P to glycogen because they were also acceptors of maltose, and they caused production of larger sized radioactive maltosaccharides. When maltotetraose was the acceptor, over 90% of the 14C-labeled product was maltohexaose, and no radioactivity was in maltopentaose, demonstrating that maltose was transferred intact. Stoichiometry showed that 0.89 μmol of inorganic phosphate was produced for each micromole of maltose transferred to glycogen, and 56% of the added maltose-1-P was transferred to glycogen. This enzyme has been named α1,4-glucan:maltose-1-P maltosyltransferase (GMPMT). Transfer of maltose to glycogen was inhibited by micromolar amounts of inorganic phosphate or arsenate but was only slightly inhibited by millimolar concentrations of glucose-1-P, glucose-6-P, or inorganic pyrophosphate. GMPMT was compared with glycogen phosphorylase (GP). GMPMT catalyzed transfer of [14C]maltose-1-P, but not [14C]glucose-1-P, to glycogen, whereas GP transferred radioactivity from glucose-1-P but not maltose-1-P. GMPMT and GP were both inhibited by 1,4-dideoxy-1,4-imino-d-arabinitol, but only GP was inhibited by isofagomine. Because mycobacteria that contain trehalose synthase accumulate large amounts of glycogen when grown in high concentrations of trehalose, we propose that trehalose synthase, maltokinase, and GMPMT represent a new pathway of glycogen synthesis using trehalose as the source of glucose.
Keywords: Bacteria, Carbohydrate Biosynthesis, Enzymes, Glycogen, Metabolism, Glycogen, Regulatory Mechanism, Stress Response, Trehalose
Introduction
Trehalose (α-d-glucose(1→1)α-d-glucose) and glycogen are both important stores of glucose, which can be called upon as needed for energy, for carbon and building blocks, or for other functions (1, 2). For example, trehalose is produced in large amounts in response to various environmental stresses such as heat or desiccation (3), and it serves to protect proteins and membranes from denaturation or disruption (4). Trehalose is also an essential component of various cell wall glycolipids in mycobacteria and related organisms (5, 6), and it may also serve as a regulator of metabolism in yeast (7) and other organisms (8). In mycobacteria, there are three different biosynthetic pathways for making trehalose (9–11), and mutants that lack all three pathways cannot grow unless trehalose is present in the growth media (12, 13). One of these pathways involves the enzyme trehalose synthase (TreS),2 which interconverts trehalose and maltose (α-d-glucose(1→4)α-d-glucose) (14). We recently demonstrated that TreS also has amylase activity that can convert glycogen to maltose, suggesting that this enzyme alone provides another pathway to convert glycogen to trehalose (15).
Recently, we found that Mycobacterium smegmatis accumulated large amounts of glycogen when grown in a mineral salts medium with 2%, or higher, concentrations of trehalose. Importantly, this accumulation only occurred in wild type cells or in mutant cells that contained TreS. Cells deleted in the gene for TreS did not accumulate increased amounts of glycogen in the presence of trehalose or any other sugars (see Table 1). These experiments indicated that TreS was somehow directly involved in the conversion of trehalose to glycogen and might also function as a sensor and/or regulator of trehalose levels within the cell.
TABLE 1.
Requirement for TreS and exogenous trehalose for accumulation of glycogen
Cells were grown in mineral salts medium with various amounts of trehalose as the carbon source. Cells were harvested after 40 h of growth and ruptured by sonication, and the glycogen was isolated from the cytoplasmic fraction. Cells are as follows: B11, wild type (WT); 47, trehalose-P-phosphatase negative mutant; 74, TreY negative mutant; 80, triple mutant missing trehalose-phosphate phosphatase; TreS, TreY, and unable to synthesize trehalose; mutant 91, missing TreS.
| Amount of trehalose in medium | Amount of glycogen in wild type and mutants |
||||
|---|---|---|---|---|---|
| WT (B11) | Mutant (47) | Mutant (74) | Mutant (80) | Mutant (91) | |
| % | nmol glucose/mg dry cells | ||||
| 0.1 | 15 | 31 | 13 | 15 | 15 |
| 2 | 341 | 188 | 315 | 4 | 13 |
| 4 | 514 | 370 | 317 | 7 | 11 |
| Presence of TreS in cells | + | + | + | − | − |
To determine how trehalose could be converted to glycogen, we examined the fate of maltose that was produced from trehalose by TreS. These studies revealed the presence of a kinase in crude extracts of M. smegmatis that phosphorylates maltose in the one position to produce maltose 1-phosphate.3
This enzyme is highly specific for maltose as the phosphate acceptor and ATP as the phosphate donor. Maltokinase has previously been characterized and purified from extracts of Actinoplanes species, but in that case the enzyme was proposed to be involved in uptake and utilization of maltose as a carbon and/or energy source (17, 18). Using the partially purified mycobacterial maltokinase, [14C]maltose-1-P was synthesized, purified, and characterized.3 When this radiolabeled substrate was incubated with a crude mycobacterial extract, radioactivity was transferred into a product that was insoluble in 75% methanol and that had properties similar to those of glycogen. In the study presented here, we demonstrate that the enzyme that catalyzes the above transfer is a maltosyltransferase that we have named α1,4-glucan:maltose-1-phosphate maltosyltransferase (GMPMT). This enzyme catalyzes the transfer of maltose from maltose-1-P to the nonreducing ends of the highly branched glycogen molecule, with the release of inorganic phosphate. GMPMT has many properties that are similar to those of GP (19), such as the inhibition of the synthetic reaction by inorganic phosphate and arsenate. Interestingly, GP is inhibited by isofagomine (20) and also by 1,4-dideoxy-1,4-imino-d-arabinitol (DIA) (21), whereas GMPMT is also inhibited to the same extent as GP by DIA but is not susceptible to inhibition by isofagomine. Thus, these two enzymes appear to be mechanistically related but clearly distinct proteins.
EXPERIMENTAL PROCEDURES
Materials
Trehalose, maltose, isomaltose, malto-oligosaccharides, and other sugars were purchased from Sigma. Rabbit muscle GP, DEAE-cellulose, and various other chromatographic resins for protein purification, molecular markers for gel filtration, and buffers were also obtained from Sigma. Bio-Rad protein reagent and DE52 were from Bio-Rad. Trypticase soy broth was from BD Biosciences. [U-14C]Maltose was from American Radiolabel Co (600 mCi/mmol).
Preparation of [14C]Maltose 1-Phosphate
Radioactive maltose 1-phosphate was prepared using a partially purified maltokinase from M. smegmatis (ATCC 14468). Incubation mixtures contained 0.04 μCi of [14C]maltose, 1 μmol of ATP, 1.5 μmol of MgCl2, 20 μmol of Tris-HCl buffer, pH 8.2, and enzyme, all in a final volume of 100 μl. Usually 25–50 individual incubations, as described above, were set up and incubated at 50 °C for 20 min. Although this synthesis could be done in a single large scale incubation mixture, we found that the yield of maltose-1-P was better when a number of smaller incubations were used rather than a single large scale incubation. Multiple small incubations also allowed us to remove single incubations at various times to examine the progress of the reaction or for other analytical purposes.
