

Abstract
Alzheimer's disease (AD) is the most common form of dementia and is pathologically characterized by senile plaques, neurofibrillary tangles, synaptic disruption and loss, and progressive neuronal deficits. The exact mechanism(s) of AD pathogenesis largely remain unknown. With advances in technology diagnosis of a pre-AD stage referred to as amnestic mild cognitive impairment (MCI) has become possible. Amnestic MCI is characterized clinically by memory deficit, but normal activities of daily living and no dementia. In the present study, compared to controls, we observed in hippocampus from subjects with MCI a significantly decreased level of PSD95, a key synaptic protein, and also decreased levels of two proteins associated with PSD95, the N-methyl-D-aspartate receptor, subunit 2A (NR2A) and the low-density lipoprotein receptor-1 (LRP1). PSD95 and NR2A are involved in long-term potentiation, a key component of memory formation, and LRP1 is involved in efflux of amyloid beta-peptide (1-42). Aβ(1-42) conceivably is critical to the pathogenesis of MCI and AD, including the oxidative stress under which brain in both conditions exist. The data obtained from the current study suggest a possible involvement of these proteins in synaptic alterations, apoptosis and consequent decrements in learning and memory associated with the progression of MCI to AD.
Keywords: Alzheimer's disease, mild cognitive impairment, hippocampus, post synaptic density protein 95 (PSD95), low density lipoprotein receptor (LRP1), N-methyl D-aspartate receptor-2A (NR2A)
Introduction
Alzheimer's disease (AD) is the most common form of dementia in the elderly. Pathologically AD is characterized by senile plaques (SP), neurofibrillary tangles (NFT), synapse loss, and progressive neuronal deficits (Hardy and Selkoe 2002; Wenk 2006). In addition to the formation of SP and NFT, gliosis, chronic inflammatory reactions, excitotoxic damage and oxidative stress all appear to contribute to the progression of AD (Aisen 2008; Butterfield et al. 2001; Butterfield et al. 2006a; Butterfield and Stadtman 1997; Dalle-Donne et al. 2006; Jacob et al. 2007; Lovell et al. 2001). There is an abundance of evidence suggesting that oxidative stress and N-methyl-D-aspartate (NMDA) receptor-associated Ca2+-mediated excitotoxicity are involved in the pathogenesis of AD; however, the exact mechanisms operative in these effects largely remain unknown. Amnestic mild cognitive impairment (MCI) is a pre-AD stage, the study of which conceivably can provide insights into progression of MCI to AD.
MCI individuals are broadly grouped into 2 subtypes referred to as: amnestic (memory-affecting) MCI or non-amnestic MCI (no change in memory) (Petersen 2004; Portet et al. 2005). Pathologically, MCI brain shows mild degradation of the hippocampus, sulci, and gyri using magnetic resonance imaging technology (Fleisher et al. 2008; Jack et al. 1999). The rate of amnestic MCI conversion to AD is roughly 10-15% per year; however, in some cases MCI individuals can revert to normal (Petersen 2000).
Brain from subjects with MCI, like AD, contains pathological hallmarks of AD, including SP and NFT and also like AD, has elevated indices of oxidative stress (Butterfield et al. 2007a; Butterfield et al. 2006c; Butterfield et al. 2007b; Keller et al. 2005; Small et al. 2006). The main component of SP is amyloid beta-peptide (1-42) [Aβ (1-42)]. Under both in vitro and in vivo conditions Aβ (1-42) is known to cause increased oxidative stress (Boyd-Kimball et al. 2005; Opii et al. 2008; Yatin et al. 1999). This observation is consistent with the notion that Aβ (1-42) plays a causal role in the development and progression of AD (Selkoe 2000). Further, a number of studies suggest that the small oligomers of Aβ are the actual toxic species of this peptide (Aksenov et al. 2001; Drake et al. 2003; Lambert et al. 2001; Murphy et al. 2007). Many MCI patients showed low levels of Aβ in cerebrospinal fluid, in contrast to increased levels of Aβ deposits in the brain (Andreasen and Blennow 2005; Andreasen et al. 1999), suggesting a diminished clearance mechanism of Aβ from brain. Current research identified a number of proteins that could be potentially involved in the efflux of Aβ from brain, including low-density lipoprotein-related receptor-1 (LRP1) (Ito et al. 2007; Jaeger and Pietrzik 2008), a protein that is investigated in this current study.
Recent reports have shown that protein synthesis in MCI brain may also be decreased in response to oxidative damage to mRNA, which could lead to decreased levels of important proteins that could be involved in the progression of disease (Cenini et al. 2007; Ding et al. 2005; Sultana et al. 2008). Hence, in hippocampus obtained from MCI and control subjects at short postmortem interval, we tested the hypothesis that levels of PSD95, a key synaptic protein, and two additional proteins associated with PSD95, NR2A and LRP1 had decreased levels. In addition, we tested the hypothesis that hippocampal levels of Bcl2 and Caspase-3 were elevated. PSD95 and NR2A are involved in learning and memory via Ca2+ influx into neurons and as noted above LRP1 is involved with clearance from brain of Aβ (1-42). The data obtained from the current study suggest a possible involvement of these proteins in this prodromal stage of AD.
