

Abstract
Acute coronary occlusion is the leading cause of death in the Western world. There is an unmet need for the development of treatments to limit the extent of myocardial infarction (MI) during the acute phase of occlusion. Recently, investigators have focused on the use of a chemokine, CXCL12, the only identified ligand for CXCR4, as a new therapeutic modality to recruit stem cells to individuals suffering from MI. Here, we examined the effects of overexpression of CXCR4 by gene transfer on MI. Adenoviruses carrying the CXCR4 gene were injected into the rat heart one week before ligation of the left anterior descending coronary artery followed by 24 hours reperfusion. Cardiac function was assessed by echocardiography couple with 2,3,5-Triphenyltetrazolium chloride staining to measure MI size. In comparison with control groups, rats receiving Ad-CXCR4 displayed an increase in infarct area (13.5% ± 4.1%) and decreased fractional shortening (38% ± 5%). Histological analysis revealed a significant increase in CXCL12 and tumor necrosis factor-α expression in ischemic area of CXCR4 overexpressed hearts. CXCR4 overexpression was associated with increased influx of inflammatory cells and enhanced cardiomyocyte apoptosis in the infarcted heart. These data suggest that in our model overexpressing CXCR4 appears to enhance ischemia/reperfusion injury possibly due to enhanced recruitment of inflammatory cells, increased tumor necrosis factor-α production, and activation of cell death/apoptotic pathways.
Coronary artery disease is a leading cause of death in the United States. Acute myocardial infarction (MI) is a catastrophic manifestation of coronary artery disease that strikes nearly one million Americans each year.1,2,3 The mainstay of current therapy in acute MI is the restoration of blood flow (reperfusion) to the affected area through thrombolytic therapy or angioplasty. This reperfusion, although key to myocyte survival, is also associated with myocardial injury.2 One component of reperfusion is the influx of inflammatory cells into the heart. These inflammatory cells are believed to cause further damage to the heart by their release of a variety of enzymes and other factors.4,5 However, in recent years, there has been an effort to maximize stem cell recruitment to speed repair of injured myocardium. Investigators have focused on the use of a chemokine, CXCL12 (also known as stromal cell–derived factor-1), which is the main functional ligand for CXCR4, as a new therapeutic modality. However, to date, several human clinical trials of stem cell therapy have shown limited cardiac benefit to individuals suffering from MI.6
Chemokines are a super family of low-molecular-weight proteins (8 to 10 kDa) that have been subdivided into families on the basis of the position of their cysteine residues. There are currently 46 ligands that bind to 18 G protein–coupled receptors.7 Although chemokines are key regulators of leukocyte migration and host defense pathways, excessive production of chemokines has been implicated in the inflammatory components of numerous diseases including chronic heart failure and ischemia-induced myocardial injury.8,9,10
CXCR4 is a 37-kDa G protein–coupled receptor, located principally at the plasmalemma of cardiac myocytes. CXCL12, the predominant CXCR4 ligand, is constitutively expressed in myocardium, detectable in the serum, and is increased early post myocardial infarction.11 Expression of CXCR4 is increased in the myocardium of patients with heart failure.9 Both CXCR4 and CXCL12 are expressed and functional in cardiomyocyte.12 Whereas the significance of CXCR4 and CXCL12 in cardiac development has already been established,13,14 there is a paucity of information regarding chemokine receptor signaling on adult cardiac myocytes. It was recently demonstrated that activation of CXCR4 results in a direct negative inotropic modulation of cardiac myocyte function in vitro implicating an important role for CXCR4 and its ligand, CXCL12, in heart function.15 In the present study, we explored the effects of CXCR4 gene transfer in a rat cardiac ischemia-reperfusion (IR) injury model. We hypothesized that overexpression of CXCR4 in the heart will increase the influx of inflammatory cells into ischemic tissue, enhance host response to oxygen free radicals, and impair the cardiac pump function. In support of this hypothesis, we examined the mechanistic role of CXCR4 in the pathophysiology of myocardial ischemic injury in vivo. Our data demonstrated that CXCR4 overexpression exacerbates the hemodynamic dysfunction and structural deterioration in a rat model of ischemic reperfusion injury. CXCR4 gene transfer increased the infarct size and decreased the cardiac pump function. Collectively, we show that overexpression of CXCR4 plays a potentially critical role dictating the outcome of ischemic reperfusion injury.
Materials and Methods
Recombinant Adenoviral Vectors
Recombinant adenoviruses were used in these studies. Ad-EGFP-β-gal contains cytomegalovirus promoter, expression cassettes for β-galactosidase (β-gal), and enhanced green fluorescent protein (EGFP).16 The backbone vector, which contains most of the adenoviral genome (pAd.EASY1), was used and the recombination performed in Escherichia coli. CXCR4 cDNA was subcloned into the adenoviral shuttle vector (pAd.TRACK), using the cytomegalovirus long-terminal repeat as a promoter. pAd.TRACK also has a concomitantly expressed green fluorescent protein (GFP) under the control of a separate cytomegalovirus promoter.15 Ad-β-gal/GFP and Ad-CXCR4 virus were propagated in HEK293 cells. Viral titer was determined by plaque assay. Stock titers were ≈1010 pfu/ml for each vector with a particle/pfu ratio of approximately ≈102. Contamination with wild-type adenovirus was ruled out by the absence of real time PCR-detectable E1 sequences.
