

Abstract
Background
Gabapentin recruits descending inhibition to produce analgesia after nerve injury, but whether this is a local action in the brainstem is unknown. The authors hypothesized that gabapentin activates noradrenergic neurons in the locus coeruleus (LC) by a local action.
Methods
Male rats underwent L5-L6 spinal nerve ligation (SNL) and received drugs by intra-LC or systemic routes for behavior testing, immunohistochemistry in the LC, and microdialysis in the spinal dorsal horn. In other studies, brainstem slices from normal and SNL animals were used for immunohistochemistry.
Results
SNL increased phosphorylated cyclic adenosine monophosphate response element binding protein (pCREB)-expressing nuclei bilaterally in the LC, and increased noradrenaline release in the spinal dorsal horn. Gabapentin, whether in isolated brainstem slices or in conscious or anesthetized animals, increased pCREB-expressing nuclei in the LC. The net increase in pCREB expression by gabapentin did not differ between normal and SNL conditions. This gabapentin-induced pCREB activation in LC neurons was abolished by an AMPA receptor antagonist, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX). Intra-LC-injected gabapentin reduced hypersensitivity in SNL rats in a dose-dependent manner. Both intra-LC coadministration of CNQX and intrathecal administration of the α2-adrenoceptor antagonist idazoxan blocked antihypersensitivity by intra-LC gabapentin. Intravenous gabapentin induced noradrenaline release in the spinal dorsal horn. The net amount of noradrenaline release by gabapentin is larger in SNL rats compared with the normal condition, although the percentage increases from the baseline were the same.
Conclusions
These results suggest that gabapentin acts directly in the brainstem via a glutamate-dependent mechanism to stimulate descending inhibition to produce antihypersensitivity after peripheral nerve injury.
PERIPHERAL nerve injury can result in chronic pain, hyperalgesia, and allodynia, which respond poorly to nonsteroidal antiinflammatory drugs. Although opioids are effective acutely,1 their chronic use is complicated by tolerance and limiting side effects. For this reason, alternatives to opioids have been sought for decades, but only a few have shown efficacy in the clinic. Gabapentin was licensed as an antiepileptic drug in 1993 and has subsequently been recognized as a first-line drug for the treatment of various chronic pain conditions.2 Because gabapentin relies on interaction with α2δ subunits of calcium channels that are up-regulated in primary afferents and spinal cord after nerve injury,3,4 most studies have focused on mechanisms of gabapentin action at the spinal level. Recently, however, we and others proposed that gabapentin also acts on supraspinal structures to stimulate bulbospinal descending inhibition to alleviate neuropathic pain.5,6
As an important endogenous analgesic in the spinal cord, noradrenaline is released by bulbospinal noradrenergic axons that originate from the locus coeruleus (LC) and adjacent nuclei in the brainstem.7,8 Noradrenaline suppresses the neurotransmission of pain in the spinal cord via activation of α2 adrenoceptors.7,8 In rats, both systemic and intracerebroventricular administration of gabapentin produce analgesia that can be blocked by intrathecal α2-adrenoceptor antagonists,5,6 consistent with its activation of this noradrenergic pathway. Gabapentin likely acts similarly in humans, because its oral administration, in a dose that produces postoperative analgesia, increases noradrenaline concentration in cerebrospinal fluid.9
The mechanism by which gabapentin activates descending inhibition is unclear. Takasu et al.10 recently reported that gabapentin reduces γ-aminobutyric acid-mediated synaptic transmission in LC neurons through presynaptic mechanisms, indicating that gabapentin may reduce inhibitory input on noradrenergic neurons, leading to their activation. Yet whether this local mechanism is relevant in the intact nervous system of the behaving animal has not been examined.
The aim of the current study is to determine whether gabapentin acts on local circuits in the brainstem to stimulate LC neurons and spinal noradrenaline release, to produce antinociception in normal animals and anti-hypersensitivity in animals with nerve injury. First, we used brainstem slice preparations from normal and spinal nerve-ligated (SNL) rats to examine whether gabapentin activates noradrenergic neurons within the LC and whether gabapentin differentially acts in normal and chronic pain states. Second, we examined whether systemically administered gabapentin activates LC neurons in normal and SNL rats. Third, we examined the significance of local actions of gabapentin in the LC for the pain modulation by testing the effects of intra-LC-injected gabapentin on withdrawal threshold to pressure applied to the hind paw in normal and SNL rats. Finally, we examined whether systemically administered gabapentin affects noradrenaline release in the spinal dorsal horn in normal and SNL rats. These studies provide important insight into the regulation of descending inhibition and the consequences of its action in the presence and absence of peripheral nerve injury.
Materials and Methods
Animals
Male Sprague-Dawley rats (weighing 180-250 g) from Harlan Industries (Indianapolis, IN) and Japan SLC Inc. (Hamamatsu, Japan) were used. Microdialysis and all other experiments were performed under Wake Forest University (Winston-Salem, North Carolina) and Gunma University Graduate School Medicine (Gunma, Japan) Animal Care and Use Committee approval and guidelines on the ethical use of animals, respectively. Animals were housed under a 12-h light-dark cycle, with food and water ad libitum.
Surgical Preparations
Spinal Nerve Ligation
As previously described,11 animals were anesthetized with inhalational 2% isoflurane in oxygen, the lateral laminae of the L6 and S1 vertebrae were exposed, the right L6 transverse process was removed, and the right L5 and L6 spinal nerves were tightly ligated using 5-0 silk suture. Animals were allowed to recover for 2 weeks.
Intrathecal Catheterization
Animals were anesthetized with 2% isoflurane, and intrathecal catheterization was performed as previously described.12 A small puncture was made in the atlanto-occipital membrane of the cisterna magnum, and a polyethylene catheter (Re-CathCO LLC, Allison Park, PA), 7.5 cm, was inserted so that the caudal tip reached the lumbar enlargement of the spinal cord. Animals were allowed at least 5 days to recover from the surgery.