At the end of the reaction time, incubations were pooled and subjected to chromatography on a column of DE52, which had been washed with water. After application of the sample, columns were washed with water to remove any remaining [14C]maltose, and the [14C]maltose-1-P was eluted with a 0–250 mm gradient of ammonium bicarbonate. The ammonium bicarbonate was removed by evaporation in the presence of triethylamine.
Preparation of [14C]Glycogen
Radioactive glycogen was prepared by incubating liver or oyster glycogen with [14C]maltose-1-P, Tris-HCl buffer, pH 7.0, and GMPMT, and the glycogen was isolated by methanol precipitation as in the typical incubations. The precipitate was isolated by centrifugation, dissolved in water, and again precipitated through several cycles of dissolving in water and precipitating with methanol, until the methanol supernatant liquid was free of radioactivity.
Bacterial Strains and Culture Conditions
M. smegmatis (ATCC 14468) was obtained from the American Type Culture Collection. It was grown as described previously in trypticase soy broth or in a mineral salts medium containing trehalose or glucose as the carbon source (14, 15). Briefly, a starter culture was prepared by inoculating a 125-ml flask of trypticase soy broth with a culture of the organism to be used. The starter culture was grown at 37 °C on a rotary shaker. After 48 h of growth, 5 ml of this culture was transferred to 2-liter flasks containing 1 liter of trypticase soy broth. These flasks were then shaken on the rotary shaker at 37 °C for various times, but usually 48 h, which corresponded to mid-logarithmic phase cells. Cells were harvested by centrifugation, washed with PBS, and stored as a paste at −20 °C until needed. A variety of mutants of the wild type were prepared previously by allele replacement mutagenesis. Some of these were single deletion mutations that were missing one of the essential enzymes in one of the three trehalose metabolic pathways as well as that specific pathway (9), double mutants missing two of the pathways, and a triple mutant missing all three trehalose biosynthetic pathways and requiring exogenous trehalose for growth (11, 15).
Partial Purification of Maltosyltransferase Activity
The enzymatic activity was partially purified using the steps outlined below. Twenty g of cell paste was suspended in 100 ml of 50 mm Tris-HCl buffer, pH 7.0, and subjected to sonic oscillation to rupture the cells. The sonicate was then centrifuged at high speed in a Beckman J-25 refrigerated centrifuge. The supernatant liquid contained most of the desired activity, although some activity (10%) was also found in the pellet. The enzymatic activity, called GMPMT, was isolated from the crude extract by ammonium sulfate fractionation, taking the protein fraction that precipitated between 35 and 70% saturation. This precipitate was isolated by centrifugation, suspended in Tris-HCl buffer, and dialyzed overnight against 4 liters of the same buffer. The enzyme fraction was removed from the dialysis tubing and applied to a DE52 column (2.5 × 7 cm), and the column was washed with 100 ml of 50 mm Tris buffer, pH 7.0. The column was then eluted batchwise with 100 ml of each of the following concentrations of NaCl: 0.1, 0.2, 0.3, 0.4, and 0.5 m in the same buffer. Most of the enzyme emerged in the 0.3 m elution. The protein in this fraction was precipitated by adding ammonium sulfate to 70% saturation, isolated by centrifugation, dissolved in a minimal volume of buffer, and dialyzed overnight as above. The dialyzed fraction was next placed on a column of Sephacryl S-100 (1.5 × 120 cm), which was then eluted with buffer. Three-ml fractions were collected, and an aliquot of every third fraction was assayed for GMPMT activity. The enzymatic activity was found in fractions 35–50, which is the area where proteins of Mr ∼60,000–70,000 molecular weight emerge. The GMPMT in this fraction was also isolated by ammonium sulfate precipitation and used in the various experiments.
Assay of GMPMT Activity
The GMPMT activity was assayed by determining the amount of radioactivity incorporated into methanol-insoluble product in the presence of added glycogen and [14C]maltose-1-P. Routine assays were performed in a final volume of 300 μl and contained the following: 25 mm Tris-HCl buffer, pH 7.0, 200–500 μg of liver or oyster glycogen, 125,000 cpm of [14C]maltose-1-P (12.5 nmol per incubation, final concentration of 4 × 10−5 m), water as necessary, various amounts of enzyme, and appropriate amounts of other components to be tested as indicated. In experiments to determine kinetic constants or the amount of inhibition or activation by phosphate, arsenate, or other compounds, the assay mixtures were similar, but the maltose-1-P concentration was increased to 5 × 10−4 m (250,000–500,000 cpm) The order of mixing did not appear to be critical, but the last two additions to the reaction mixtures were always the radioactive maltose-1-P and then enzyme. Incubations were done at 37 °C for various times, but usually 20 min was appropriate for most studies. Reactions were terminated by heating in a boiling water bath for 3 min, and after cooling, 3 volumes (900 μl) of methanol were added. The tubes were vigorously shaken on a Vortex mixer and placed in the −20 °C freezer for about 30 min to help precipitate the glycogen. The precipitate was isolated by centrifuging in a microcentrifuge, dissolved in water, and subjected to scintillation counting.
When maltosaccharides were used as acceptors, methanol was still added at the end of the incubation, and the precipitate was removed by centrifugation. However, in this case, the supernatant liquid was saved and taken to dryness. The residue was dissolved in a small volume of water and treated with mixed-bed ion-exchange resin (equal mixture of Dowex-50-H+ and Dowex-1-CO32−) to remove positively and negatively charged molecules. The deionized fraction was subjected to paper chromatography as discussed below to identify the newly formed 14C-oligosaccharides.
Kinetic Properties and Stoichiometry
The optimum maltose-1-P concentration for GMPMT was determined using a 3 mm solution of maltose-1-P that contained 1000 cpm/nmol. Various amounts of this mixture from 5 to 150 μl (15–450 nmol per incubation) were added to standard assay mixtures of 0.3 ml. After a 20-min incubation, reactions were stopped by heating. The glycogen was then isolated, and its radioactive content was determined as in previously described assays. The data from this experiment were plotted according to the method of Lineweaver and Burk (22).
To determine the stoichiometry of the reaction, four large scale reaction mixtures were prepared that contained 1 mg of oyster glycogen and 0.9 μmol of radioactive maltose-1-P (120,000 cpm) as well as 25 mm Tris-HCl buffer and 50 μg of GMPMT from the Sephacryl G-100 column, all in a final volume of 0.75 ml. Because all of the GMPMT preparations contained amylase activity that produced some maltose from the glycogen, and therefore made quantitation of maltose-1-P disappearance difficult, we also added 25 nmol of acarbose to these incubations to inhibit the amylase (15). Preliminary experiments showed that this amount of acarbose inhibited most of the amylase activity but did not inhibit GMPMT activity. One of the four incubation mixtures served as a 0 time control and was subjected to heat denaturation as soon as the enzyme was added. The other three assay mixtures were incubated for 15, 30, and 60 min. Each assay was stopped by heating, and after cooling the glycogen was precipitated by adding 3 volumes of methanol.