Gene Transfer
Male Sprague-Dawley (SD) (Charles River Laboratories, Wilmington, MA) rats were anesthetized with pentobarbital (60 mg/kg) i.p. and placed on a ventilator. The chest was accessed from the right side through the third intercostal space. The pericardium was opened and a 7-0 suture was placed at the apex of the left ventricle. A 22G catheter containing 200 μl of adenovirus was advanced from the apex of the left ventricle (LV) to the aortic root. The aorta and pulmonary artery were clamped distal to the site of the catheter and the solution was injected. The clamp was held for 10s while the heart pumped against a closed system (isovolumically). The solution containing the adenovirus circulated through the coronary arteries without further manipulation, as described previously.16 All procedures were approved by and performed in accordance with the Institutional Animal Care and Use Committee of the Mount Sinai School of Medicine. The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Ischemia-Reperfusion in Rats and Echocardiographic Measurements
One week after adenoviruses (Ad-) administration, a left thoracotomy was performed, and the left anterior descending coronary artery (LAD) was ligated with 6-0 silk suture ≈4 mm from its origin with a slipknot. Successful ligation of the LAD was verified by visual inspection of the left ventricular apex. After 30 minutes, the LAD ligature was released and reperfusion was visually confirmed.17 The chest was closed with continuous 2-0 silk suture. After 24 hours reperfusion, rats were re-anesthetized with 50 mg/kg of Ketamine and echocardiograms were performed using a VIVID 7 general electric device with a 14-MHZ probe as previously described.18 LV cavity size, wall motion, fractional shortening, and wall thickening were evaluated on M-mode echocardiography. They were measured from a short-axis view at the level of the papillary muscles. In addition to animals receiving Ad-CXCR4, controls included: 1) Blank control with no IR (Blank), 2) Saline-injected control group who had IR (Control), and 3) Ad–β-gal–injected animals who were also exposed to IR (β-gal).
Evaluation of Risk Area and Infarct Size
Histological analysis of infarct size was performed by 2,3,5-Triphenyltetrazolium chloride (TTC) staining. Briefly, after echocardiogranphic measurements, heparin (1000 u/ml) 0.4 ml was injected intravenously. The hearts were excised and mounted on a Langendorff apparatus and perfused for 3 minutes under constant pressure (100 cmH20) with K-H buffer at room temperature. The LAD was re-occluded. The hearts were perfused with 5% solution of Phthalo blue dye in normal saline over 3 minutes. The right ventricle of each heart was excised. The hearts were then frozen at −20°C for 20 minutes, followed by transverse sectioning into 2-mm slices. Sections were then incubated in 15 ml of 1.5% TTC for 20 minutes at 37°C. The sections were fixed in 10% formaldehyde. Twenty-four hours later, the slices were weighed and photographed. Color digital images of each transverse slice were obtained using a digital camera (Canon 640A). The blue regions represents nonischemic normal tissue, red regions represents risk area (ischemic but noninfarcted), and unstained pale white regions represent infarct tissue. The blue, red, and white areas were outlined on each color image and measured using Image Plus. On each side, the fraction of the LV area representing infarct-related tissue (average of 2 images) was multiplied by the weight of the section to determine the absolute weight of infarct-related tissue.17,19
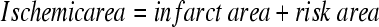 |
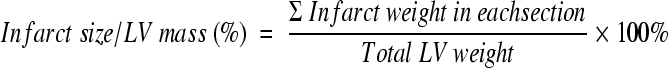 |
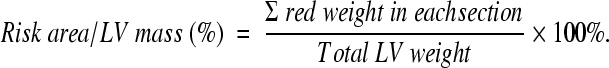 |
Evaluation of Gene Transfer Method Using X-Gal Staining
After gene transfer and ischemia/reperfusion, the hearts were harvested, cut across the suture, and embedded in OCT. The tissues were cryosectioned (5- to 10-μm sections). Subsequently, β-gal/lacZ gene expression was determined as previously described.16
Histological Assessment
Frozen tissue blocks from the above hearts were cut into 5- to 10-μm sections, and parallel sections were prepared for hematoxylin and eosin staining and immunohistochemistry. Immunohistochemical analyses included staining for CXCL12, CXCR4, and tumor necrosis factor (TNF)-α, as well as for macrophages and pan leukocytes markers. The following antibodies were used: (1) rabbit anti-mouse/rat CXCL12 (eBiosciences; San Diego, CA); (2) goat anti-rat CXCR4 antibody (Torrey Pines Biolabs; East Orange, NJ); (3) goat anti-mouse/rat TNF- α, (R&D), (4) mouse anti-rat leukocyte antibody (BD Pharmingen; Franklin Lakes, NJ); and (5) mouse anti- rat macrophage marker (VP-M640, Vector Laboratories; Burlingame, CA) Sections from the hearts were fixed and blocked with the normal serum and incubated with primary antibody for 2 hours at room temperature. For visualization, slides were developed using either the Vectastain ABC kit with DAB as a substrate,20 or with secondary fluorescent conjugated antibodies; with using a BP530/30 filter for FITC and/or a LP590 filter for Texas red. Images were taken with Axioplan2IE or Zeiss LSM510 META confocal microscope.