Intra-LC Cannulation
Animals were anesthetized with intraperitoneal sodium pentobarbital (50 mg/kg) and placed securely in a stereotaxic frame (KOPF, Tujunga, CA). A sterile stainless steel guide cannula (26-gauge needle shaft; Plastics One, Roanoke, VA) was implanted into the right LC. The coordinates for placement of the tip of the guide cannula were 9.8 mm posterior and 1.4 mm lateral to the bregma, and 7.0 mm ventral from the surface of the dura mater, according to the rat brain atlas.13 The cannula was secured using dental resin along with a self-tapping screw and capped with a dummy cannula. Animals were allowed at least 5 days to recover from the surgery. After the experiment, rats received a methylene blue injection (1 μl) into the LC and were killed by pentobarbital (150 mg/kg intraperitoneal) injection. The brainstem was removed, post-fixed with 8% buffered paraformaldehyde, and sectioned, and the placement of the cannula was verified microscopically. In all cases, the LC was stained by methylene blue.
Immunohistochemistry
Brainstem Slice Study
Brainstem slice experiments were performed as previously reported,14 with minor modifications to assess the local effects of gabapentin in the LC. We used dopamine-β-hydroxylase (DβH) as a marker for noradrenergic neurons15 and phosphorylated cyclic adenosine monophosphate response element binding protein (pCREB) for neuronal activation.14 During deep anesthesia with inhalation of 5% isoflurane, animals were killed by decapitation to remove brainstem. Transverse brainstem slices containing the LC (600-μm thickness) were obtained from normal and L5-L6 SNL animals using a vibratome (VT1000S; Leica Mircosystems, Nussloch, Germany) and perfused in the chamber with artificial cerebrospinal fluid (2 ml/min, 37° ± 0.5°C, 95% O2-5% CO2 saturated). The composition of artificial cerebrospinal fluid was 126.0 mM NaCl, 2.5 mM KCl, 1.3 mM MgSO4, 2.5 mM CaCl2, 12 mM glucose, 1.2 mM NaH2PO4, and 25 mM NaHCO3. Gabapentin (Toronto Research Chemicals Inc., North York, Ontario, Canada) was dissolved in saline, and kainic acid (Cayman Chemical, Ann Arbor, MI) and 6-cyano-7-nitroquinoxaline-2,3-dione disodium salt (CNQX-2Na; Sigma Chemical Co., St. Louis, MO) were dissolved in dimethyl sulfoxide and diluted with artificial cerebrospinal fluid. After 3 h of baseline perfusion, brainstem slices were transferred to the drug challenge chamber and incubated with drugs for 30 min and then fixed with 4% paraformaldehyde for 1 h. The fixed tissues were then cryoprotected with 30% sucrose for 48 h and sectioned (16 μm). Because of potential damage of tissue, the first and last seven sections from the tissue surface were discarded, and the rest were used for the immunocytochemistry. For DβH and pCREB staining, four brainstem sections were randomly selected from each animal. Sections were pretreated with 1.5% normal donkey serum (Vector, Burlingame, CA) and then incubated for 24 h at 4°C with a mouse monoclonal anti-DβH antibody (1:1,000, MAB308; Chemicon International Inc., Temecula, CA) and a rabbit anti-pCREB antibody (1:1,000, No. 06-519; Upstate, Lake Placid, NY) diluted in phosphate-buffered saline containing 0.3% Triton X-100 and 1.5% normal donkey serum. Subsequently, the sections were incubated for 1 h with a donkey anti-mouse fluorescein (1:100; Chemicon International Inc.) and then incubated with a donkey anti-rabbit rhodamine (1:100; Chemicon International Inc.). Finally, DβH and pCREB immunostaining were visualized under an inverted microscope (200× magnification) with standard fluorescein isothiocyanate and tetramethylrhodamine isothiocyanate filters. Images of both sides of LCs in SNL rats and randomly selected right or left side of LC in normal rats were captured using a digital charge-coupled device camera with a consistent setting. Cells with DβH and pCREB immunostaining were counted in the entire LC in each section. In each animal, 72-112 DβH-immunoreactive LC cells with visible nuclei were counted. The person performing immunohistochemistry and counting cells was blinded to drug and treatment.
In Vivo Study
Anesthetized (2% isoflurane) or conscious animals received an intravenous injection of saline or gabapentin (50 mg/kg) through the tail vein. Animals were killed by decapitation 30 min after injection, and the brainstem was collected and fixed overnight in 4% paraformaldehyde. The fixed tissues were then cryoprotected with 30% sucrose for 72 h and sectioned for immunocytochemistry as described in the previous paragraph.
Behavioral Tests
The person performing the behavioral test was blinded to drug and dose. Withdrawal threshold to pressure applied to the hind paw, expressed in grams, was measured using an analgesimeter (Ugo Basile, Comerio, Italy) as previously described.16 The device applies increasing pressure to the hind paw. When the animal withdrew the paw or vocalized, the pressure was immediately released, and the withdrawal threshold read on a scale. Training of animals for this test was performed for 3-5 days before the drug treatment. A cutoff of 250 g was used to avoid potential tissue injury. We used these animals two or three times on different days. Experiments in the same animals were separated by at least 6 days. Drugs and the doses were randomly assigned. For intra-LC injection, gabapentin and CNQX-2Na were dissolved in artificial cerebrospinal fluid, and a volume of 1 μl solution was injected at the speed of 0.5 μl/min using a syringe pump (model 200; KD Scientific, Holliston, MA) 15 min before the behavioral measurement. For intrathecal administration, idazoxan hydrochloride (Sigma Chemical Co.) was dissolved in saline and injected in a volume of 10 μl followed by 10 μl saline 30 min before the measurement. To calculate the effective dose to produce a 50% maximum effect (ED50) of gabapentin, the response threshold data were converted to a percentage of return to presurgery threshold according to the following formula: % return to presurgery threshold = (postdrug threshold - baseline predrug threshold)/(pre-SNL threshold - baseline predrug threshold) × 100. Predrug threshold was the withdrawal threshold after SNL. ED50 was determined using linear regression.