Materials and Methods
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) with exceptions of nitrocellulose membranes (Bio-Rad, Hercules, CA), electrophoretic transfer system (Trans-blot Semi-dry Transfer Cell; Bio-Rad).
Control and MCI brains
The Rapid Autopsy Program of the University of Kentucky Alzheimer's Disease clinical (UK ADC) provided frozen hippocampus samples from six each of MCI and age-matched controls for the present study. All subjects came from the longitudinally followed normal control group that had annual neuropsychological testing and neurological and physical examinations every 2 years. Control subjects had no cognitive complaints, normal cognitive test scores, especially objective memory test scores, and normal neurological examinations. Patients with amnestic MCI met the criteria described by Petersen (Petersen, 2004), which include the following: a memory complaint corroborated by an informant; objective memory test impairment (age and education adjusted); general normal global intellectual function; Clinical Dementia Rating score of 0.0–0.5 (no dementia); and a clinical evaluation that revealed no other cause for memory decline. The post mortem intervals of the samples used in the present study were extremely short, i.e., 2.9 ± 0.5 h for controls and 3.1 ± 0.4 h for MCI (Table I).
Sample preparation
The brain tissues (hippocampus) from control or MCI subjects were homogenized in a lysis buffer (10mM HEPES, 137mM NaCl, 4.6 mM KCl, 1.1mM KH2PO4, 0.6 mM MgSO4) containing the protease inhibitors [leupeptin (0.5 mg/mL), pepstatin (0.7mg/mL), trypsin inhibitor (0.5 mg/mL), and PMSF (40 mg/mL)]. Homogenates were centrifuged at 15,800×g for 10 min to remove debris. The supernatant was used to determine the total protein concentration by the BCA method (Pierce, Rockford, IL).
Western blot analysis
For Western blot analysis 50 μg of hippocampus brain sample from controls and MCI were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by transfer of the proteins to nitrocellulose membranes at 160 mA/gel for 2 h. The nitrocellulose membranes were blocked for 1 h at room temperature in fresh blocking buffer (10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.05% Tween 20, pH 7.4, containing 5% BSA). This procedure was then followed by incubation of the membrane with primary antibodies against anti-PSD95 [1:1000] (Cell signaling, Danvers, MA), anti-LRP1 [1:1000] (Santa Cruz Biotech, CA), anti-NR2A [1:1000] (Sigma-Aldrich, St. Louis, MO), anti-BCl2 [1:1000] (Calbiochem, NJ), or anti-caspase-3 [1:1000] Calbiochem, NJ) prepared fresh in wash blot (10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.05% Tween 20, pH 7.4) with 3% BSA. Following three washes with wash blot, the blots were incubated with secondary antibody conjugated with horse radish peroxidase in wash blot for 1 h at room temperature. The blots were washed again three times in wash blot, and the bands were visualized by enhanced chemiluminescence (ECL; Amersham, Life Sciences Inc.).
Image analysis
After immunodetection of proteins of interest the membranes were completely dried at room temperature and were then scanned using a Microtek Scanmaker 4900 scanner. Images were saved as tiff files and the intensity of the proteins were quantified using scion image analysis software.
Statistical analysis
The results are presented as means ± SD. Statistical evaluation was performed using a Student's t-test. Differences were considered to be significant at p < 0.05.
Results
Loss of synapses is one of the key pathological hallmarks of AD. In the current study, we measured the protein levels of some key synaptic proteins in the hippocampal region from subjects with MCI and from age-matched controls. Western blot analyses of hippocampal samples probed with anti-PSD95 showed a significant (p<0.05) decrease in the levels of this protein in MCI hippocampus compared to the age-matched controls. The PSD 95 blot was stripped with stripping buffer (Sigma-Aldrich, St. Louis, MO) and probed with anti-actin antibody (1:1000) as a loading control (Figure 1C). No significant difference in actin levels was observed between the control and MCI hippocampus. Further, we also measured the levels of post synaptic associated proteins, i.e., NR2A and LRP-1, using anti-NR2A or anti-LRP-1 antibodies, respectively, and both these proteins showed significant decreased levels (p<0.05) in MCI brain compared to control brain (Fig 2 and Fig 3, respectively). Proteins sometimes used as makers of the cell death, such as BCl2 and caspase-3, showed a significantly increased level (p<0.05) in MCI hippocampus compared to age-matched control (Fig 4 and 5, respectively). Table II shows the percent control of PSD95, NR2A, LRP-1, BCl2 and caspase-3 protein levels in MCI hippocampus relative to normal control values.
Figure 1.
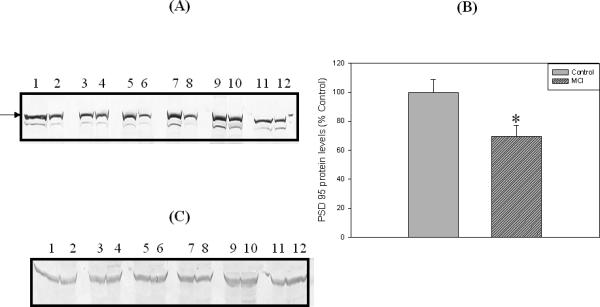
‘A’ is a representative Western blot of hippocampal PSD95 protein levels in MCI and age-matched controls. ‘B’ is the bar graph of the PSD95 Western blot results. An arrow indicate the position of PSD 95 on the blot. Control and MCI hippocampal samples were loaded in adjacent lanes. Lanes 1, 3, 5, 7, 9, 11 represent control samples, and lanes 2, 4, 6, 8, 10, 12 represent MCI samples. See text for methods and further details. Data are shown as Mean ±S.D. n=6 , * p<0.05. “C” shows actin levels in Control and MCI samples, and analysis shows no significant difference in actin in MCI hippocampus compared to control hippocampus. See text.