The glycogen was isolated by centrifugation and subjected to scintillation counting. The methanol supernatant liquids were poured into clean tubes and concentrated to dryness under a stream of air at 50 °C. One ml of water was added to each tube, and several aliquots (usually 25, 50, and 100 μl) from each time point were removed, and the amount of maltose-1-P remaining was measured by the anthrone method (23). This is a colorimetric method for hexose determination, which is done in strong H2SO4 that hydrolyzes maltose-1-P to glucose and then determines the amount of glucose. A second series of aliquots was removed for the measurement of inorganic phosphate using the arsenomolybdate colorimetric method (24). This experiment was repeated several times with very similar results, and 50 to 65% of the maltose-1-P added to the incubations was converted to glycogen as indicated by the loss of maltose-1-P as well as by the amount of radioactivity incorporated into glycogen (see data in Table 4).
TABLE 4.
Stoichiometry of GMPMT reaction
Incubations were as described in the text and contained 900 nmol of maltose-1-P (120,000 cpm). Incubations were for 15, 30, or 60 min, and the methanol supernatant was taken to dryness, suspended in water, and analyzed for its content of maltose-1-P remaining, as well as inorganic phosphate produced. The methanol precipitate was resuspended in water, and aliquots were taken for determination of radioactive content. Ratio of maltose incorporated into glycogen/inorganic phosphate produced was 1.0:0.89.
| Incubation time | Radioactivity incorporated into glycogen |
Amount of Malt-1-P used |
Amount of inorganic phosphate appearing, nanomoles produced | ||
|---|---|---|---|---|---|
| Total cpm | % total radioactivity added | Nanomoles disappearing | % of total maltose-P added | ||
| min | |||||
| 0 | 3400 | ||||
| 15 | 25,000 | 21 | 150 | 16 | 200 |
| 30 | 42,000 | 35 | 300 | 33 | 250 |
| 60 | 67,000 | 56 | 600 | 66 | 450 |
Separation and Identification of Sugars
Radioactive malto-oligosaccharides were isolated and identified by descending paper chromatography. Unknown sugars were applied to Whatman 3MM paper in a 2- or 3-inch band near the top of the paper, and a variety of standard sugars such as maltose, maltotriose, maltopentaose, and so on were applied to each side of the paper. Papers were usually run in either of the following two solvents: solvent A, ethyl acetate/pyridine/water (12:5:4, v/v), or solvent B, n-butyl alcohol, pyridine, 0.1 n HCl (5:3:2). Radioactive sugars were identified by cutting a section of the band from the top of the paper to the bottom into 1-cm strips and counting each strip in the scintillation counter. The location of the standard sugars was determined by exposing the section of the paper that contained the standard sugars to staining with silver nitrate (25).
Other Methods
Protein was measured with the Bio-Rad protein reagent using bovine serum albumin as the standard. Hexoses were quantitated by the anthrone colorimetric method (23). Inorganic phosphate was measured colorimetrically using the arsenomolybdate method (24). The molecular mass of the native GMPMT was estimated by gel filtration on Sephacryl S-100. Molecular mass standards included α-amylase (200 kDa), serum albumin (66 kDa), carbonic anhydrase (29 kDa), and cytochrome c (12.5 kDa). SDS-PAGE was performed according to Laemmli (26) in 10% polyacrylamide gel. The gels were stained with 0.5% Coomassie Blue in 10% acetic acid.
RESULTS
Evidence for a Maltose to Glycogen Transferase
As shown in Table 1, when wild type or mutant cells of M. smegmatis are grown in a mineral salts medium with 2% or higher concentrations of trehalose, these cells increase their content of glycogen at least 10–30-fold over normal amounts. Table 1 also shows that this accumulation only occurs in cells that contain the enzyme, TreS, such as wild type (B11), or mutants 47 (trehalose-phosphate phosphatase−) or 74 (trehalose-phosphate synthase−, trehalose-phosphate phosphatase−, and malto-oligosyltrehalose synthase−), indicating that both TreS and trehalose are necessary ingredients for production of large amounts of glycogen by this organism. Based on these results, experiments were initiated to determine the mechanism of this glycogen accumulation. Initial experiments showed the presence of a maltokinase that produced maltose-1-P from maltose and ATP,3 and also an enzyme that transferred the maltose from its phosphorylated derivative to high molecular weight glycans. This latter activity is further characterized in this study.
Table 2 shows the requirements for incorporation of radioactive maltose from [14C]maltose-1-P into the methanol-insoluble product. Incubations were as described under “Experimental Procedures.” As shown in Table 2, the complete incubation mixture showed the greatest incorporation of radioactivity into the methanol-insoluble material. However, when glycogen was omitted from the incubation mixture, there was very little incorporation of radioactivity into product with either enzyme preparation, even if glycogen was added just before the reactions were stopped. However, in a few cases, the crude extract did show some activity in the absence of added glycogen, but following ammonium sulfate fractionation, the requirement for glycogen was almost absolute. With the partially purified GMPMT, the incorporation of radioactivity into product by these extracts was proportional to the amount of enzyme and somewhat proportional to the amount of glycogen added. Table 2 also shows that no activity was observed with either extract when they were heated in a boiling water bath for 3 min.
TABLE 2.
Requirements for maltosyltransferase activity
| Incubations mixtures | Radioactivity in methanol-insoluble product |
|
|---|---|---|
| Crude extract (not dialyzed) | (NH4)2SO4 fraction (dialyzed) | |
| cpm | ||
| Completea | 8200 | 9600 |
| Heat-killed enzyme | 1300 | 1200 |
| Without glycogen | 1100 | 1400 |
| Plus 300 nm Pi | 6000 | 6600 |
| 3 mm Pi | 2000 | 2800 |
| Plus 300 nm arsenate | 4100 | 2900 |
| 3 mm arsenate | 1500 | 1300 |
| Plus 300 mm Glc-1-P | 6300 | 8700 |
| 3 mm Glc-1-P | 4900 | 7300 |
| Plus 3 mm ATP | 1000 | 1000 |
a Complete reaction contained 150 μl of 50 mm Tris buffer, pH 7.0, 50 μl of glycogen (10 mg/ml), 25 μl of [14C]maltose-1-P (12.5 nmol, 125,000 cpm), enzyme (25–50 μg of protein), and other components as shown. Incubations were for 20 min and were stopped by placing tubes in a boiling water bath for 3 min. Then 900 μl of methanol were added, and tubes were vortexed vigorously and placed in a −20 °C freezer for 30 min. The precipitates were isolated by centrifugation, suspended in water, and subjected to scintillation counting.
Table 2 also demonstrates that conversion of radioactive maltose into glycogen was inhibited by addition of inorganic phosphate or arsenate to the incubation mixtures but was not inhibited by inorganic pyrophosphate or other salts (see below for more details). Both inorganic phosphate and arsenate also inhibit glycogen formation from glucose-1-P by glycogen phosphorylase (19). Incorporation of maltose from [14C]maltose-1-P into glycogen was not significantly inhibited by adding unlabeled glucose-1-P but was inhibited by millimolar concentrations of ATP.