Measurements of Spinal Noradrenaline Release
Microdialysis studies were performed with normal and SNL rats as previously described,17 with minor modifications. Anesthesia was induced with urethane (1.2-1.5 g/kg intraperitoneal) and then maintained with 0.5% isoflurane in 100% oxygen through a nose cone. The left femoral vein was cannulated for drug injections. The rectal temperature was kept at 37°-38°C by a heating pad placed beneath the animal. The L3-L5 level of spinal cord was exposed by a thoracolumbar laminectomy, and then the rat was placed in a stereotaxic apparatus. After opening the dura, a dorsal root that enters the spinal cord above the level of recording sites was lifted using a glass retractor, so that a microdialysis probe was able to advance into the superficial layer of the dorsal horn. The probe was inserted from just lateral to the dorsal root and advanced at an angle of 15° at a depth of 1 mm using a micromanipulator (model WR-88; Narishige, Tokyo, Japan). The surface of the spinal cord was covered with mineral oil. Microdialysis probes consisted of a 1-mm length of hairpin-shaped dialysis membrane (OD = 0.22 mm, ID = 0.20 mm), and the membrane was attached to a 1-cm silica double-lumen tube (OD = 0.35 mm; Eicom Co., Kyoto, Japan). The microdialysis probe was perfused with Ringer's solution (147 mM NaCl, 4 mM KCl, 2.3 mM CaCl2) at a constant flow rate (1 μl/min) using a syringe pump (ESP-64; Eicom Co.). After 120 min of constant perfusion, two consecutive samples were collected to determine basal noradrenaline concentrations in the dialysate. Saline (0.5 ml) or gabapentin (50 mg/kg intravenous) was administered, and the sample was collected into an auto injector (EAS-20; Eicom Co.) and analyzed for noradrenaline concentration using high-performance liquid chromatography with electrochemical detection by an HTEC-500 analyzing system (Eicom Co.). The chromatographic conditions are as follows: The mobile phase consisted of 0.1 M ammonium acetate buffer (pH 6.0) and methanol (7:3 vol/vol) containing 0.05 M sodium sulfonate, and 50 mg/l EDTA-2Na, and the column was an EICOMPAC CAX (2.0 mmφ × 200 mm; Eicom Co.). The limit of detection of this assay in the current study was 62 fg per injection.
Statistical Analyses
Unless otherwise stated, data were normally distributed and are presented as mean ± SE. Differences among groups were determined using one- or two-way analysis of variance as appropriate. P < 0.05 was considered significant.
Results
Brainstem Slice Studies
Figures 1 and 2 depict DβH and pCREB expression in the LC of normal and SNL animals, respectively. Initially, we verified the ability of the brainstem slice preparation to respond to excitation by exposure to the AMPA receptor agonist kainate (50 μM). Kainate increased the percentage of pCREB-immunoreactive cells in DβH-immunoreactive cells in the LC from normal animals compared with vehicle from 5.0 ± 0.6 to 45 ± 6.9% (n = 4 each; P < 0.05; fig. 1G-I). Kainate also induced pCREB expression in brainstem slices from SNL animals, with 64 ± 8.3% DβH-immunoreactive cells in the LC ipsilateral to SNL expressing pCREB immunoreactivity after kainate (n = 4) compared with 21 ± 4.1% after vehicle (n = 5; P < 0.05).
Fig. 1.
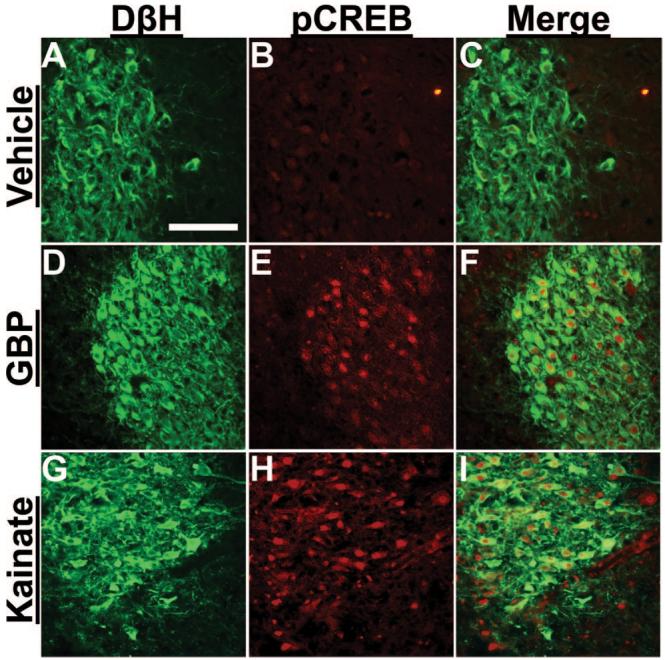
Photomicrographs of dopamine-β-hydroxylase (DβH)- and phosphorylated cyclic adenosine monophosphate response element binding protein (pCREB)-immunoreactive neurons in brainstem slices from normal rats. Brainstem slices treated with vehicle (A-C), 100 μM gabapentin (GBP; D-F), and 50 μM kainate (G-I) for 30 min were stained with antibodies for DβH (green) and pCREB (red). Note that GBP (G) and kainate (I) increased pCREB immunoreactivity in DβH-immunoreactive neurons. Scale bar = 100 μm.
Fig. 2.