Figure 2.
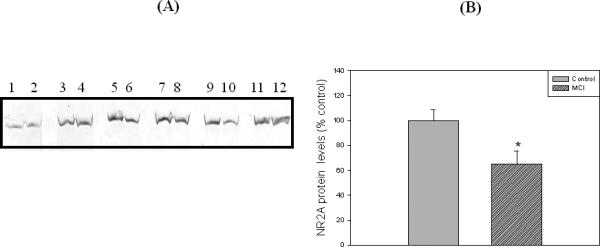
‘A’ is a representative Western blot of hippocampal NR2A protein levels in MCI and age-matched controls. ‘B’ is the bar graph of the NR2A Western blot results. Control and MCI hippocampal samples were loaded in adjacent lanes. Lanes 1, 3, 5, 7, 9, 11 represent control samples, and lanes 2, 4, 6, 8, 10, 12 represent MCI samples. See text for details. Data are shown as Mean ±S.D. n=6 , * p<0.05.
Figure 3.
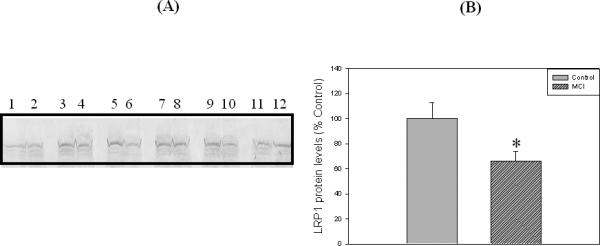
‘A’ is a representative Western blot of hippocampal LRP1 protein levels in MCI and age-matched controls. ‘B’ represents the histogram of the LRP1 Western blot results. Control and MCI hippocampal samples were loaded in adjacent lanes. Lanes 1, 3, 5, 7, 9, 11 represent control samples, and lanes 2, 4, 6, 8, 10, 12 represents MCI samples. See text for details. Data are shown as Mean ±S.D. n=6 , * p<0.05.
Figure 4.
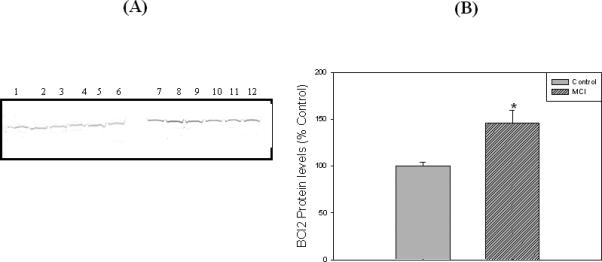
‘A’ represents Western blot of hippocampal BCl2 protein levels in MCI and age-matched controls. ‘B’ represents histogram of BCl2 blot. Lanes 1-6 represent control hippocampus, and lanes 7-12 represent MCI hippocampus. See text for details. Data are shown as Mean ±S.D. n=6 , * p<0.05.
Figure 5.
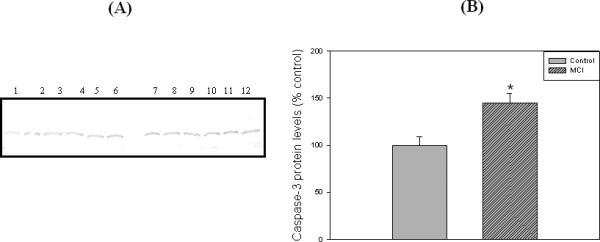
‘A’ represents Western blot of hippocampal caspase-3 protein levels in MCI and age-matched controls. ‘B’ represents the bar graph of the caspase-3 blot. Lanes 1-6 represent control hippocampus, and lanes 7-12 represent MCI hippocampus. See text for details. Data are shown as Mean ±S.D. n=6 , * p<0.05.
Discussion
In the present study we measured the protein levels of PSD95, LRP-1, NR2A, caspase-3, and Bcl2 in MCI and control hippocampus mainly for two reasons: first, hippocampus is one of the regions of the brain that is involved in learning and memory and is known to be significantly involved in AD and amnestic MCI; second, these proteins are directly or indirectly involved in learning and memory processes, which are severely affected in amnestic MCI and AD. Synaptic communications are important in the process of learning and memory; however, in AD there is clear evidence of loss of synaptic connections in the neocortex and hippocampus, and this loss strongly correlated with the cognitive decline observed in AD patients (Masliah et al. 1994; Scheff and Price 2003). PSD95, NR2A, and LRP1 proteins are localized at synapses, and the maintenance of the levels of these proteins is important for structure and function of brain cells (Martin et al. 2008; Niethammer et al. 1996). PSD95 is a neuronal scaffolding protein that contains several domains, including three PDZ/DHR (PSD-95, Dlg, ZO-1/Dlg-homologous region) domains, an SH3 (Src-homology-3) domain and a guanylate kinase (GK)-homology domain. PSD95 domains participate in protein–protein interactions; for example, among other proteins, these PSD95 domains are reported to bind to the C-terminus of NR2 subunits (Niethammer et al. 1996) and LRP1 protein (Martin et al. 2008). Hence, PSD95 is important in regulating the functions of other proteins. In the present study, we observed a significant decrease in the level of PSD95 in MCI hippocampus compared to that of control. These results are consistent with the two previous reports of decreased levels of PSD95 in AD brain (Gylys et al. 2004; Love et al. 2006). In contrast, recently another group reported an increased level of PSD95 in AD (Leuba et al. 2008).