Western Blot Analysis
Rats’ hearts were transected along the LAD ligature to separate ischemic tissue and remote myocardium. The ischemic and remote tissues were lysed in lyses buffer. Western blot analysis was performed using procedures established in our laboratory. For these studies the following primary polyclonal antibodies were used: (1) anti–CXCR4 (Torrey Pines Biolabs; East Orange, NJ); (2) anti-CXCL12 (eBiosciences; San Diego, CA); (3) anti-Caspase 3 (Cell Signaling; Danvers, MA); or (4) anti-Glyceraldehyde 3-phosphate dehydrogenase (anti-GAPDH, IMgenex; San diego, CA). A secondary antibody conjugated with alkaline phosphatase (Sigma; St. Louis, MO). The bands were visualized using the enhanced chemiluminescence method according to the manufacturer’s instructions (Pierce; Rockford, IL).21,22
Inflammatory Cells Count
For each of the hearts examined, the number of inflammatory cells was quantified in the infarcted zone (LV anterior wall), in a blinded manner. Briefly, frozen tissue blocks were cut into 5- to 10-μm sections, stained with hematoxylin and eosin, and examined by light microscopy (×10, ×40, and ×63 objective). For each section, the number of inflammatory cells per mm2 was quantified.17 Three sections were assessed per heart. Ten fields were randomly chosen per section, four section per animal, a total of 40 fields per heart (n = 3 per treatment group) were counted using a defined rectangular field area (×40 objective). The number of monocytes and macrophages was also counted under high-power field (×40) using monocyte/macrophage specific antibody (VP-M640, Vector Laboratories; Burlingame, CA).
Morphological Examination of Apoptosis
For each of the hearts examined, myocardial cell apoptosis was quantified using a commercially available in situ cell death detection kit (Roche Applied Science, Indianapolis, IN) to find DNA strand breaks using the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) reagent according to the manufacturer’s protocol. Briefly, a double-staining technique was used (ie, TUNEL staining by using an In Situ Cell Death Detection Kit [Roche, USA] for apoptotic cell nuclei and 4′,6-diamidino-2-phenylindole [DAPI] staining for all cell nuclei). An additional staining was performed with a monoclonal antibody against α-sarcomeric actinin (Sigma; St. Louis, MO) for the identification of myocytes. Cardiomyocytes from at least four slides per block were selected and evaluated immunohistochemically to determine the number and percentage of cells exhibiting positive staining for apoptosis. For each slide, 10 fields were randomly chosen, and a total of 100 to 150 myocytes per field were counted using a defined rectangular field area (×20 and ×40 objective). Apoptotic index was determined (number of TUNEL-positive myocytes/total number of myocytes stained with anti–α-actinin × 100) from a total of 40 fields per heart. Assays were performed in a blinded manner.
Quantitative RT-PCR
CXCL12 mRNA levels were determined by QRT-PCR using a QuantiTect SYBR Green RT-PCR Kit (Qiagen Ltd; Valencia CA) on a LightCycler (Roche Diagnostics Ltd., Lewes, UK). Total RNA was isolated from samples with Trizol reagent (Gibco BRL; Carlsbad, CA) according to the manufacturer’s instructions. Primers were designed to generate short amplification products. The sequences of the specific primers were: CXCL12 5′-TTGCCAGCACAAAGACACTCC-3′ and 5′-CTCCAAAGCAAACCGAATACAG-3′; TNF-α 5′-CTCTGCTTGGTGGTTTGCTA-3′ and 5′-CAAGGAGGAGAAGTTCCCAA-3′ 18S: 5′-GTTGGTTTTCGGAACTGAGGC-3′ and 5′- GTCGGCATCGTTTATGGTCG-3′. RT-PCR was performed in 20-μl reaction volumes using 10 pmol of primers. Reverse transcription was performed at 50°C for 20 minutes, and cDNA was amplified for 37 cycles: 94°C for 10 s, 57°C for 15 s, and 72°C for 5 seconds. The relative quantity of gene expression was calculated according to the manufacturer’s recommendations. 18S was used as an internal control to calculate the relative abundance of CXCL12 and TNF- α mRNAs.
Statistics
All data are expressed as SD (±SD). Differences between groups were determined with either a paired t test and/or two-tailed Student t test and/or a one-way analysis of variance followed by a Newman–Keuls post hoc test. Probability values of P < 0.05 were considered to be significant.
Results
Myocardial CXCR4-Overexpression in Rats Exhibit Larger Infarcts and Worsening Cardiac Function After Ischemia-Reperfusion in Vivo
MI in rats was induced by LAD ligation (30 minutes followed by 24 hours reperfusion). Histological analysis of infarct size was performed by TTC staining. Figure 1, A–D shows that the infarct area as a percentage of LV was significantly increased in CXCR4 group (13.5% ± 4.2%) in comparison with β-gal/GFP group (7.6% ± 3.4%) and saline group (control) (8.0% ± 2.0%). Although the risk area/LV was slightly increased in CXCR4 group, there was no significant statistical difference when it was compared with β-gal/GFP and saline groups. But the infarct/ischemia ratio was significantly increased in CXCR4 group (46% ± 7%) as compared with 36% ± 11% in β-gal/GFP group or 39% ± 8% in the saline group. The infarct/risk ratio was also significantly increased in CXCR4 group (89% ± 30%) as compared with β-gal/GFP control (60% ± 28%, P = 0.02). Cardiac function was also measured by echocardiography. Figure 2A illustrates that movement of the LV anterior wall was diminished in CXCR4 group. The left ventricle internal diastolic/systolic diameter and end diastolic/systolic volume were increased in CXCR4 IR group. Factional shortening was also decreased significantly in CXCR4 group as compared with β-gal/GFP and saline groups (control); (Figure 2B; see Supplemental Table S2, at http://ajp.amjpathol.org). Cardiac CXCR4 overexpression did not have any effect on myocardial function of noninfarcted rats (no IR); (Figure 2C; see Supplemental Table S253 at http://ajp.amjpathol.org).
Figure 1.