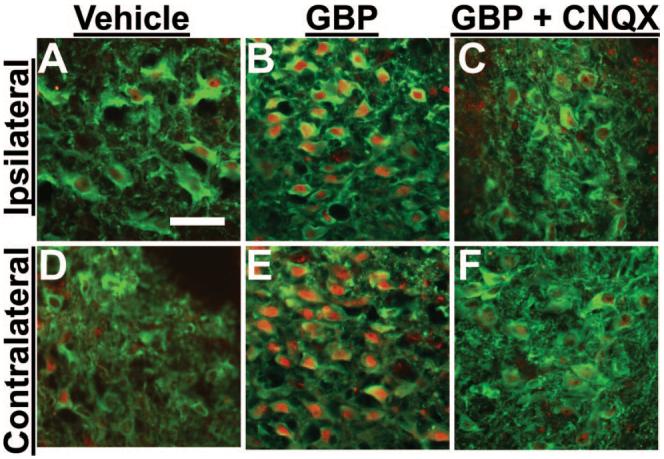
Photomicrographs of dopamine-β-hydroxylase (DβH)- and phosphorylated cyclic adenosine monophosphate response element binding protein (pCREB)-immunoreactive neurons in brainstem slices from spinal nerve-ligated rats. Brainstem slices were treated with vehicle (A and D), 100 μM gabapentin (GBP; B and E), and 100 μM GBP with 50 μM CNQX (C and F) for 30 min. The locus coeruleus ipsilateral (A-C) and contralateral (D-F) to nerve injury was stained with antibodies for DβH (green) and pCREB (red). Note that GBP bilaterally increased pCREB immunoreactivity in DβH-immunoreactive neurons (B and E), and coincubation of GBP with CNQX blocked this effect (C and F). Scale bar = 50 μm.
Effect of Nerve Injury on pCREB Expression
As shown in figure 1B, few cells expressed pCREB in brainstem slices from normal animals. In contrast, 2 weeks after SNL surgery, pCREB nuclei were commonly observed in the LC both ipsilateral and contralateral to SNL (figs. 2A and D). Quantitatively, SNL increased the percentage of pCREB-immunoreactive cells in DβH-immunoreactive cells in the LC both ipsilateral (21 ± 4.1%, n = 5) and contralateral (18 ± 5.5%, n = 5) to injury compared with normal animals (5.0 ± 0.6%, n = 4; P < 0.05; fig. 3A). These results demonstrate that unilateral peripheral nerve injury resulted in bilateral activation of noradrenergic neurons in the LC.
Fig. 3.
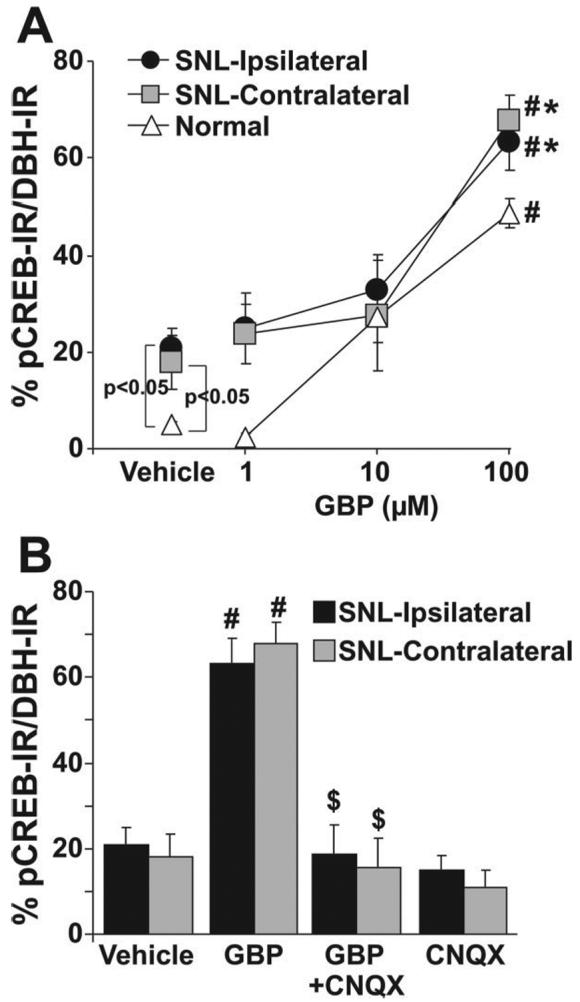
Quantification of gabapentin (GBP) concentration response and effect of AMPA antagonist on phosphorylated cyclic adenosine monophosphate response element binding protein (pCREB) activation in brainstem slices from normal and spinal nerve-ligated (SNL) rats. Data are presented as percentage of pCREB immunoreactivity (IR) in dopamine-β-hydroxylase-immunoreactive (DBH-IR) neurons. (A) Brainstem slices from normal (n = 4) and SNL (n = 4 or 5) rats were treated with vehicle and GBP (1-100 μM) for 30 min. *P < 0.05 versus normal by two-way analysis of variance. #P < 0.05 versus vehicle by one-way analysis of variance. (B) Brainstem slices from SNL rats were treated with vehicle (n = 5), GBP (100 μM, n = 5), CNQX (50 μM, n = 4), and GBP plus CNQX (n = 4) for 30 min. # P < 0.05 versus vehicle. $ P < 0.05 versus GBP.
Gabapentin-induced pCREB Activation
Gabapentin increased the percentage of pCREB-immunoreactive nuclei in DβH-immunoreactive cells in brainstem slices from both normal and SNL animals in a concentration-dependent manner (1-100 μM; figs. 1F, 2B, 2E, and 3A. The absolute concentration responses of gabapentin for pCREB immunoreactivity colocalization with DβH immunoreactivity in the brainstem slices differ significantly between normal and SNL animals (P < 0.05). However, the net increases of pCREB immunoreactivity colocalization with DβH immunoreactivity with exposure to gabapentin compared with vehicle were similar between SNL (ipsilateral 43%, contralateral 49%) and normal (44%) groups. The AMPA receptor antagonist CNQX (50 μM) did not affect basal pCREB activity but abolished gabapentin-induced pCREB activation (figs. 2C, 2F, and 3B), demonstrating a glutamatergic dependence to gabapentin's effect.