In vivo studies in mice reported decreased PSD95 levels could lead to learning and memory impairments, in addition to locomotive impairment (Migaud et al. 1998; Nyffeler et al. 2007). A previous study showed that mice with a mutated form of PSD95, lacking the last PDZ domain, had impaired learning and LTP (Mattar et al. 2005; Migaud et al. 1998).
Learning and memory impairment reported previously can be explained based on the alterations in synaptic signaling possibly through NMDA receptors or through interaction with dopamine receptors or perhaps by altering the interplay of glutamatergic and dopaminergic axons on postsynaptic dendritic spines. NMDA receptors and PSD-95 co-cluster in cotransfected cells (Kim et al. 1996). Furthermore, PSD95 and NMDA receptors were reported to be co-localized in primary neurons (Kornau et al. 1995). The molecules involved in regulation of the receptor function appear to be recruited to the proximity of the receptor at synapses by PSD95 (Shiraishi et al. 2003).
In the present study, we also found that the levels of NR2A are decreased in amnestic MCI hippocampus compared to that of control. As mentioned previously, PSD95 interacts with the NR2A subunit of the NMDA receptor, a heteromeric complex that is composed of the NR1 subunit combined with various types of NR2 subunits, e.g., NR2A, NR2B, NR2C, and NR2D. Consistent with the interaction of PSD95 with NR2A, an in vitro study in fibroblasts showed that PSD-95 may be required for the localization of NMDA receptors to synapses (Buller and Monaghan 1997; Hoe et al. 2006).
The decreased levels of NR2A protein together with decreased levels of PSD95 could possibly make the NMDA receptor less functional with consequent reduced LTP associated with the NMDA receptors . Previous studies showed conflicting results regarding the levels of these various NMDA receptors in AD brain, and the binding sites for NMDA receptors have been shown to be either reduced or stable (Mishizen-Eberz et al. 2004; Scheuer et al. 1996; Sze et al. 2001). The mRNA levels are decreased in the AD brain (Hynd et al. 2003). One study showed reduction of NR1/NR2B receptor expression levels with the progression of AD (Mishizen-Eberz et al. 2004). An in vivo study showed reduced number of cell surface NMDA receptors in mice carrying the familial Swedish APP mutation, suggesting the involvement of Aβ (Snyder et al. 2005). Based on the above-mentioned reasons, we are tempted to speculate that the decreased levels of NR2A found in brain of subjects with amnestic MCI in the current study could be due to reduced levels of PSD95. This suggestion could result in keeping NR2A at the cell surface, which could lead to activation of the NMDA channel, with consequent elevated intracellular Ca2+ and increased oxidative stress and excitotoxicity. Other explanations for lower levels of NR2A in MCI brain could include truncation of the C-terminus of NR2A with consequent lack of recognition of this subunit by the anti-NR2A antibody (Sultana and Babu 2003). Some findings demonstrate reduce synaptic plasticity and glutamatergic transmission is associated with the non-toxic amounts of secreted Aβ peptide. However, the exact role of NMDA receptor activation in AD pathogenesis is still not fully elucidated.
Low-density lipoprotein receptor-related protein 1 (LRP1) is a large, endocytic receptor that mediates the cellular uptake of a great variety of ligands,among which are ApoE-containing lipoproteins, lipoprotein lipase, complexes of proteinases-proteinase inhibitors, and hormones and growth factors such as insulin and platelet-derived growth factor (PDGF) (Croy et al. 2004; Martin et al. 2008; May et al. 2005). LRP1 is also involved in phagocytosis and signaling pathways in addition to many other functions that are still to be discovered (Zilberberg et al. 2004). LRP1 is present both on glial and neuronal cells, and an in vitro study suggests involvement of LRP1 in different aspects of neuronal metabolism (May et al. 2004). Further, LRP1 has been shown to interact with amyloid precursor protein (APP) and also is involved in the regulation of proteolytic processing and the production of the Aβ peptide (Goto and Tanzi 2002; Shi et al. 2006). In addition, LRP1 has been reported to be as one of the mechanism(s) of Aβ clearance from brain, and down-regulation of LRP1 has been reported in human AD brain and endothelial cells (Pritchard et al. 2005).