Partial Purification of GMPMT
The enzymatic activity was partially purified using the steps described under “Experimental Procedures” and outlined in Table 3. This procedure gave a purification from crude extracts of about 90-fold with a recovery of enzyme of about 10%. The enzyme that emerged from the DE52 column, after precipitation with ammonium sulfate and dialysis, was stable to storage at −20 °C for at least 3 months and was also stable to multiple rounds of freezing and thawing. The enzyme could also be kept on ice for at least 1 month with no apparent loss of activity. After gel filtration, the enzyme was somewhat more susceptible to inactivation by multiple rounds of freezing and thawing but was still stable to storage at −20 °C for at least a month.
TABLE 3.
Purification of GMPMT from M. smegmatis
| Purification steps | Activity | Specific activity | Purification |
|---|---|---|---|
| unitsa | units/mg protein | -fold | |
| Crude extract | 116,000 | 170 | 1 |
| Ammonium sulfate (35–75% fraction) | 85,000 | 290 | 2 |
| DE52 column (0.3 m NaCl) | 51,000 | 1,500 | 9 |
| Sephacryl-100 column | 15,000 | 15,200 | 89 |
a One unit of enzyme is defined as 1 nmol of maltose converted to glycogen in a 20-min incubation.
Requirement for an α-1,4-Glucan as the Acceptor
The partially purified enzyme required the addition of a high molecular weight acceptor to catalyze transfer of maltose from maltose-1-P into the methanol-insoluble product. Fig. 1A shows that the branched α1,4-,α1,6-glucans, such as mycobacterial glycogen (■) as well as oyster or liver glycogen (▴), were the most effective acceptors, whereas amylopectin (▾) was still a good acceptor but somewhat less effective. However, the linear α1,4-glucan, amylose (Fig. 1A, ♦), was a much poorer acceptor of maltose. With each of the good acceptors, the incorporation of radioactivity into insoluble product was proportional to the amount of acceptor added to about 200–300 μg of polysaccharide per incubation mixture. However, when mycobacterial glycogen and liver glycogen were compared in a more careful experiment, the glycogen isolated and purified from M. smegmatis appeared to be somewhat more effective as the acceptor (Fig. 1B).
FIGURE 1.

Effect of concentration of acceptor molecule on the incorporation of radioactivity from [14C]maltose-1-P into methanol-insoluble product. A, incubations contained 25 mm Tris-HCl buffer, pH 7.0, 125,000 cpm of maltose-1-P (12.5 nmol), various amounts of either mycobacterial glycogen (■), liver glycogen (▴), amylopectin (▾), or amylose (♦), and 25 μg of protein from the DE52 extract in a final volume of 300 μl. After an incubation of 20 min, reactions were stopped by placing tubes in a boiling water bath, and after cooling, 900 μl of methanol were added, and tubes were placed in a −20 °C freezer for 30 min. The precipitated glycogen was isolated by centrifugation, and its radioactive content was determined. B, incubations were as in A but contained various amounts of either mycobacterial glycogen (■) or liver glycogen (▴).
Kinetics and Stoichiometry
The Km value for maltose-1-P was determined as shown in Fig. 2. Incubations were as described under “Experimental Procedures.” The data were also plotted by the method of Lineweaver and Burk as shown by Fig. 2, inset, and the Km value was determined to be 2.5 × 10−4 m, although the Vmax value was calculated as 0.63 nmol/min. This experiment was done several times with maltose-1-P solutions of different specific activities, but the calculated Km value was essentially the same.
FIGURE 2.

Effect of concentration of maltose 1-phosphate on the incorporation of radioactive maltose into glycogen. Incubations contained 25 mm Tris-HCl buffer, pH 7.0, various amounts of maltose-1-P from 15 to 450 nmol (specific activity of 1000 cpm/nmol), 100 μg of oyster glycogen, and 25 μg of enzyme from DE52, all in a final volume of 0.3 ml. After an incubation of 20 min, glycogen was isolated as described in the text, and its radioactive content was determined. The data were also plotted by the method of Lineweaver and Burk as shown by the inset.
The stoichiometry of the reaction was also determined at several different times over a 1-h time course as shown in Table 4. These incubations contained 900 nmol of maltose-1-P and 120,000 cpm of radioactivity in a final volume of 0.75 ml. Table 4 presents the data from this experiment. It can be seen that the amount of radioactivity transferred to the methanol-insoluble material was 56% of that added (i.e. 67,000 cpm out of 120,000), although the amount of inorganic phosphate released was 50% of the maltose-1-P added (i.e. 450 nmol out of 900 nmol added). Thus, the ratio of maltose incorporated into glycogen to inorganic phosphate released is 1.0:0.89. The amount of maltose-1-P used up in the reaction was also determined, and at 60 min, 600 of the 900 nmol added had disappeared as determined by the anthrone method (66% of that added or a ratio of 1.17). This number is not as reliable because the extract has other activities such as amylase and probably glycosidases that can affect the anthrone reactivity. Nevertheless, the stoichiometry indicates that one Pi is released for each maltose-1-P used and each maltose incorporated into glycogen.
Inhibition of Incorporation of Maltose-1-P by Other Acceptors
The addition of maltose to the incubation mixtures inhibited the incorporation of radioactivity from maltose-1-P into the methanol-insoluble product. This prompted us to test a number of maltosaccharides, including maltotriose (Fig. 3, ■), maltopentaose (▾), and maltohexaose (▴) as inhibitors of the reaction. Fig. 3 shows that all of these maltosaccharides were almost equally effective on a molar basis in inhibiting this reaction. The methanol-soluble supernatant liquid from each of these incubations was taken to dryness, dissolved in a small volume of water, deionized with mixed-bed resin, and subjected to paper chromatography in solvent A to determine the nature of the products.
FIGURE 3.