CXCR4-overexpressed rats exhibit larger infarcts after ischemia-reperfusion in vivo. One week after CXCR4 gene transfer, myocardial infarction was induced by LAD ligation (30 minutes followed by 24 hours reperfusion) in rats in vivo. A: Histological analysis of infarct size was performed by TTC staining, white = infarct area; red = risk area; blue = normal area. B–D: Infarct size as a percentage of risk area (B) and evaluation of risk area and infarct size: quantitative assessment (mean ± SD) of risk area as a percentage of LV (C) for control groups (β-gal and control) and the CXCR4-overexpressed group. Although risk areas are similar for all groups, infarct sizes as a percentage of risk area are statistically different and are larger in CXCR4 group. Infarct data: infarct/ischemic area = infarct area/(infarct area + risk area) × 100. P > 0.05 in risk area, *P < 0.05 in infarct and ischemic area, compared with β-gal and control, **P < 0.05 compared with β-gal control. Mean ± SD.
Figure 2.

CXCR4-overexpressed rats exhibit worsening cardiac function after ischemia-reperfusion in vivo. A: Echocardiography was performed on rats’ hearts post 30 minutes ischemia and 24 hours reperfusion. Short axis. Left, blank control without IR; Middle, β-gal gene transfer with IR; Right, CXCR4 gene transfer with IR. B: Percentage of fractional shortening (%) was calculated: *P < 0.05 compared with blank, **P < 0.05 compared with β-gal and control. Mean ± SD. C: Echocardiography was performed on rats’ hearts before Ad-CXCR4 gene transfer and one week after injection in an absence of any ischemic injury.
Myocardial CXCR4-Overexpression Augments CXCL12 Expression in Ischemic Myocardium in the Rat
Gene transfer was assessed by quantifying expressions of β-gal and CXCR4. β-gal and CXCR4 proteins expression were overexpressed in ischemic myocardium after gene transfer (Figure 3, A–D). These results were confirmed by Western blotting (Figure 3E). The CXCR4-overexpressed group exhibited increased CXCR4 protein expression in ischemic myocardium as compared with β-gal and control (saline injected) groups. Next, we assessed whether changes in CXCR4 expression could effect concomitant expression of its ligand, CXCL12. Interestingly, we found a marked increased expression of CXCL12 in hearts injected with Ad-CXCR4 and exposed to IR as compared with β-gal and control groups as were assessed by immunofluoresence staining (Figure 4A), Western blot (Figure 4B), and quantitative real-time PCR (Figure 4C).
Figure 3.

CXCR4 protein expression on myocardium after one week gene transfer and ischemia 30 minutes followed by 24 hours reperfusion. A: X-gal staining for β-gal expression, blue color is positive, (B) H-E staining, (C–D) immunostaining (DAB) for CXCR4 protein expression. C: CXCR4 group. D: β- gal control. E: CXCR4-overexpressed rats exhibit increased CXCR4 protein expression in ischemic myocardium after IR in vivo. Protein lysates were prepared from rats’ hearts one week after CXCR4 gene transfer followed by 30 minutes LAD ligation and 24 hours reperfusion. The ischemic and remote tissues were separated, lysed, and analyzed by Western blot. CXCR4 Mr; 40 to 47 kDa. Representative gel of three independent experiments is shown. Densitometric analysis of data from three different experiments is shown. *P < 0.05.
Figure 4.

CXCL12 is upregulated in ischemic myocardium of CXCR4-overexpressed rat. A: CXCL12 protein expression was assessed in hearts injected with either CXCR4 or β-gal and/or control (saline-injected; with IR) by immunofluorescence staining. Frozen sections were fixed and stained with anti-CXCL12 (green) and anti–α actinin (red). Primary Abs visualized with FITC or Texas Red conjugate. Nuclei were stained with DAPI. Images are taken with confocal microscopy. B: Protein lysates were prepared from rats’ hearts one week after CXCR4 gene transfer followed by 30 minutes LAD ligation and 24 hours reperfusion. The ischemic and remote tissues were lysed and analyzed by Western blot. Representative gel of three independent experiments is shown. C: CXCL12 mRNA levels were determined by quantitative real-time PCR (QRT-PCR) using a QuantiTect SYBR Green RT-PCR Kit and using specific primers for CXCL12 and 18S. Primers were designed to generate short amplification products. Densitometric analysis of data from three different experiments is shown. *P < 0.05.
CXCR4-Overexpression Leads to a Significant Increase in Inflammatory Cell Infiltration after Ischemia-Reperfusion in Vivo
Because inflammatory cells are the important modulators of ischemic injury, we assessed the presence of inflammatory cells in rats overexpressing CXCR4 in the heart. Our data demonstrate that hearts injected with CXCR4 and exposed to IR have significantly more inflammatory cellular infiltrate in the ischemic region (Figure 5A). The number of inflammatory cells was quantified per mm2 (Figure 5B). Importantly, there were no significant differences in monocytes/macrophages migrating into the ischemic area of CXCR4, β-gal, and saline injected groups (Figure 5, C and D). The number of monocytes and macrophages was counted in high power fields (×40) using cell type–specific mouse anti-rat macrophage marker (VP-M640) antibody. Importantly, CXCR4 overexpression did not seem to mediate inflammatory reactions in the absence of IR (see Supplemental Figure S1, at http://ajp.amjpathol.org).
Figure 5.