In Vivo Effect of Gabapentin on pCREB Expression in LC Neurons
Consistent with observations in the slice preparation, the number of pCREB-immunoreactive nuclei in the LC of SNL animals was increased compared with normal animals (figs. 4 and 5). Intravenous administration of gabapentin (50 mg/kg; a dose that results in an approximately 50% reversal of mechanical withdrawal threshold in rats after peripheral nerve injury18) during isoflurane anesthesia significantly increased the number of pCREB-immunoreactive nuclei in the LC compared with saline in both normal and SNL animals (fig. 4). pCREB expression under vehicle and gabapentin treatment in the LC of SNL animals did not differ between anesthetized and conscious conditions (figs. 4 and 5), demonstrating that 2% isoflurane anesthesia did not influence LC neuronal activation, at least as assessed by this marker.
Fig. 4.

Photomicrograph of effect of systemic administered gabapentin (GBP) on phosphorylated cyclic adenosine monophosphate response element binding protein (pCREB) activation in the locus coeruleus. Normal and spinal nerve-ligated (SNL) rats received saline (A-C) or GBP (50 mg/kg intravenous; D-F) while conscious or during 2% isoflurane anesthesia, and then brainstems were collected 30 min after injection for dopamine-β-hydroxylase (green) and pCREB (red) staining. In the SNL group (B, C, E, and F), photomicrographs of brainstems ipsilateral to nerve injury are presented. Scale bar = 50 μm.
Fig. 5.

Quantification of phosphorylated cyclic adenosine monophosphate response element binding protein immunoreactivity (pCREB-IR) in dopamine-β-hydroxylase-immunoreactive (DBH-IR) neurons in the brainstem from normal and spinal nerve-ligated (SNL) rats after systemic administration of saline or gabapentin (GBP, 50 mg/kg intravenous). See conditions in figure 4. Data from four rats per group are presented as the percentage of pCREB-IR in DBH-IR neurons. *P < 0.05 versus saline. #P < 0.05 versus normal.
Behavioral Effect of Intra-LC Injection of Gabapentin in Hypersensitivity after Nerve Injury
Spinal nerve ligation strongly decreased the withdrawal threshold of the hind paw ipsilateral to SNL from 140 ± 12 g to 67 ± 11 g (mean ± SD, n = 31; P < 0.0001). Intra-LC injection of gabapentin increased withdrawal threshold in the hind paw ipsilateral to SNL in a dose-dependent manner (fig. 6A). Gabapentin showed significant antihypersensitivity effects from 0.3 to 3 μg compared with vehicle (P < 0.05). The peak effect of gabapentin was observed 15 min after injection. The ED50 value (95% confidence interval) of gabapentin calculated at the 15-min time point was 0.54 μg (0.18-0.98 μg). Intrathecal administration of the α2-adrenoceptor antagonist idazoxan (30 μg) or intra-LC injection of CNQX (0.3 μg), neither of which affected withdrawal threshold alone, completely blocked the antihypersensitivity effect of intra-LC gabapentin (1 μg; figs. 6B and C). In contrast to its effect in SNL animals, intra-LC injection of gabapentin (3 μg) did not affect withdrawal threshold in normal animals (fig. 6A), indicating that gabapentin is unlikely to induce severe sedation, which impairs motor function, at least the dose we used in the current study.
Fig. 6.

Intra-locus coeruleus (LC)-injected gabapentin (GBP) reduced mechanical hypersensitivity after spinal nerve ligation (SNL) surgery. (A) Normal (n = 6) and SNL rats (n = 7 or 8) received intra-LC injection of vehicle or GBP (0.1-3 μg). The mechanical withdrawal threshold to pressure to the paw in normal rats or ipsilateral to SNL is presented over time. * P < 0.05 versus time 0. In SNL rats, 0.3-3 μg GBP groups differ significantly from the vehicle group by two-way repeated-measures analysis of variance (P < 0.05). (B) Intrathecal (IT) saline or idazoxan (Ida, 30 μg) was administered followed in 30 min by vehicle or 1 μg GBP injection in the LC, and the postdrug measurement (Post) was obtained 15 min later in SNL rats (n = 7). * P < 0.05 versus predrug (Pre). (C) 1 μg GBP, 0.3 μg CNQX, or their mixture was injected in the LC 15 min before the measurement (Post) in SNL rats (n = 8). * P < 0.05 versus predrug (Pre).
Effect of Gabapentin on Spinal Noradrenaline Release in Normal and SNL Animals
There was a small but statistically significant increase in noradrenaline concentration in the dialysates from SNL animals (0.50 ± 0.07 pg/15 μl, n = 12) compared with normal animals (0.34 ± 0.04 pg/15 μl, n = 12; P < 0.05) in the absence of drug treatment. In the saline-treated group, a slight decrease in noradrenaline concentrations in the dialysates was observed over time (fig. 7). In the gabapentin (50 mg/kg intravenous)-treated group, noradrenaline concentrations increased in a time-dependent manner after drug injection and significantly differed from saline treatment in both normal and SNL animals (P < 0.05). Although the percentage increases from the baseline did not differ in normal and SNL animals, the total noradrenaline content in the dialysates collected from 15-90 min after gabapentin administration was significantly greater in SNL animals (3.5 ± 0.6 pg/90 μl, n = 6) than in normal animals (2.0 ± 0.4 pg/90 μl, n = 6; P < 0.05).
Fig. 7.

Microdialysis for spinal noradrenaline release. Normal (n = 6) and spinal nerve-ligated (SNL; n = 6) rats received intravenous saline or gabapentin (GBP, 50 mg/kg). Data are presented over time as percentage of baseline. GBP groups significantly differ from saline group by two-way repeated-measures analysis of variance (P < 0.05). * P < 0.05 versus time 0 by one-way analysis of variance.