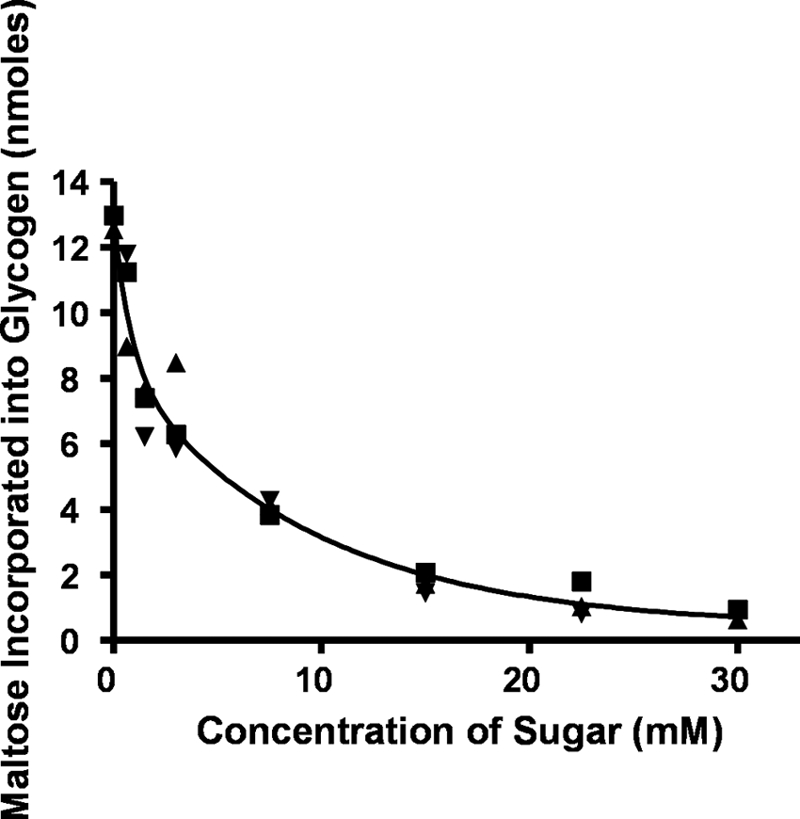
Effect of maltose and higher maltosaccharides on the transfer of radioactive maltose to the glycogen acceptor. Incubation mixtures containing buffer, oyster glycogen, [14C]maltose-1-P (125,000 cpm, 12.5 nmol), and enzyme (25 μg) were the same as described in Fig. 1 but contained various amounts of maltose (■), maltotriose (▴), maltopentaose (▾), or maltohexaose (data not shown). After an incubation of 20 min, the glycogen was isolated as described in Fig. 1 and subjected to scintillation counting.
Interestingly, a major radioactive band that migrated more slowly than the original saccharide was found in each case. For example, as shown in Fig. 4, when maltotetraose was added as the acceptor (2 mg or about 6 mm), one major radioactive band was detected on the paper chromatograms that migrated in the same area as the maltohexaose standard. The significance of this chromatogram is that no radioactivity was detected in the maltopentaose or maltotriose area of the papers strongly implying that each transfer was of a two-hexose piece (i.e. maltose) rather than of a single monosaccharide. In these incubations, the initial acceptor, such as maltotetraose, was added in excess to prevent the resulting product from also being an acceptor. Similar results were observed when maltopentaose was used as the acceptor; the major radioactive peak corresponded to maltoheptaose, and very little radioactivity was in the maltohexaose area of the paper, although when maltohexaose was the acceptor, the major peak was in the area where malto-octaose would be expected to run (data not shown). This data and the fact that glucose-1-P does not inhibit the transfer of maltose-1-P to glycogen (see below) indicates that this enzyme is a maltosyltransferase rather than a glucosyltransferase.
FIGURE 4.

Identification of maltohexaose produced using maltotetraose as the acceptor. A large scale incubation was prepared with maltotetraose (2 mg), 50 nmol of [14C]-maltose-1-P (150,000 cpm), 25 mm Tris buffer, and enzyme (50 μg) in a final volume of 0.5 ml. After an incubation of 60 min, reactions were stopped by heating, and the methanol supernatant liquid was taken to dryness. The residue was dissolved in 2 ml of water and treated with mixed-bed ion-exchange resin to remove salt and other charged molecules. The neutral fraction was subjected to paper chromatography on Whatman 3MM paper in solvent A. Standards of various maltooligosaccharides were run on the same papers to determine the size of any newly formed malto-oligosaccharides. The radioactive band was cut into 1-cm strips and subjected to scintillation counting. Standard sugars were as follows: maltohexose (HEX), maltopentose (PEN), maltotetraose (TET), and maltotriose (TRI).
Inhibition of the Enzyme by Inorganic Phosphate and Arsenate
The transfer of radioactivity from maltose-1-P to glycogen was inhibited by adding either inorganic phosphate or arsenate to the incubation mixtures. Fig. 5A shows the results of increasing amounts of inorganic phosphate, inorganic pyrophosphate, or arsenate on the production of radioactive glycogen. It can be seen that inorganic phosphate (Fig. 5A, ■) caused 50% inhibition at a concentration of about 3 × 10−4 m, whereas inorganic pyrophosphate (Fig. 5A, ▾) was much less effective and required much higher concentrations for significant inhibition. On the other hand, Fig. 5A shows that arsenate (▴) was somewhat more effective as an inhibitor than was inorganic phosphate and caused 50% inhibition at about 1 × 10−4 m.
FIGURE 5.

Inhibition of maltose incorporation into glycogen by inorganic phosphate and arsenate. A, incubation mixtures contained buffer, glycogen, maltose-1-P, and enzyme as in Figs. 1 and 2 but also contained various amounts of sodium phosphate (■), sodium arsenate (▴), or sodium pyrophosphate (▾). After an incubation of 20 min, reactions were stopped with heating, and glycogen was isolated, and its radioactive content was determined. B, the incubations were similar to those in A except that the maltose-1-P concentration was increased 10-fold to 5 × 10−4 m to be above the Km value for this substrate. Both high (□) and low (●) maltose-1-P concentrations were run with arsenate as the inhibitor.
These initial studies were done at low substrate concentrations (5 × 10−5 m) before the Km value for maltose-1-P was determined. However, they do provide a reasonable comparison of the relative effectiveness of arsenate as an inhibitor as compared with phosphate and pyrophosphate.
The effects of arsenate and phosphate were also determined at substrate concentrations above the Km (5 × 10−4 m maltose-1-P) to compare the extent of inhibition at high and low maltose-1-P concentrations. The results of this experiment with arsenate are shown in Fig. 5B. It can be seen that the shapes of the curves at high and low substrate concentration were very similar, and the arsenate concentration for 50% inhibition at higher substrate concentrations was about 2 × 10−4 m. The inhibition by inorganic phosphate was also examined at high and low maltose-1-P concentrations, and those inhibition curves also had a very similar shape and slope (data not shown). The Ki value for Pi in both cases was seen at about 4 × 10−4 m.
Based on studies that have been done with glycogen phosphorylase (19), it seemed likely that the effects of phosphate and arsenate were the results of their causing the phosphorolysis or arsenolysis of glycogen and that the GMPMT reaction was also reversible. To determine whether this were the case, experiments were set up to determine what products were produced when radioactive glycogen was incubated with partially purified GMPMT in the presence of either inorganic phosphate or arsenate. A number of incubations were prepared that contained radioactive glycogen (100,000 cpm), prepared as described under “Experimental Procedures,” Tris buffer, and GMPMT from the DE52 column. Three of these incubations contained increasing amounts of inorganic phosphate; the fourth contained arsenate; the fifth had no other additions, and the sixth contained heat-killed enzyme. After an incubation of 60 min, the glycogen in each incubation was removed by precipitation with methanol. The methanol supernatant liquids were taken to dryness, dissolved in water, and applied to columns of DE52 to bind negatively charged molecules. The columns were washed with water to remove uncharged molecules and then eluted with 0.2 n HCl to elute phosphorylated sugars.