CXCR4-overexpressed rats exhibit increased leukocyte counts in ischemic myocardium after IR in vivo. A: Frozen sections from hearts either injected with Ad-CXCR4, or β-gal and/or control (saline injected) hearts (with IR) stained with hematoxylin and eosin, and examined by light microscopy at ×10 and ×40 objectives for the presence of inflammatory cells. B: The number of inflammatory cells was quantified per mm2. Inflammatory infiltrate is significantly increased in CXCR4-overexpressed hearts (*P < 0.05). C and D: The number of monocytes and macrophages were also assessed by DAB staining using cell specific antibody (VP-M640). Number of monocyte/macrophages were counted in high-power field (HPF; ×40), and average cell count was graphed (n = 3).
CXCR4-Overexpressed Rats Exhibit Significant Increase in Cell Death/Apoptosis After Ischemia-Reperfusion in Vivo
Cardiomyocyte necrosis and apoptosis play important roles in the pathology of ischemic/reperfusion injury. To determine myocardial apoptosis, in situ detection of apoptotic myocytes was performed by using TUNEL assay. Positive apoptotic nuclei were stained green in infarcted myocardial tissue, and healthy nuclei were stained blue (DAPI staining). In Figure 6A we show there are significantly higher number of TUNEL-positive cells in CXCR4 group in comparison with controls (β-gal–injected and/or saline-injected). Representative pictures were taken at ×40 objective. The number of TUNEL-positive myocytes/total number of myocytes is presented as a bar graph of apoptotic index (Figure 6B). To further corroborate our TUNEL data, we assessed the activation of caspase 3, an established member of the caspase family known to be involved in the final execution phase of apoptosis. Caspase-3 activation has also been implicated in ischemic reperfusion injuries.23 In Figure 6C, we present representative Western blot data showing a marked increase in caspase 3 activity in the CXCR4 group (Figure 6C). Becasue inflammatory cytokines such as TNF-α have been implicated in inflammatory-mediated ischemic injury, we next evaluated TNF-α expression using immunofluorescence and QRT-PCR assay. Representative analyses are presented in Figure 6, D and E. There was a significant increase in TNF-α expression at the level of protein and mRNA in ischemic area in the CXCR4 group as compared with controls.
Figure 6.

CXCR4-overexpressed rats exhibit significant increase in cell death/apoptosis after ischemia-reperfusion in vivo. A: Increased cardiomyocyte susceptibility to ischemic injury was examined by assessing apoptosis using TUNEL assay. A double-staining technique was used (ie, TUNEL staining by using an In Situ Cell Death Detection Kit [Roche, USA] for apoptotic cell nuclei and DAPI staining for all cell nuclei. Representative images are shown. B: Apoptotic index was determined (ie, number of TUNEL-positive myocytes/total number of myocytes stained with anti-α-actinin × 100) from a total of 40 fields per heart. Assays were performed in a blinded manner. C: Increased cardiomyocyte susceptibility to ischemic injury was examined by assessing apoptosis using Western blot analysis of caspase 3 activation. Densitometric analysis of data from three different experiments is shown. *P < 0.05. D and E: The expression of inflammatory cytokine TNF-α was assessed in those hearts using immunofluorscence and QRT-PCR assay using a QuantiTect SYBR Green RT-PCR Kit. Specific primers for TNF-α and 18S were used. Primers were designed to generate short amplification products. Densitometric analysis of data from three different experiments is shown. *P < 0.05.
Discussion
The role of inflammation in the development, progression, and destabilization of coronary artery disease is becoming increasingly recognized.24,25,26 Elevated peripheral levels of cytokines have been demonstrated in patients with CHF and are associated with a worse prognosis. Recent studies have indicated that IR increases the production and release of proinflammatory cytokines such as TNF-α, which can play a key role in causing contractile dysfunction, apoptosis, and remodeling in heart.27,28,29 TNF-α is also implicated in recruitment of lymphocytes in inflammation mediated injury.30,31 Inflammation is a complex biological response. Thus, gaining a better understanding of its role in cardiac injury, in particular during reperfusion after myocardial ischemia, is important. The viable “border zone” appears to have the highest density of inflammatory cells such as neutrophils and lymphocytes.32,33 The presence of contraction bands and an inflammatory infiltrate characterized by intense margination in the small veins is a hallmark of early reperfusion.33,34,35 Administration of granulocyte-colony stimulating factor (G-CSF) after MI improves LV function and reduces LV remodeling and infarct size.36,37 These beneficial effects of G-CSF were entirely abolished by the specific CXCR4 antagonist AMD3100. These results suggested that the CXCR4/CXCL12 axis plays a critical role in the beneficial effects of G-CSF via recruitment of CXCR4+ progenitor cells into the infarcted myocardium.36,37 However, we have also demonstrated that activation of CXCR4 decreased papillary muscle and cardiac myocyte contractility in response to Ca2+ and isoproterenol in vitro.15 The negative inotropic effects on the heart may benefit cardiomyocyte survival and preserve heart function in ischemia and reperfusion injury.38 Given the previous emphasis on the role of CXCR4 mediated migration of hematopoietic progenitor cells in the heart, we wished to determine the effect of increased expression of CXCR4 in the heart and IR-related injury. We have previously reported that the catheter-based adenoviral technique induces global gene transfer in rat hearts with an expression pattern that is grossly homogeneous throughout the ventricles.16,39 Furthermore, the cardiomyocytes isolated from these rat hearts after in vivo transduction with Ad.β-Gal demonstrate specific nuclear β-Gal activity,39 reflecting transgene expression in these myocytes. In this report, we have also used catheter-based adenoviral technique to induce CXCR4 gene transfer in rat hearts and used a model of myocardial IR injury to determine the effects of cardiac CXCR4 function. We have show that CXCR4 overexpression by gene transfer with adenovirus have increased myocardial infarct size and impaired cardiac pump function. The presence of inflammatory cells was increased in CXCR4 gene transfer hearts post ischemia and reperfusion, suggesting that upregulation of CXCR4 may worsen IR injury.