The down regulation of LRP1 in brain of subjects with amnestic MCI reported here could lead to accumulation of Aβ in the brain resulting in increased SP formation and eventually to the progression to AD pathology. Further, the increased levels of Aβ could also increase the degradation of LRP1 by enhancing the proteasome degradation of LRP1. Since LRP1 is involved in APP processing and the clearance of Aβ, decreased levels of LRP1, as observed in MCI brain in the current study compared to the control, suggest that LRP1 may be involved in the development of SP in AD and other secondary consequences (Aisen 2008; Butterfield et al. 2006a; Hardy and Selkoe 2002; Lovell et al. 2001; Wenk 2006). LRP1 and the NMDA receptor have been assumed from immunohistochemical and co-immunoprecipitation experiments to interact at synapses (Martin et al. 2008). Moreover, LRP1 has been found to regulate calcium influx into neurons after stimulation with the glutamate receptor agonist NMDA (Martin et al. 2008). Since LRP1 has been shown to interact with the postsynaptic density protein PSD-95 and NMDA receptor either directly or indirectly, alteration in the levels of all these three proteins may be able to modulate the conductance of Ca2+ through NMDA channel. Hence, decreased levels of these proteins observed in the present study could have consequent detrimental physiological and pathological effects in MCI and could be related to synapse and memory loss, Aβ (1-42) accumulation, and neurodegeneration.
Brains from subjects with MCI show morphological changes that are indicative of cell loss; however, to the present the exact mechanism(s) of cell loss in not clear, but a role of oxidative stress and altered levels of calcium homeostasis have been proposed (Mattson et al. 1993; Mattson and Chan 2001). One of the proteins that is involved in calcium homeostasis is Bcl2, a member of the Bcl2 family that plays an important role as an anti-apoptotic and cell survival protein. Alterations in the levels of Bcl2 together with NMDA receptors could lead to altered calcium homeostasis and consequently to activation of apoptotic proteins like caspases-3. Previous studies showed an upregulation of Bcl2 protein levels in the hippocampus and entorhinal cortex of AD brains (Satou et al. 1995; Su et al. 1996). Consistent with and confirming our previous finding of the increased levels of Bcl2 in the inferior parietal lobule of subjects with amnestic MCI (Bader Lange et al. 2008) , we observed an increased level of Bcl2 in hippocampus of amnestic MCI subjects. Further, we also observed the upregulation of caspase-3 in MCI subjects, suggesting that apoptosis might be initiated in MCI hippocampus. The upregulation of the Bcl2 conceivably can be considered as a cellular compensatory mechanism to protect the cells loss.
In conclusion, the hippocampus is one of the areas of the brain that was reported previously to show pronounced neuronal and synaptic loss in AD. Our current findings, suggesting decreased levels of PSD95, NR2A, and LRP-1, with elevated levels of caspase-3 and Bcl2 proteins, may reflect or contribute to neuronal and synaptic loss in the MCI hippocampus. Since PSD95 has been reported as an intracellular linker between LRP1 and the NMDA receptor and all of these proteins are localized at synapses, their decreased levels may alter neuronal functions, consequently leading to the synaptic loss and neurodegeneration. Further molecular dissection of these proteins may be helpful in unraveling the mechanism of conversion of MCI to AD, and may help in finding a therapeutic approach to delay or prevent this devastating disease.
Acknowledgements
: The authors thank the UK ADC for providing the brain specimens used for this study. This research was supported in part by NIH grants to D.A.B [AG-029839; AG-05119, AG-10836] and to W.A.B. [AG-029839].
Footnotes
Conflict of interest: There is no conflict of interest for any author concerning this manuscript.
References
- Aisen PS. The inflammatory hypothesis of Alzheimer disease: dead or alive? Alzheimer Dis Assoc Disord. 2008;22(1):4–5. doi: 10.1097/WAD.0b013e318166ca4c. [DOI] [PubMed] [Google Scholar]
- Aksenov MY, Aksenova MV, Butterfield DA, Geddes JW, Markesbery WR. Protein oxidation in the brain in Alzheimer's disease. Neuroscience. 2001;103(2):373–383. doi: 10.1016/s0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- Andreasen N, Blennow K. CSF biomarkers for mild cognitive impairment and early Alzheimer's disease. Clin Neurol Neurosurg. 2005;107(3):165–173. doi: 10.1016/j.clineuro.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Andreasen N, Minthon L, Vanmechelen E, Vanderstichele H, Davidsson P, Winblad B, Blennow K. Cerebrospinal fluid tau and Abeta42 as predictors of development of Alzheimer's disease in patients with mild cognitive impairment. Neurosci Lett. 1999;273(1):5–8. doi: 10.1016/s0304-3940(99)00617-5. [DOI] [PubMed] [Google Scholar]