As seen in Table 5, the incubations that contained inorganic phosphate had a considerable amount of radioactivity in the 0.2 n HCl elution, whereas the incubation without any additions had very little radioactivity either in the water wash or in the HCl elution. In fact, most of the radioactivity remained in the glycogen. On the other hand, the incubation with arsenate had a considerable amount of radioactivity in the water wash and very little that bound to the DE52 column. The water fraction from the arsenate incubation was deionized with mixed-bed resin and subjected to paper chromatography. As shown in Fig. 6, the major radioactive peak was found to be in maltose, which would be the expected product of an arsenolysis by GMPMT (based on glycogen phosphorylase). However, a second band of radioactivity was also detected that corresponded to the maltotriose standard, and this peak also contained a significant amount of radioactivity. Thus, GMPMT also appears to be able to catalyze arsenolysis of glycogen with release of malto-oligosaccharides or at least maltotriose Importantly, no radioactivity was found in glucose in any of these reverse reactions.
TABLE 5.
Formation of maltose-1-P from glycogen by mycobacterial enzyme fraction
Each incubation mixture contained 25,000 cpm of [14C]glycogen, Tris buffer, and enzyme (25 μg of protein) from the DE52 column. Various amounts of inorganic phosphate or arsenate were added, and incubations were for 30 min. The amount of radioactivity binding to DE52 (i.e. sugar-P) was determined. The DE52 eluted radioactivity was treated with alkaline phosphatase to remove covalently bound phosphate, and the neutralized sugars were subjected to paper chromatography as shown in Fig. 6.
| Additions to incubation | Radioactivity bound to DE52 |
|---|---|
| None | 460 |
| 0.5 mmol of Pi | 2100 |
| 1 mmol of Pi | 6600 |
| 5 mmol of Pi | 6500 |
| 2.5 mmol of A5O4 | 480 |
| Heat-killed enzyme + 2.5 mmol of Pi | 610 |
FIGURE 6.

Paper chromatographic identification of radioactive maltose resulting from arsenolysis of [14C]glycogen by GMPMT. GMPMT (50 μg) was incubated with 100,000 cpm of radioactive glycogen in the presence of 10 μmol of arsenate. Glycogen was precipitated with methanol, and the methanol supernatant liquid was taken to dryness, suspended in water, and applied to a column of DE52. The column was washed with water, and most of the radioactivity in the methanol supernatant fraction emerged in the water wash. This fraction was deionized with mixed-bed resin, and the radioactive sugar(s) was identified by paper chromatography as in Fig. 4. Standard sugars were as follows: maltotriose (TRI), maltose (MALT), and glucose (GLC).
The HCl elutions from the incubations with inorganic phosphate were taken to dryness and evaporated several times in the presence of methanol to remove HCl. This fraction was dissolved in glycine buffer, pH 9.0, and treated with alkaline phosphatase to remove phosphate groups from any phosphorylated sugars. After this treatment, the fractions were deionized and subjected to paper chromatography. Most of the radioactivity in these fractions migrated in the maltose area of the paper (data not shown). However, a small amount of radioactivity (10 or 15%) was also found in the glucose area but very little activity in higher maltooligosaccharides. This radioactive glucose may be the result of hydrolysis of maltose or maltose-1-P by α-glucosidases, or it may also be due to the presence of contaminating glycogen phosphorylase. Regardless, the presence of radioactive maltose from [14C]glycogen in the presence of arsenate and radioactive maltose-1-P in the presence of inorganic phosphate are strong indications of the reversibility of the GMPMT and its difference as well as similarity to glycogen phosphorylase.
Effect of Glucose 1-Phosphate or Other Phosphorylated Sugars on the Reaction
To be certain that the GMPMT extract was actually transferring maltose from maltose-1-P rather than converting maltose-1-P to glucose-1-P, and then using the glucose-1-P as the substrate for production of glycogen, nonradioactive glucose-1-P, glucose-6-P, and maltose-1-P were tested as inhibitors of the transfer of [14C]maltose from [14C]maltose-1-P to glycogen. The results of this experiment are shown in Fig. 7. It can be seen that neither glucose-1-P nor glucose-6-P were strong inhibitors of maltose transfer, whereas maltose-1-P was an effective inhibitor of this reaction. The data suggest that glucose-1-P is not a substrate nor an intermediate in the reaction. Additional evidence that GMPMT does not transfer glucose to glycogen is indicated below.
FIGURE 7.

Effect of unlabeled glucose 1-phosphate, glucose 6-phosphate, or maltose 1-phosphate on the incorporation of radioactive maltose from [14C]maltose 1-phosphate into glycogen. Standard assay mixtures were prepared as indicated in Figs. 1–3 but also had various amounts of nonradioactive glucose-1-P or glucose-6-P (■) or maltose-1-P (□) added to determine whether these sugar phosphates would inhibit incorporation of radioactivity into glycogen. This experiment was done at low maltose-1-P concentrations, but essentially the same results were obtained at the high maltose-1-P concentration.
Comparison of Substrate Specificities of GMPMT and Glycogen Phosphorylase
As further evidence that GMPMT is not a glycogen phosphorylase, but rather is a maltosyltransferase, we compared the substrate specificities of GMPMT with those of commercially available rabbit muscle glycogen phosphorylase (Sigma). In this experiment, various amounts of either GMPMT or glycogen phosphorylase were incubated with either [14C]glucose-1-P or [14C]maltose-1-P and liver glycogen for 15 or 30 min, and the amount of radioactivity in the methanol-insoluble product was measured. Fig. 8A shows that GMPMT had strong activity for transferring radioactive maltose to glycogen, and this activity increased with increasing amounts of enzyme and with time of incubation. On the other hand, the enzyme did not show any catalytic activity for the transfer of glucose into glycogen. Importantly, glycogen phosphorylase showed the opposite activity profiles with these two substrates as shown in Fig. 8B. It can be seen that the glycogen phosphorylase was very effective in transferring glucose to glycogen, and this activity was reasonably linear with respect to enzyme concentration (Fig. 8B). However, when maltose-1-P was incubated with this enzyme, there was no transfer of radioactivity into methanol-insoluble product. This experiment is additional evidence that the mycobacterial enzyme is distinct from glycogen phosphorylase, although the two do share some similarities such as inhibition of the forward reaction in the presence of inorganic phosphate and arsenate.
FIGURE 8.

Comparison of the substrate specificity of GMPMT and glycogen phosphorylase. A, 25 μg of GMPMT eluted from DE52 was incubated with [14C]maltose-1-P for 15 (□) or 30 min (○) in the standard assay mixture described in other figures. Also shown in this figure are results when GMPMT was incubated with [14C]glucose-1-P (125,000 cpm, 10 nmol) for 15 (■) or 30 min (●) in similar incubation mixtures that also contained buffer, glycogen, and GMPMT. In all cases, glycogen was isolated by methanol precipitation, and its radioactive content was determined. B, a similar set of experiments using [14C]glucose-1-P for 15 (■) or 30 min (●) and [14C]maltose-1-P for 15 (□) or 30 min (○) as substrates for glycogen biosynthesis by glycogen phosphorylase. Incubation mixtures in this case were the same as in A and contained 20 μg of protein.