Previous studies by Rokosh et al have shown that CXCL12 administration before coronary occlusion followed by 4 hours of reperfusion resulted in decreased infarct size, and they concluded that CXCL12 pretreatment promoted an antiapoptotic program that confers protection against ischemia/reperfusion damage.40 There are some fundamental differences between their model and ours that might explain the contrasting results. First, in our model hearts undergo reperfusion over 24 hours. During this time internal proinflammatory cytokines and chemokines can accumulate at the site of the injury. Chemokine-mediated chemotaxis of neutrophils is known to be important in the pathogenesis of ischemia-reperfusion injury.41 The actions of CXCR4 in myocardial infarction may not be limited to leukocyte chemotaxis and might have a direct myocardial cytoprotection as was suggested by Hu et al.40 However, this mechanism could be likely masked by the recruitment of inflammatory cells as seen in our model. Efficient delivery of proteins is challenged by barriers including the rapid diffusion of chemokines and their degradation by proteases activated in injured tissues. CXCL12 is cleaved by matrix metalloproteinase−2.42 This proteolytic activity will likely limit the effectiveness of CXCL12 within the inflammatory environment of infarcted myocardium. Therefore, injection of CXC4 by adenoviral delivery provides a different molecular mechanism of action.
In our studies, we demonstrated that there were no significant changes in cardiac function following CXCR4 gene transfer in an absence of ischemic reperfusion injury (no IR). However, after IR there were significant morphological and functional defects in CXCR4-overexpressed hearts. CXCR4-overexpressed rats exhibited larger ischemic injury. This was also confirmed with significant decline in their cardiac function. To demonstrate the underlying mechanisms, first we assessed whether myocardium CXCL12 production is altered as a result of overexpression of its receptor CXCR4. CXCL12 is the main ligand for CXCR4, and it is a critical chemokine modulating T cell migration. We found a significant increase in expression of CXCL12 in hearts injected with Ad-CXCR4 and exposed to IR. The CXCR4 group was also distinguished by a marked accumulation of inflammatory cells in the myocardium. This could be in part attributable to higher CXCL12 expression. CXCL12 overexpression could lead to more T-cell recruitment. There is also evidence in the literature that in ischemic injury T-lymphocytes mediate the ischemia/reperfusion inflammatory response by stimulating neutrophil migration. T-cells are suggested to play a role in initiating IR-induced inflammatory responses.43 Additionally, we did not see significant differences in the number of monocytes/macrophages. This also is in agreement with the literature because monocytes and macrophages are involved in the chronic stages of IR.44 Finally, CXCR4 overexpression in an absence of IR did not seem to mediate inflammatory responses, which might be attributable to lack of CXCL12 overexpression in an absence of IR. In this model CXCL12 expression seems to be playing an important role in enhancing the IR injury.
It is now generally accepted that extensive programmed cell death occurs not only during cardiac ischemia but also during reperfusion after an ischemic episode (reviewed in 45). Here we examined the mechanism(s) through which CXCR4/CXCL12 upregulation in the infarct area induces apoptosis. Increased cardiomyocyte susceptibility to ischemic injury was examined by assessing 1) apoptosis using TUNEL assay, and 2) measuring inflammatory cytokines such as TNF-α in CXCR4 overexpressed heart. Recent studies indicating that CXCR4 activation induces apoptosis in the CD4+ Jurkat T cells through the death receptor pathway.46 Next, we examined the effect of CXCR4 activation and overexpression on inflammatory mediators (eg, TNF-α) implicated in ischemic reperfusion injury. TNF-α could interact directly with cardiac myocytes resulting in activation of apoptotic pathways or indirectly by acting on migrating leukocytes to exacerbate myocardial injury. TNF-α is known activator of the cell death pathway, and its contribution to ischemic injury has been studied extensively.47,31 We observed a significant increase in TNF-α production in our IR injury model thus contributing to the underlying mechanisms of increased susceptibility to ischemic injury in heart overexpressing CXCR4. TNF-α is also implicated in recruitment of lymphocytes in inflammation mediated injury.30 TNF-α upregulation not only increases apoptosis but also might be contributing to an increased inflammatory infiltrate on ischemic injury. Interestingly, CXCL12 has been implicated in induction of TNF-α production in the context of astrocyte cell death.48 In our IR model, hearts infected with Ad-CXCR4 showed both an increase in TNF-α expression and in TUNEL-positive cells post IR. TNF-α is also implicated to inhibit CXCL12 production,49 therefore upregulation of TNF-α in addition to promoting cell death may act via a negative feedback loop to control CXCL12. In this model there is a significant amount of caspase 3 activation accompanying delivery of Ad-CXCR4, thus it is possible that activation of apoptotic pathways is in part attributable to TNF-α production or that CXCL12 has apoptotic effects independent of TNF-α. Further clarification awaits additional experiments.
Collectively, in this study we have demonstrated that overexpression of CXCR4 by gene transfer increased myocardial infarct size and worsened cardiac function in rats post-IR injury. Leukocytes attracted by CXCL12, the ligand for CXCR4, may impact ischemic reperfusion injury. Myocardium overexpressing CXCR4 exhibit significantly increased cell death/apoptosis after IR in vivo. These results suggest that the net effect of CXCR4 overexpression in the heart of an intact animal during MI is enhancement of cardiomyocyte injury. As noted in this model, this is likely attributable to enhanced recruitment of inflammatory cells, increased TNF-α production, and cell death/apoptosis.