- Bader Lange ML, Cenini G, Piroddi M, Abdul HM, Sultana R, Galli F, Memo M, Butterfield DA. Loss of phospholipid asymmetry and elevated brain apoptotic protein levels in subjects with amnestic mild cognitive impairment and Alzheimer disease. Neurobiology of disease. 2008;29(3):456–464. doi: 10.1016/j.nbd.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd-Kimball D, Sultana R, Poon HF, Lynn BC, Casamenti F, Pepeu G, Klein JB, Butterfield DA. Proteomic identification of proteins specifically oxidized by intracerebral injection of amyloid beta-peptide (1-42) into rat brain: implications for Alzheimer's disease. Neuroscience. 2005;132(2):313–324. doi: 10.1016/j.neuroscience.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Buller AL, Monaghan DT. Pharmacological heterogeneity of NMDA receptors: characterization of NR1a/NR2D heteromers expressed in Xenopus oocytes. Eur J Pharmacol. 1997;320(1):87–94. doi: 10.1016/s0014-2999(96)00880-1. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7(12):548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Perluigi M, Sultana R. Oxidative stress in Alzheimer's disease brain: new insights from redox proteomics. Eur J Pharmacol. 2006a;545(1):39–50. doi: 10.1016/j.ejphar.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiol Dis. 2006b;22(2):223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Reed T, Newman SF, Sultana R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment. Free Radic Biol Med. 2007a;43(5):658–677. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Reed T, Perluigi M, De Marco C, Coccia R, Cini C, Sultana R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci Lett. 2006c;397(3):170–173. doi: 10.1016/j.neulet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Reed TT, Perluigi M, De Marco C, Coccia R, Keller JN, Markesbery WR, Sultana R. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: implications for the role of nitration in the progression of Alzheimer's disease. Brain Res. 2007b;1148:243–248. doi: 10.1016/j.brainres.2007.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Stadtman ER. Protein Oxidation processes in aging brain. Adv Cell Aging Gerontol. 1997;2:161–191. [Google Scholar]
- Cenini G, Sultana R, Memo M, Butterfield DA. Elevated Levels of Pro-apoptotic p53 and Its Oxidative Modification by the Lipid Peroxidation Product, HNE, in Brain from Subjects with Amnestic Mild Cognitive Impairment and Alzheimer's Disease. J Cell Mol Med. 2007 doi: 10.1111/j.1582-4934.2008.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croy JE, Brandon T, Komives EA. Two apolipoprotein E mimetic peptides, ApoE(130-149) and ApoE(141-155)2, bind to LRP1. Biochemistry. 2004;43(23):7328–7335. doi: 10.1021/bi036208p. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Scaloni A, Butterfield DA. Redox Proteomics. Wiley; New York: 2006. [Google Scholar]
- Ding Q, Markesbery WR, Chen Q, Li F, Keller JN. Ribosome dysfunction is an early event in Alzheimer's disease. J Neurosci. 2005;25(40):9171–9175. doi: 10.1523/JNEUROSCI.3040-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake J, Link CD, Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer's disease amyloid beta-peptide (1-42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging. 2003;24(3):415–420. doi: 10.1016/s0197-4580(02)00225-7. [DOI] [PubMed] [Google Scholar]
- Fleisher AS, Sun S, Taylor C, Ward CP, Gamst AC, Petersen RC, Jack CR, Jr., Aisen PS, Thal LJ. Volumetric MRI vs clinical predictors of Alzheimer disease in mild cognitive impairment. Neurology. 2008;70(3):191–199. doi: 10.1212/01.wnl.0000287091.57376.65. [DOI] [PubMed] [Google Scholar]
- Goto JJ, Tanzi RE. The role of the low-density lipoprotein receptor-related protein (LRP1) in Alzheimer's A beta generation: development of a cell-based model system. J Mol Neurosci. 2002;19(1-2):37–41. doi: 10.1007/s12031-002-0008-4. [DOI] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer's disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165(5):1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Pocivavsek A, Chakraborty G, Fu Z, Vicini S, Ehlers MD, Rebeck GW. Apolipoprotein E receptor 2 interactions with the N-methyl-D-aspartate receptor. J Biol Chem. 2006;281(6):3425–3431. doi: 10.1074/jbc.M509380200. [DOI] [PubMed] [Google Scholar]
- Hynd MR, Scott HL, Dodd PR. Quantitation of NMDA receptor NR2 mRNA transcripts in human brain by competitive RT-PCR. Brain Res Brain Res Protoc. 2003;11(1):67–79. doi: 10.1016/s1385-299x(03)00017-5. [DOI] [PubMed] [Google Scholar]
- Ito S, Ohtsuki S, Kamiie J, Nezu Y, Terasaki T. Cerebral clearance of human amyloid-beta peptide (1-40) across the blood-brain barrier is reduced by self-aggregation and formation of low-density lipoprotein receptor-related protein-1 ligand complexes. J Neurochem. 2007;103(6):2482–2490. doi: 10.1111/j.1471-4159.2007.04938.x. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr., Petersen RC, Xu YC, O'Brien PC, Smith GE, Ivnik RJ, Boeve BF, Waring SC, Tangalos EG, Kokmen E. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology. 1999;52(7):1397–1403. doi: 10.1212/wnl.52.7.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob CP, Koutsilieri E, Bartl J, Neuen-Jacob E, Arzberger T, Zander N, Ravid R, Roggendorf W, Riederer P, Grunblatt E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer's disease. J Alzheimers Dis. 2007;11(1):97–116. doi: 10.3233/jad-2007-11113. [DOI] [PubMed] [Google Scholar]