Discussion
Peripheral nerve injury leading to hypersensitivity is associated with a plethora of changes in sensory processing at all levels, from the primary afferent to the spinal cord to supraspinal and cortical responses. The gate control theory of pain includes regulation of spinal cord processing of sensory information by descending influences.19 Although descending inhibitory mechanisms have been probed for several decades,20 there is some evidence that patients with chronic pain have a reduced ability to physiologically recruit descending inhibition.21 This and more recent work on descending facilitation22 has shifted focus away from descending inhibition in chronic hypersensitivity states. Yet several approved treatments for neuropathic pain in patients, including noradrenaline reuptake inhibitors, the α2-adrenoceptor agonist clonidine, and gabapentin, as suggested in the current report, interact with or mimic activation of bulbospinal noradrenergic pathways to produce analgesia.5,23,24 Therefore, the current study provides novel evidence regarding a mechanism of action of one of the widest prescribed treatments for neuropathic pain and for the relevance of descending inhibition to its efficacy. Because orally administered gabapentin increases noradrenaline concentration in lumbar cerebrospinal fluid in patients,9 we believe it is likely that gabapentin acts supraspinally to engage descending noradrenergic inhibition not only in animals, but also in humans.
The regulation of descending modulation of sensory transmission is poorly understood, in part because of the complexity of interacting supraspinal circuits and in part because of the lack of probes to selectively and locally affect descending pathways. The current study suggests that gabapentin represents one such probe for descending noradrenergic inhibition. Although gabapentin reduces exaggerated primary afferent and spinal cord responses to sensory stimuli after peripheral nerve injury,18,25 the relevance of these sites of action after systemic administration is uncertain. We and others previously reported that systemic and intracerebroventricular administration of gabapentin reduced hypersensitivity after peripheral nerve injury5,6 in a manner that was blocked by spinal injection of α2-adrenoceptor antagonists, consistent with an enhancement by gabapentin of descending inhibition. The current study extends these observations and supports this hypothesis in several ways. Intra-LC injection of gabapentin also inhibited hypersensitivity in a manner that was blocked by spinal injection of an α2-adrenoceptor antagonist. Systemic gabapentin increased noradrenergic neuronal activity in the LC, as measured by pCREB expression, and this occurred similarly in the anesthetized as well as the conscious state. Lack of effect of anesthesia is important because the microdialysis experiments, which demonstrated increased spinal release of noradrenaline after systemic gabapentin, were performed by necessity in anesthetized animals. Finally, although not only the LC but also other adjacent nuclei in the brainstem send noradrenergic axons to the spinal dorsal horn to induce descending inhibition,7,8 activation of descending noradrenergic inhibition by gabapentin most likely reflects local circuits within the LC, because noradrenergic neuronal activation also occurred with in vitro drug exposure.
The mechanism by which gabapentin locally activates noradrenergic neurons in the LC is only partially explained by the current study. There are three likely possibilities: (1) Gabapentin directly activates LC neurons, (2) gabapentin directly increases glutamate release upon LC neurons, or (3) gabapentin decreases γ-aminobutyric acid release and indirectly increases the influences and/or release of glutamate on LC neurons. Because gabapentin-induced LC neuronal activation was completely blocked by an AMPA receptor blocker in the current study, hypothesis 1 is unlikely. A major glutamatergic input to the LC arises from the nucleus paragigantocellularis and activates LC neurons via AMPA receptors,26 and terminals of this projection in our slice preparation may have provided the substrate for gabapentin action. On the other hand, studies in the spinal cord27,28 and brain29,30 show that gabapentin reduces rather than increases glutamate release, and if this is similarly true in the LC, mechanism 2 is unlikely. Takasu et al.10 recently reported that gabapentin reduces γ-aminobutyric acid-mediated synaptic transmission in the LC neurons through presynaptic mechanisms in mice with peripheral nerve injury. This observation suggests that gabapentin may increase glutamatergic tone in the LC by disinhibition. We therefore consider hypothesis 3 as the most likely mechanism of gabapentin action.
Several of the observations in the current study may seem paradoxical. Although another study showed that SNL did not alter spontaneous discharge rate of LC neurons,31 we showed that unilateral SNL significantly increases pCREB expression in noradrenergic neurons bilaterally in the LC in the slice preparation and in vivo, and also increases basal noradrenaline release in the spinal dorsal horn. This difference may be due to the differing measures of neuronal activity (electrical activity vs. pCREB expression and noradrenaline release). In the current study, we did not study sham-operated animals. Because SNL is invasive surgery and likely to induce inflammation, we cannot state unequivocally that this bilateral pCREB activation and increased spinal noradrenaline release were due solely to nerve ligation or from other aspects of this invasive surgery. However, the results in the current study are consistent with the bilateral input and output of the LC7,8,26,32 and with previous reports that bilateral activation of the LC neurons was induced by unilateral peripheral nerve injury but not by sham surgery33 and that SNL induced brain-derived neurotrophic factor-dependent noradrenergic axon sprouting in the spinal dorsal horn, whereas sham surgery did not alter brain-derived neurotrophic factor content in the spinal cord.15 Although Takeuchi et al.34 reported that spinal noradrenaline turnover after intracerebroventricular injection of gabapentin increased in mice with partial sciatic nerve ligation but did not change in sham-operated mice, the current study showed that gabapentin induced spinal noradrenaline release in both normal and SNL animals, in accord with activation of the LC neurons. This discrepancy may be due to differences of species, noradrenaline release measurements, or site of gabapentin injection.
Interestingly, although gabapentin activated descending noradrenergic pathways, LC-injected gabapentin did not affect withdrawal threshold in normal rats, consistent with previous reports of a lack of effect in the absence of hypersensitivity.6,18 Perhaps this disconnection between activation of descending noradrenergic pathway and antinociception on the normal animal reflects relatively low spinal noradrenaline release or differences between normal and neuropathic states in the location and function of α2 adrenoceptors in the spinal cord. In support of the latter, intrathecal injection of clonidine demonstrates increased potency and efficacy in neuropathic pain states in animals35 and humans.23 Several potential causes for this plasticity after nerve injury have been demonstrated in animals, including increased expression of inhibitory α2 adrenoceptors on calcitonin gene-related peptide expressing afferents,36 increased G protein-coupling efficiency of spinal α2 adrenoceptors,37 and increased α2 adrenoceptor-mediated activation of inhibitory cholinergic interneurons associated with up-regulation of inhibitory M2 muscarinic receptors in the primary sensory neurons.38,39 Consistent with the increased basal noradrenaline release and noradrenergic axon sprouting15 in the spinal dorsal horn after SNL, the current study also demonstrated that gabapentin induced more spinal noradrenaline release in SNL animals compared with normal animals, although the percentage increases from the baseline were the same. These findings suggest that peripheral nerve injury enhances efficacy and release of noradrenaline and that gabapentin uses these plastic changes to induce more spinal noradrenaline release after nerve injury than in the normal condition.