Footnotes
Address reprint requests to Dr. Sima T. Tarzami or Dr. Roger J. Hajjar, Mount Sinai School of Medicine, 1 Gustave L Levy Place, Box 1030, New York, NY 10029. E-mail: sima.tarzami@mssm.edu or roger.hajjar@mssm.edu.
Supported in part by the American Heart Association (grant AHA0735576T to S.T.T.) and the National Institutes of Health (grant R01 HL050000 to R.J.H.).
R.J.H. and S.T.T. contributed equally to this work.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Tissier R, Berdeaux A, Ghaleh B, Couvreur N, Krieg T, Cohen MV, Downey JM. Making the heart resistant to infarction: how can we further decrease infarct size? Front Biosci. 2008;13:284–301. doi: 10.2741/2679. [DOI] [PubMed] [Google Scholar]
- Yellon DM, Baxter GF. Protecting the ischaemic and reperfused myocardium in acute myocardial infarction: distant dream or near reality? Heart. 2000;83:381–387. doi: 10.1136/heart.83.4.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carden DL, Granger DN. Pathophysiology of ischaemia-reperfusion injury. J Pathol. 2000;190:255–266. doi: 10.1002/(SICI)1096-9896(200002)190:3<255::AID-PATH526>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Vermeiren GL, Claeys MJ, Van Bockstaele D, Grobben B, Slegers H, Bossaert L, Jorens PG. Reperfusion injury after focal myocardial ischaemia: polymorphonuclear leukocyte activation and its clinical implications. Resuscitation. 2000;45:35–61. doi: 10.1016/s0300-9572(00)00168-4. [DOI] [PubMed] [Google Scholar]
- Gao L, Yin H, Smith S RJ, Chao L, Chao J. Role of kallistatin in prevention of cardiac remodeling after chronic myocardial infarction. Lab Invest. 2008;88:1157–1166. doi: 10.1038/labinvest.2008.85. [DOI] [PubMed] [Google Scholar]
- Abbott JD, Huang Y, Liu D, Hickey R, Krause DS, Giordano FJ. Stromal cell-derived factor-1alpha plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation. 2004;110:3300–3305. doi: 10.1161/01.CIR.0000147780.30124.CF. [DOI] [PubMed] [Google Scholar]
- Luster AD. Chemokines–chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- Pease JE, Williams TJ. The attraction of chemokines as a target for specific anti-inflammatory therapy. Br J Pharmacol. 2006;147 Suppl 1:S212–S21. doi: 10.1038/sj.bjp.0706475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damas JK, Eiken HG, Oie E, Bjerkeli V, Yndestad A, Ueland T, Tonnessen T, Geiran OR, Aass H, Simonsen S, Christensen G, Froland SS, Attramadal H, Gullestad L, Aukrust P. Myocardial expression of CC- and CXC-chemokines and their receptors in human end-stage heart failure. Cardiovasc Res. 2000;47:778–787. doi: 10.1016/s0008-6363(00)00142-5. [DOI] [PubMed] [Google Scholar]
- Sasayama S, Okada M, Matsumori A. Chemokines and cardiovascular diseases. Cardiovasc Res. 2000;45:267–269. doi: 10.1016/s0008-6363(99)00381-8. [DOI] [PubMed] [Google Scholar]
- Ma J, Ge J, Zhang S, Sun A, Shen J, Chen L, Wang K, Zou Y. Time course of myocardial stromal cell-derived factor 1 expression and beneficial effects of intravenously administered bone marrow stem cells in rats with experimental myocardial infarction. Basic Res Cardiol. 2005;100:217–223. doi: 10.1007/s00395-005-0521-z. [DOI] [PubMed] [Google Scholar]
- Segret A, Rucker-Martin C, Pavoine C, Flavigny J, Deroubaix E, Chatel MA, Lombet A, Renaud JF. Structural localization and expression of CXCL12 and CXCR4 in rat heart and isolated cardiac myocytes. J Histochem Cytochem. 2007;55:141–150. doi: 10.1369/jhc.6A7050.2006. [DOI] [PubMed] [Google Scholar]
- Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T, Bronson RT, Springer TA. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci USA. 1998;95:9448–9453. doi: 10.1073/pnas.95.16.9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- Pyo RT, Sui J, Dhume A, Palomeque J, Blaxall BC, Diaz G, Tunstead J, Logothetis DE, Hajjar RJ, Schecter AD. CXCR4 modulates contractility in adult cardiac myocytes. J Mol Cell Cardiol. 2006;41:834–844. doi: 10.1016/j.yjmcc.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar RJ, Schmidt U, Matsui T, Guerrero JL, Lee KH, Gwathmey JK, Dec GW, Semigran MJ, Rosenzweig A. Modulation of ventricular function through gene transfer in vivo. Proc Natl Acad Sci USA. 1998;95:5251–5256. doi: 10.1073/pnas.95.9.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarzami ST, Miao W, Mani K, Lopez L, Factor SM, Berman JW, Kitsis RN. Opposing effects mediated by the chemokine receptor CXCR2 on myocardial ischemia-reperfusion injury: recruitment of potentially damaging neutrophils and direct myocardial protection. Circulation. 2003;108:2387–2392. doi: 10.1161/01.CIR.0000093192.72099.9A. [DOI] [PubMed] [Google Scholar]