- Jaeger S, Pietrzik CU. Functional role of lipoprotein receptors in Alzheimer's disease. Curr Alzheimer Res. 2008;5(1):15–25. doi: 10.2174/156720508783884675. [DOI] [PubMed] [Google Scholar]
- Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64(7):1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- Kim E, Cho KO, Rothschild A, Sheng M. Heteromultimerization and NMDA receptor-clustering activity of Chapsyn-110, a member of the PSD-95 family of proteins. Neuron. 1996;17(1):103–113. doi: 10.1016/s0896-6273(00)80284-6. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269(5231):1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Mann DM, Harris JM, Chartier-Harlin MC, Cumming A, Coates J, Lemmon H, StClair D, Iwatsubo T, Lendon C. The -48 C/T polymorphism in the presenilin 1 promoter is associated with an increased risk of developing Alzheimer's disease and an increased Abeta load in brain. J Med Genet. 2001;38(6):353–355. doi: 10.1136/jmg.38.6.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuba G, Walzer C, Vernay A, Carnal B, Kraftsik R, Piotton F, Marin P, Bouras C, Savioz A. Postsynaptic density protein PSD-95 expression in Alzheimer's disease and okadaic acid induced neuritic retraction. Neurobiol Dis. 2008;30(3):408–419. doi: 10.1016/j.nbd.2008.02.012. [DOI] [PubMed] [Google Scholar]
- Love S, Siew LK, Dawbarn D, Wilcock GK, Ben-Shlomo Y, Allen SJ. Premorbid effects of APOE on synaptic proteins in human temporal neocortex. Neurobiol Aging. 2006;27(6):797–803. doi: 10.1016/j.neurobiolaging.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Xie C, Markesbery WR. Acrolein is increased in Alzheimer's disease brain and is toxic to primary hippocampal cultures. Neurobiol Aging. 2001;22(2):187–194. doi: 10.1016/s0197-4580(00)00235-9. [DOI] [PubMed] [Google Scholar]
- Martin AM, Kuhlmann C, Trossbach S, Jaeger S, Waldron E, Roebroek A, Luhmann HJ, Laatsch A, Weggen S, Lessmann V, Pietrzik CU. The functional role of the second NPXY motif of the LRP1 beta-chain in tissue-type plasminogen activator-mediated activation of N-methyl-D-aspartate receptors. J Biol Chem. 2008;283(18):12004–12013. doi: 10.1074/jbc.M707607200. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R. Synaptic and neuritic alterations during the progression of Alzheimer's disease. Neurosci Lett. 1994;174(1):67–72. doi: 10.1016/0304-3940(94)90121-x. [DOI] [PubMed] [Google Scholar]
- Mattar PA, Holmes KD, Dekaban GA. The NR1-4 C-terminus interferes with N-methyl-D-aspartate receptor-mediated excitotoxicity: evidence against a typical T/SXV-PDZ interaction. Neuroscience. 2005;132(2):281–298. doi: 10.1016/j.neuroscience.2004.11.049. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Barger SW, Cheng B, Lieberburg I, Smith-Swintosky VL, Rydel RE. beta-Amyloid precursor protein metabolites and loss of neuronal Ca2+ homeostasis in Alzheimer's disease. Trends Neurosci. 1993;16(10):409–414. doi: 10.1016/0166-2236(93)90009-b. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Chan SL. Dysregulation of cellular calcium homeostasis in Alzheimer's disease: bad genes and bad habits. J Mol Neurosci. 2001;17(2):205–224. doi: 10.1385/JMN:17:2:205. [DOI] [PubMed] [Google Scholar]
- May P, Herz J, Bock HH. Molecular mechanisms of lipoprotein receptor signalling. Cell Mol Life Sci. 2005;62(19-20):2325–2338. doi: 10.1007/s00018-005-5231-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May P, Rohlmann A, Bock HH, Zurhove K, Marth JD, Schomburg ED, Noebels JL, Beffert U, Sweatt JD, Weeber EJ, Herz J. Neuronal LRP1 functionally associates with postsynaptic proteins and is required for normal motor function in mice. Mol Cell Biol. 2004;24(20):8872–8883. doi: 10.1128/MCB.24.20.8872-8883.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396(6710):433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Mishizen-Eberz AJ, Rissman RA, Carter TL, Ikonomovic MD, Wolfe BB, Armstrong DM. Biochemical and molecular studies of NMDA receptor subunits NR1/2A/2B in hippocampal subregions throughout progression of Alzheimer's disease pathology. Neurobiol Dis. 2004;15(1):80–92. doi: 10.1016/j.nbd.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Beckett TL, Ding Q, Patel E, Markesbery WR, St Clair DK, LeVine H, 3rd, Keller JN. Abeta solubility and deposition during AD progression and in APPxPS-1 knock-in mice. Neurobiol Dis. 2007;27(3):301–311. doi: 10.1016/j.nbd.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Kim E, Sheng M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci. 1996;16(7):2157–2163. doi: 10.1523/JNEUROSCI.16-07-02157.