In summary, the current study suggests that descending inhibition can play an important role in analgesia in neuropathic pain and that a commonly used treatment in this condition relies heavily on this mechanism rather than on actions in the periphery or spinal cord. These results imply an important role of descending inhibition in modulation of hypersensitivity after peripheral nerve injury and provide a strong rationale to explore combination drug therapy, which targets this pathway, such as gabapentin and noradrenaline transporter inhibitors. Finally, these data suggest that gabapentin is a useful probe to understand the local regulation of glutamate in the LC and activation of descending noradrenergic inhibition.
Acknowledgments
Supported by grant Nos. NS59574 (to Dr. Eisenach) and DA024826 (to Dr. Hayashida) from the National Institutes of Health, Bethesda, Maryland.
Footnotes
Michael M. Todd, M.D., served as Handling Editor for this article. Dr. Eisenach was not involved in the decision-making process.
References
- 1.Raja SN, Haythornthwaite JA, Pappagallo M, Clark MR, Travison TG, Sabeen S, Royall RM, Max MB. Opioids versus antidepressants in postherpetic neuralgia: A randomized, placebo-controlled trial. Neurology. 2002;59:1015–21. doi: 10.1212/wnl.59.7.1015. [DOI] [PubMed] [Google Scholar]
- 2.Laird MA, Gidal BE. Use of gabapentin in the treatment of neuropathic pain. Ann Pharmacother. 2000;34:802–7. doi: 10.1345/aph.19303. [DOI] [PubMed] [Google Scholar]
- 3.Gee NS, Brown JP, Dissanayake VU, Offord J, Thurlow R, Woodruff GN. The novel anticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J Biol Chem. 1996;271:5768–76. doi: 10.1074/jbc.271.10.5768. [DOI] [PubMed] [Google Scholar]
- 4.Luo ZD, Calcutt NA, Higuera ES, Valder CR, Song YH, Svensson CI, Myers RR. Injury type-specific calcium channel alpha 2 delta-1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. J Pharmacol Exp Ther. 2002;303:1199–205. doi: 10.1124/jpet.102.041574. [DOI] [PubMed] [Google Scholar]
- 5.Hayashida K, Parker R, Eisenach JC. Oral gabapentin activates spinal cholinergic circuits to reduce hypersensitivity after peripheral nerve injury and interacts synergistically with oral donepezil. ANESTHESIOLOGY. 2007;106:1213–9. doi: 10.1097/01.anes.0000267605.40258.98. [DOI] [PubMed] [Google Scholar]
- 6.Tanabe M, Takasu K, Kasuya N, Shimizu S, Honda M, Ono H. Role of descending noradrenergic system and spinal alpha2-adrenergic receptors in the effects of gabapentin on thermal and mechanical nociception after partial nerve injury in the mouse. Br J Pharmacol. 2005;144:703–14. doi: 10.1038/sj.bjp.0706109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aston-Jones G, Shipley MT, Chouvet G, Ennis M, van Bockstaele E, Pieribone V, Shiekhattar R, Akaoka H, Drolet G, Astier B. Afferent regulation of locus coeruleus neurons: Anatomy, physiology and pharmacology. Prog Brain Res. 1991;88:47–75. doi: 10.1016/s0079-6123(08)63799-1. [DOI] [PubMed] [Google Scholar]
- 8.Pertovaara A. Noradrenergic pain modulation. Prog Neurobiol. 2006;80:53–83. doi: 10.1016/j.pneurobio.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 9.Hayashida K, DeGoes S, Curry R, Eisenach JC. Gabapentin activates spinal noradrenergic activity in rats and humans and reduces hypersensitivity after surgery. ANESTHESIOLOGY. 2007;106:557–62. doi: 10.1097/00000542-200703000-00021. [DOI] [PubMed] [Google Scholar]
- 10.Takasu K, Ono H, Tanabe M. Gabapentin produces PKA-dependent presynaptic inhibition of GABAergic synaptic transmission in LC neurons following partial nerve injury in mice. J Neurochem. 2008;105:933–42. doi: 10.1111/j.1471-4159.2008.05212.x. [DOI] [PubMed] [Google Scholar]
- 11.Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–63. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 12.Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–6. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- 13.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; San Diego: 2005. [Google Scholar]
- 14.Kawasaki Y, Kohno T, Zhuang ZY, Brenner GJ, Wang H, Van Der Meer C, Befort K, Woolf CJ, Ji RR. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci. 2004;24:8310–21. doi: 10.1523/JNEUROSCI.2396-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayashida KI, Clayton BA, Johnson JE, Eisenach JC. Brain derived nerve growth factor induces spinal noradrenergic fiber sprouting and enhances clonidine analgesia following nerve injury in rats. Pain. 2008;136:348–55. doi: 10.1016/j.pain.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther. 1957;111:409–19. [PubMed] [Google Scholar]
- 17.Li X, Eisenach JC. Nicotinic acetylcholine receptor regulation of spinal norepinephrine release. ANESTHESIOLOGY. 2002;96:1450–6. doi: 10.1097/00000542-200206000-00026. [DOI] [PubMed] [Google Scholar]
- 18.Pan HL, Eisenach JC, Chen SR. Gabapentin suppresses ectopic nerve discharges and reverses allodynia in neuropathic rats. J Pharmacol Exp Ther. 1999;288:1026–30. [PubMed] [Google Scholar]
- 19.Melzack R, Wall PD. Pain mechanisms: A new theory. Science. 1965;150:971–9. doi: 10.1126/science.150.3699.971. [DOI] [PubMed] [Google Scholar]