Effect of Glycogen Phosphorylase Inhibitors on GMPMT
Several sugar analogues have been shown to be inhibitors of glycogen phosphorylase (24), and two of these compounds were tested as inhibitors of the mycobacterial enzyme as compared with their activity on glycogen phosphorylase. Fig. 9A compares the effect of isofagomine on GMPMT and on glycogen phosphorylase. It can be seen that isofagomine was a reasonably good inhibitor of glycogen phosphorylase but a poor inhibitor of GMPMT.
FIGURE 9.

Comparison of inhibition of GMPMT and glycogen phosphorylase by glycogen phosphorylase inhibitors, isofagomine and DIA. Incubations for measuring activity of glycogen phosphorylase were as described in Fig. 8 but contained various amounts of either isofagomine (A) or DIA (B). Incubations for measuring activity of GMPMT were as described in Fig. 8 but also contained various amounts of either isofagomine (A) or DIA (B). A, glycogen phosphorylase (■) activity and GMPMT (□) activity was measured and compared in the presence of various amounts of isofagomine. B, both glycogen phosphorylase (■) and GMPMT (□) were assayed in the presence of increasing amounts of DIA.
On the other hand as shown in Fig. 9B, DIA inhibited both enzymes almost to the same extent, i.e. the Ki value for inhibition of GMPMT was about 1 mm, and for GP it was slightly higher (2 mm). Thus, the mycobacterial maltosyltransferase does share some properties in common with glycogen phosphorylase such as the effects of Pi, arsenate, and DIA, but these two enzymes differ in terms of substrate specificity. They probably also differ in terms of the physiological direction of the reaction and its function to the cell. That point is considered in more detail below.
Characterization of the Product Produced by GMPMT
The methanol-insoluble material was isolated by centrifugation, and the pellet was resuspended and reprecipitated several times to remove any soluble radioactivity. The radioactive product was subjected to paper chromatography in solvents A and B, and the radioactivity was found to remain at the origin, even when the maltohexaose standard had moved halfway down the paper. This radioactive product was incubated at pH 5.0 with β-amylase for several hours, and the remaining glycogen was precipitated with methanol. The supernatant fraction was taken to dryness, taken up in water, and deionized with mixed-bed resin. The neutral fraction was then subjected to paper chromatography in solvent A. ∼100% of the radioactivity was found in the maltose area of the paper. The methanol-insoluble product was also susceptible to hydrolysis by α-amylase, but in this case the liberated radioactivity was found 30% in maltose as well as in higher malto-oligosaccharides (53% tri-, 15% tetra-, and 2% penta-oligosaccharides). Finally, the radioactivity was released from the insoluble product upon treatment with α-glucosidase. These properties indicate that the radioactivity is linked in α1,4-glucosidic linkages to the glycogen acceptor.
DISCUSSION
The studies described here demonstrate a new enzymatic activity that transfers maltose from maltose-1-P to glycogen. We propose that this reaction is the last, or next to the last, reaction in a pathway that allows the mycobacterial cells to convert excess trehalose to glycogen. This pathway is proposed to be as shown in Scheme 1,
 |
The supposition that trehalose can be converted to glycogen is based on previous studies from this laboratory showing that when M. smegmatis was grown in a mineral salts medium with 2% or higher amounts of trehalose as the carbon source, the cells accumulated 10–30 times as much glycogen as cells grown in trypticase soy broth or in a mineral salts medium with high concentrations of glucose or other sugars. However, glycogen accumulation did not occur in any mutant cells in which the TreS gene had been deleted, even with 4 or 5% concentrations of trehalose in the medium. Thus, in order for these cells to accumulate large amounts of glycogen, they require both a high concentration of trehalose and the presence of active TreS.
Why would trehalose levels in the cell become so high? In many organisms, trehalose can serve as a protectant of proteins and membranes during periods of stress, such as from desiccation, heat shock, freezing, and so on. In fact, yeast have been shown to accelerate the production of trehalose after they have been subjected to heat shock (27). Nematodes also produce large amounts of trehalose when they are exposed to dehydration conditions (28). Thus far, this has not been demonstrated in mycobacteria, but these organisms certainly have the capacity to synthesize large amounts of trehalose from at least one, if not all three, of the pathways that can produce trehalose (9–11). Our current hypothesis is that TreS functions as a sensor and controller of trehalose levels in these cells. As such, TreS can expedite the conversion of excess trehalose to glycogen when trehalose levels become dangerously high, because there is some evidence that large amounts of trehalose can be toxic to cells (15). Why not just degrade trehalose to glucose with a trehalase and let it go at that? From an energetic standpoint, the obvious answer is that it takes energy, such as in the form of UDP-glucose or glycogen, to make trehalose, and this energy would probably be lost if trehalose were converted to glucose, because cells cannot safely store large amounts of glucose in their cytoplasm. In fact, M. smegmatis does have a potent trehalase (29) but that enzyme does not appear to play a role in controlling trehalose levels in the cell. However, corynebacteria apparently do not have a trehalase, and in that case, investigators have speculated that the role of TreS is to convert trehalose to maltose, which can then be converted to glucose via an α-glucosidase (30). Furthermore, storing trehalose in the form of glycogen makes it readily available to the cell when it needs it, because trehalose can be readily formed from glycogen as indicated below.
In terms of TreS as a sensor or controller of trehalose levels in the cell, previous studies have shown that TreS has amylase activity as well as the maltose ↔ trehalose inter-converting activity. Thus, when trehalose levels in the cell become dangerously low, we propose that TreS can expedite the conversion of glycogen to maltose via its amylase activity and then convert the maltose to trehalose (15). It is also possible that TreS or some intermediate produced by TreS, such as maltose, could have an activating effect on the other glycogen to trehalose pathway (i.e. the malto-oligosyltrehalose synthase/malto-oligosyltrehalose trehalohydrolase pathway, see Refs. 9 or 11).
Interestingly, the GMPMT described in this study is inhibited by ATP. The concentration required to inhibit the enzyme by 50% is about 1 mm, which would appear to be in the range to be of physiological significance. This could suggest an interesting and significant role for this enzyme and for the postulated pathway in the interconversion of glycogen and trehalose. We propose that under normal conditions when the cells have adequate amounts of energy and are not stressed, they synthesize glycogen and trehalose via the known ADP-glucose and UDP-glucose pathways (9–11). In this case, ATP levels would probably exceed 5 mm (16), and the pathway described here would be inhibited. However, after a heat stress, trehalose levels would be high, and ATP levels would likely be well below the inhibitory concentration. Under these conditions, the maltosyltransferase pathway would be activated, and the excess trehalose could be converted to glycogen and stored for future use. Thus, the major role of this pathway may be to convert excess trehalose to glycogen.
We explored whether there are structurally conserved regions between GP and other M. smegmatis proteins that might provide insight into which gene codes for the maltosyltransferase. We performed BLAST searches of the following proteins from M. smegmatis against the complete M. smegmatis genome as follows: glycogen synthase, glycogen phosphorylase, amylase, maltose phosphorylase, maltokinase, trehalose phosphorylase TreS, and α-glucosidase. No obvious candidate proteins were identified. Thus, we postulate that the activity that we detected for GMPMT correlates to a protein with unique structural motifs.