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyffeler M, Zhang WN, Feldon J, Knuesel I. Differential expression of PSD proteins in age-related spatial learning impairments. Neurobiol Aging. 2007;28(1):143–155. doi: 10.1016/j.neurobiolaging.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Opii WO, Joshi G, Head E, Milgram NW, Muggenburg BA, Klein JB, Pierce WM, Cotman CW, Butterfield DA. Proteomic identification of brain proteins in the canine model of human aging following a long-term treatment with antioxidants and a program of behavioral enrichment: relevance to Alzheimer's disease. Neurobiol Aging. 2008;29(1):51–70. doi: 10.1016/j.neurobiolaging.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC. Mild cognitive impairment: transition between aging and Alzheimer's disease. Neurologia. 2000;15(3):93–101. [PubMed] [Google Scholar]
- Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256(3):183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- Portet F, Ousset PJ, Touchon J. [What is a mild cognitive impairment?]. Rev Prat. 2005;55(17):1891–1894. [PubMed] [Google Scholar]
- Pritchard A, Harris J, Pritchard CW, St Clair D, Lemmon H, Lambert JC, Chartier-Harlin MC, Hayes A, Thaker U, Iwatsubo T, Mann DM, Lendon C. Association study and meta-analysis of low-density lipoprotein receptor related protein in Alzheimer's disease. Neurosci Lett. 2005;382(3):221–226. doi: 10.1016/j.neulet.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Satou T, Cummings BJ, Cotman CW. Immunoreactivity for Bcl-2 protein within neurons in the Alzheimer's disease brain increases with disease severity. Brain Res. 1995;697(1-2):35–43. doi: 10.1016/0006-8993(95)00748-f. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA. Synaptic pathology in Alzheimer's disease: a review of ultrastructural studies. Neurobiol Aging. 2003;24(8):1029–1046. doi: 10.1016/j.neurobiolaging.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Scheuer K, Maras A, Gattaz WF, Cairns N, Forstl H, Muller WE. Cortical NMDA receptor properties and membrane fluidity are altered in Alzheimer's disease. Dementia. 1996;7(4):210–214. doi: 10.1159/000106881. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. The origins of Alzheimer disease: a is for amyloid. Jama. 2000;283(12):1615–1617. doi: 10.1001/jama.283.12.1615. [DOI] [PubMed] [Google Scholar]
- Shi J, Tian J, Pritchard A, Lendon C, Lambert JC, Iwatsubo T, Mann DM. A 3'-UTR polymorphism in the oxidized LDL receptor 1 gene increases Abeta40 load as cerebral amyloid angiopathy in Alzheimer's disease. Acta Neuropathol. 2006;111(1):15–20. doi: 10.1007/s00401-005-1108-3. [DOI] [PubMed] [Google Scholar]
- Shiraishi Y, Mizutani A, Mikoshiba K, Furuichi T. Coincidence in dendritic clustering and synaptic targeting of homer proteins and NMDA receptor complex proteins NR2B and PSD95 during development of cultured hippocampal neurons. Mol Cell Neurosci. 2003;22(2):188–201. doi: 10.1016/s1044-7431(03)00037-x. [DOI] [PubMed] [Google Scholar]
- Small GW, Kepe V, Barrio JR. Seeing is believing: neuroimaging adds to our understanding of cerebral pathology. Curr Opin Psychiatry. 2006;19(6):564–569. doi: 10.1097/01.yco.0000245747.53008.e2. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8(8):1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Su JH, Satou T, Anderson AJ, Cotman CW. Up-regulation of Bcl-2 is associated with neuronal DNA damage in Alzheimer's disease. Neuroreport. 1996;7(2):437–440. doi: 10.1097/00001756-199601310-00015. [DOI] [PubMed] [Google Scholar]
- Sultana R, Babu PP. Ethanol-induced alteration in N-methyl-D-aspartate receptor 2A C-terminus and protein kinase C activity in rat brain. Neurosci Lett. 2003;349(1):45–48. doi: 10.1016/s0304-3940(03)00755-9. [DOI] [PubMed] [Google Scholar]
- Sultana R, Piroddi M, Galli F, Butterfield DA. Protein Levels and Activity of Some Antioxidant Enzymes in Hippocampus of Subjects with Amnestic Mild Cognitive Impairment. Neurochem Res. 2008 doi: 10.1007/s11064-008-9593-0. [DOI] [PubMed] [Google Scholar]
- Sze C, Bi H, Kleinschmidt-DeMasters BK, Filley CM, Martin LJ. N-Methyl-D-aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer's disease. J Neurol Sci. 2001;182(2):151–159. doi: 10.1016/s0022-510x(00)00467-6. [DOI] [PubMed] [Google Scholar]
- Wenk GL. Neuropathologic changes in Alzheimer's disease: potential targets for treatment. J Clin Psychiatry. 2006;67(Suppl 3):3–7. quiz 23. [PubMed] [Google Scholar]
- Yatin SM, Varadarajan S, Link CD, Butterfield DA. In vitro and in vivo oxidative stress associated with Alzheimer's amyloid beta-peptide (1-42). Neurobiol Aging. 1999;20(3):325–330. doi: 10.1016/s0197-4580(99)00056-1. discussion 339-342. [DOI] [PubMed] [Google Scholar]
- Zilberberg A, Yaniv A, Gazit A. The low density lipoprotein receptor-1, LRP1, interacts with the human frizzled-1 (HFz1) and down-regulates the canonical Wnt signaling pathway. J Biol Chem. 2004;279(17):17535–17542. doi: 10.1074/jbc.M311292200. [DOI] [PubMed] [Google Scholar]