- 20.Fields HL, Basbaum AI. Brainstem control of spinal pain-transmission neurons. Annu Rev Physiol. 1978;40:217–48. doi: 10.1146/annurev.ph.40.030178.001245. [DOI] [PubMed] [Google Scholar]
- 21.Witting N, Svensson P, Jensen TS. Differential recruitment of endogenous pain inhibitory systems in neuropathic pain patients. Pain. 2003;103:75–81. doi: 10.1016/s0304-3959(02)00421-9. [DOI] [PubMed] [Google Scholar]
- 22.Burgess SE, Gardell LR, Ossipov MH, Malan TP, Jr, Vanderah TW, Lai J, Porreca F. Time-dependent descending facilitation from the rostral ventromedial medulla maintains, but does not initiate, neuropathic pain. J Neurosci. 2002;22:5129–36. doi: 10.1523/JNEUROSCI.22-12-05129.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eisenach JC, DuPen S, Dubois M, Miguel R, Allin D. Epidural clonidine analgesia for intractable cancer pain. The Epidural Clonidine Study Group. Pain. 1995;61:391–9. doi: 10.1016/0304-3959(94)00209-W. [DOI] [PubMed] [Google Scholar]
- 24.Sindrup SH, Otto M, Finnerup NB, Jensen TS. Antidepressants in the treatment of neuropathic pain. Basic Clin Pharmacol Toxicol. 2005;96:399–409. doi: 10.1111/j.1742-7843.2005.pto_96696601.x. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki R, Rahman W, Rygh LJ, Webber M, Hunt SP, Dickenson AH. Spinal-supraspinal serotonergic circuits regulating neuropathic pain and its treatment with gabapentin. Pain. 2005;117:292–303. doi: 10.1016/j.pain.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 26.Singewald N, Philippu A. Release of neurotransmitters in the locus coeruleus. Prog Neurobiol. 1998;56:237–67. doi: 10.1016/s0301-0082(98)00039-2. [DOI] [PubMed] [Google Scholar]
- 27.Coderre TJ, Kumar N, Lefebvre CD, Yu JS. Evidence that gabapentin reduces neuropathic pain by inhibiting the spinal release of glutamate. J Neurochem. 2005;94:1131–9. doi: 10.1111/j.1471-4159.2005.03263.x. [DOI] [PubMed] [Google Scholar]
- 28.Shimoyama M, Shimoyama N, Hori Y. Gabapentin affects glutamatergic excitatory neurotransmission in the rat dorsal horn. Pain. 2000;85:405–14. doi: 10.1016/S0304-3959(99)00283-3. [DOI] [PubMed] [Google Scholar]
- 29.Dooley DJ, Mieske CA, Borosky SA. Inhibition of K(+)-evoked glutamate release from rat neocortical and hippocampal slices by gabapentin. Neurosci Lett. 2000;280:107–10. doi: 10.1016/s0304-3940(00)00769-2. [DOI] [PubMed] [Google Scholar]
- 30.Maneuf YP, Hughes J, McKnight AT. Gabapentin inhibits the substance P-facilitated K(+)-evoked release of [(3)H]glutamate from rat caudal trigeminal nucleus slices. Pain. 2001;93:191–6. doi: 10.1016/S0304-3959(01)00316-5. [DOI] [PubMed] [Google Scholar]
- 31.Viisanen H, Pertovaara A. Influence of peripheral nerve injury on response properties of locus coeruleus neurons and coeruleospinal antinociception in the rat. Neuroscience. 2007;146:1785–94. doi: 10.1016/j.neuroscience.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 32.Clark FM, Proudfit HK. Anatomical evidence for genetic differences in the innervation of the rat spinal cord by noradrenergic locus coeruleus neurons. Brain Res. 1992;591:44–53. doi: 10.1016/0006-8993(92)90976-g. [DOI] [PubMed] [Google Scholar]
- 33.Mao J, Mayer DJ, Price DD. Patterns of increased brain activity indicative of pain in a rat model of peripheral mononeuropathy. J Neurosci. 1993;13:2689–702. doi: 10.1523/JNEUROSCI.13-06-02689.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takeuchi Y, Takasu K, Honda M, Ono H, Tanabe M. Neurochemical evidence that supraspinally administered gabapentin activates the descending noradrenergic system after peripheral nerve injury. Eur J Pharmacol. 2007;556:69–74. doi: 10.1016/j.ejphar.2006.10.059. [DOI] [PubMed] [Google Scholar]
- 35.Paqueron X, Conklin D, Eisenach JC. Plasticity in action of intrathecal clonidine to mechanical but not thermal nociception after peripheral nerve injury. ANESTHESIOLOGY. 2003;99:199–204. doi: 10.1097/00000542-200307000-00030. [DOI] [PubMed] [Google Scholar]
- 36.Eisenach JC, Zhang Y, Duflo F. Alpha2-adrenoceptors inhibit the intracellular Ca2+ response to electrical stimulation in normal and injured sensory neurons, with increased inhibition of calcitonin gene-related peptide expressing neurons after injury. Neuroscience. 2005;131:189–97. doi: 10.1016/j.neuroscience.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 37.Bantel C, Eisenach JC, Duflo F, Tobin JR, Childers SR. Spinal nerve ligation increases alpha2-adrenergic receptor G-protein coupling in the spinal cord. Brain Res. 2005;1038:76–82. doi: 10.1016/j.brainres.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 38.Hayashida KI, Bynum T, Vincler M, Eisenach JC. Inhibitory M2 muscarinic receptors are upregulated in both axotomized and intact small diameter dorsal root ganglion cells after peripheral nerve injury. Neuroscience. 2006;140:259–68. doi: 10.1016/j.neuroscience.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 39.Obata H, Li X, Eisenach JC. α2-Adrenoceptor activation by clonidine enhances stimulation-evoked acetylcholine release from spinal cord tissue after nerve ligation in rats. ANESTHESIOLOGY. 2005;102:657–62. doi: 10.1097/00000542-200503000-00027. [DOI] [PubMed] [Google Scholar]