What is the evidence that GMPMT works in the direction of synthesis of glycogen rather than in the direction of glycogen degradation? At this time, there is no direct evidence to prove that contention. However, maltose is not readily utilized by M. smegmatis, and crude extracts of the organism do not appear to convert maltose-1-P to anything other than glycogen. Because high trehalose levels and the presence of TreS are necessary to cause the accumulation of large amounts of glycogen, and because maltose is an intermediate in this process, it seems most likely that the pathway must be favorable in the direction of synthesis of glycogen. In fact, the kinetic studies indicate that even at low maltose-1-P concentrations, much of the maltose-1-P is converted to glycogen in these incubations. It will be interesting and important to obtain some mutants deleted in the ADP-glucose → glycogen pathway and also mutants in the pathway described here. They should be valuable tools for deciphering the significance and function of each of these pathways.
This paper is dedicated to the memory of Dr. Alan Elbein, who was a leader in the field of glycobiology and a beloved colleague.
Y. T. Pan and A. D. Elbein, manuscript in preparation.
- TreS
- trehalose synthase
- DIA
- 1,4-dideoxy-1,4-imino-dd-arabinitol
- GMPMT
- α-1,4-glucan:maltose-1-phosphate maltosyltransferase
- GP
- glycogen phosphorylase.
REFERENCES
- 1.Elbein A. D. (1974) Adv. Carbohydr. Chem. Biochem. 30, 227–256 [DOI] [PubMed] [Google Scholar]
- 2.Nwaka S., Holzer H. (1998) Prog. Nucleic Acid Res. Mol. Biol. 58, 197–237 [DOI] [PubMed] [Google Scholar]
- 3.Bell W., Klaassen P., Ohnacker M., Boller T., Herweijer M., Schoppink P., Van der Zee P., Wiemken A. (1992) Eur. J. Biochem. 209, 951–959 [DOI] [PubMed] [Google Scholar]
- 4.Crowe J. H., Hoekstra F. A., Crowe L. M. (1992) Annu. Rev. Physiol. 54, 579–599 [DOI] [PubMed] [Google Scholar]
- 5.Takayama K., Armstrong E. L. (1976) Biochemistry 15, 441–446 [DOI] [PubMed] [Google Scholar]
- 6.Brennan P. J., Nikaido H. (1995) Annu. Rev. Biochem. 64, 29–63 [DOI] [PubMed] [Google Scholar]
- 7.Blázquez M. A., Lagunas R., Gancedo C., Gancedo J. M. (1993) FEBS Lett. 329, 51–54 [DOI] [PubMed] [Google Scholar]
- 8.Vogel G., Aeschbacher R. A., Müller J., Boller T., Wiemken A. (1998) Plant J. 13, 673–683 [DOI] [PubMed] [Google Scholar]
- 9.De Smet K. A., Weston A., Brown I. N., Young D. B., Robertson B. D. (2000) Microbiology 146, 199–208 [DOI] [PubMed] [Google Scholar]
- 10.Maruta K., Hattori K., Nakada T., Kubota M., Sugimoto T., Kurimoto M. (1996) Biosci. Biotechnol. Biochem. 60, 717–720 [DOI] [PubMed] [Google Scholar]
- 11.Elbein A. D., Pan Y. T., Pastuszak I., Carroll D. (2003) Glycobiology 13, 17R–27R [DOI] [PubMed] [Google Scholar]
- 12.Murphy H. N., Stewart G. R., Mischenko V. V., Apt A. S., Harris R., McAlister M. S., Driscoll P. C., Young D. B., Robertson B. D. (2005) J. Biol. Chem. 280, 14524–14529 [DOI] [PubMed] [Google Scholar]
- 13.Tropis M., Meniche X., Wolf A., Gebhardt H., Strelkov S., Chami M., Schomburg D., Krämer R., Morbach S., Daffé M. (2005) J. Biol. Chem. 280, 26573–26585 [DOI] [PubMed] [Google Scholar]
- 14.Pan Y. T., Koroth, Edavana V., Jourdian W. J., Edmondson R., Carroll J. D., Pastuszak I., Elbein A. D. (2004) Eur. J. Biochem. 271, 4259–4269 [DOI] [PubMed] [Google Scholar]
- 15.Pan Y. T., Carroll J. D., Asano N., Pastuszak I., Edavana V. K., Elbein A. D. (2008) FEBS J. 275, 3408–3420 [DOI] [PubMed] [Google Scholar]
- 16.Veech R. L., Lawson J. W., Cornell N. W., Krebs H. A. (1979) J. Biol. Chem. 254, 6538–6547 [PubMed] [Google Scholar]
- 17.Jarling M., Cauvet T., Grundmeier M., Kuhnert K., Pape H. (2004) J. Basic Microbiol. 44, 360–373 [DOI] [PubMed] [Google Scholar]
- 18.Niehues B., Jossek R., Kramer U., Koch A., Jarling M., Schröder W., Pape H. (2003) Arch. Microbiol. 180, 233–239 [DOI] [PubMed] [Google Scholar]
- 19.Rath V. L., Newgard C. B., Sprang S. R., Goldsmith E. J., Fletterick R. J. (1987) Proteins 2, 225–235 [DOI] [PubMed] [Google Scholar]
- 20.Jakobsen P., Lundbeck J. M., Kristiansen M., Breinholt J., Demuth H., Pawlas J., Candela M. P., Andersen B., Westergaard N., Lundgren K., Asano N. (2001) Bioorg. Med. Chem. 9, 733–739 [DOI] [PubMed] [Google Scholar]
- 21.Latsis T., Andersen B., Agius L. (2002) Biochem. J. 368, 309–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christensen H. N., Palmer G. A. (1974) Enzyme Kinetics, pp. 75–100, W. B. Saunders Co., Philadelphia, PA [Google Scholar]
- 23.Loewus F. (1952) Anal. Chem. 24, 219–224 [Google Scholar]
- 24.Klutts S., Pastuszak I., Edavana V. K., Thampi P., Pan Y. T., Abraham E. C., Carroll J. D., Elbein A. D. (2003) J. Biol. Chem. 278, 2093–2100 [DOI] [PubMed] [Google Scholar]
- 25.Trevelyan E. W., Proctor D. P., Harrison J. S. (1950) Nature 166, 444–445 [DOI] [PubMed] [Google Scholar]
- 26.Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 27.Hottiger T., Boller T., Wiemken A. (1987) FEBS Lett. 220, 113–115 [DOI] [PubMed] [Google Scholar]
- 28.Madin K. A. C., Crowe J. H. (1975) J. Exp. Zool. 193, 335–342 [Google Scholar]
- 29.Carroll J. D., Pastuszak I., Edavana V. K., Pan Y. T., Elbein A. D. (2007) FEBS J. 274, 1701–1714 [DOI] [PubMed] [Google Scholar]
- 30.Wolf A., Krämer R., Morbach S. (2003) Mol. Microbiol. 49, 1119–1134 [DOI] [PubMed] [Google Scholar]