- del Monte F, Lebeche D, Guerrero JL, Tsuji T, Doye AA, Gwathmey JK, Hajjar RJ. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc Natl Acad Sci USA. 2004;101:5622–5627. doi: 10.1073/pnas.0305778101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu BQ, Simonis U, Cecchini G, Zhou HZ, Li L, Teerlink JR, Karliner JS. Comparison of pyrroloquinoline quinone and/or metoprolol on myocardial infarct size and mitochondrial damage in a rat model of ischemia/reperfusion injury. J Cardiovasc Pharmacol Ther. 2006;11:119–128. doi: 10.1177/1074248406288757. [DOI] [PubMed] [Google Scholar]
- Chen J, Kuhlencordt P, Urano F, Ichinose H, Astern J, Huang PL. Effects of chronic treatment with L-arginine on atherosclerosis in apoE knockout and apoE/inducible NO synthase double-knockout mice. Arterioscler Thromb Vasc Biol. 2003;23:97–103. doi: 10.1161/01.atv.0000040223.74255.5a. [DOI] [PubMed] [Google Scholar]
- Tarzami ST, Cheng R, Miao W, Kitsis RN, Berman JW. Chemokine expression in myocardial ischemia: mIP-2 dependent MCP-1 expression protects cardiomyocytes from cell death. J Mol Cell Cardiol. 2002;34:209–221. doi: 10.1006/jmcc.2001.1503. [DOI] [PubMed] [Google Scholar]
- Chen J, Tung CH, Mahmood U, Ntziachristos V, Gyurko R, Fishman MC, Huang PL, Weissleder R. In vivo imaging of proteolytic activity in atherosclerosis. Circulation. 2002;105:2766–2771. doi: 10.1161/01.cir.0000017860.20619.23. [DOI] [PubMed] [Google Scholar]
- Zhan RZ, Wu C, Fujihara H, Taga K, Qi S, Naito M, Shimoji K. Both caspase-dependent and caspase-independent pathways may be involved in hippocampal CA1 neuronal death because of loss of cytochrome c From mitochondria in a rat forebrain ischemia model. J Cereb Blood Flow Metab. 2001;21:529–540. doi: 10.1097/00004647-200105000-00007. [DOI] [PubMed] [Google Scholar]
- Yndestad A, Damas JK, Oie E, Ueland T, Gullestad L, Aukrust P. Role of inflammation in the progression of heart failure. Curr Cardiol Rep. 2007;9:236–241. doi: 10.1007/BF02938356. [DOI] [PubMed] [Google Scholar]
- Mehra VC, Ramgolam VS, Bender JR. Cytokines and cardiovascular disease. J Leukoc Biol. 2005;78:805–818. doi: 10.1189/jlb.0405182. [DOI] [PubMed] [Google Scholar]
- Blum A, Miller H. Pathophysiological role of cytokines in congestive heart failure. Annu Rev Med. 2001;52:15–27. doi: 10.1146/annurev.med.52.1.15. [DOI] [PubMed] [Google Scholar]
- Cain BS, Meldrum DR, Dinarello CA, Meng X, Banerjee A, Harken AH. Adenosine reduces cardiac TNF-alpha production and human myocardial injury following ischemia-reperfusion. J Surg Res. 1998;76:117–123. doi: 10.1006/jsre.1998.5304. [DOI] [PubMed] [Google Scholar]
- Krown KA, Page MT, Nguyen C, Zechner D, Gutierrez V, Comstock KL, Glembotski CC, Quintana PJ, Sabbadini RA. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest. 1996;98:2854–2865. doi: 10.1172/JCI119114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94:1543–1553. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- Issekutz TB, Stoltz JM. Stimulation of lymphocyte migration by endotoxin, tumor necrosis factor, and interferon. Cell Immunol. 1989;120:165–173. doi: 10.1016/0008-8749(89)90184-6. [DOI] [PubMed] [Google Scholar]
- Issekutz TB. Effects of six different cytokines on lymphocyte adherence to microvascular endothelium and in vivo lymphocyte migration in the rat. J Immunol. 1990;144:2140–2146. [PubMed] [Google Scholar]
- Youker KA, Hawkins HK, Kukielka GL, Perrard JL, Michael LH, Ballantyne CM, Smith CW, Entman ML. Molecular evidence for a border zone vulnerable to inflammatory reperfusion injury. Trans Assoc Am Physicians. 1993;106:145–154. [PubMed] [Google Scholar]
- Youker KA, Hawkins HK, Kukielka GL, Perrard JL, Michael LH, Ballantyne CM, Smith CW, Entman ML. Molecular evidence for induction of intracellular adhesion molecule-1 in the viable border zone associated with ischemia-reperfusion injury of the dog heart. Circulation. 1994;89:2736–2746. doi: 10.1161/01.cir.89.6.2736. [DOI] [PubMed] [Google Scholar]
- Kukielka GL, Youker KA, Michael LH, Kumar AG, Ballantyne CM, Smith CW, Entman ML. Role of early reperfusion in the induction of adhesion molecules and cytokines in previously ischemic myocardium. Mol Cell Biochem. 1995;147:5–12. doi: 10.1007/BF00944777. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- Misao Y, Arai M, Ohno T, Ushikoshi H, Onogi H, Kobayashi H, Takemura G, Minatoguchi S, Fujiwara T, Fujiwara H. Modification of post-myocardial infarction granulocyte-colony stimulating factor therapy with myelo-suppressives. Circ J. 2007;71:580–590. doi: 10.1253/circj.71.580. [DOI] [PubMed] [Google Scholar]
- Misao Y, Takemura G, Arai M, Ohno T, Onogi H, Takahashi T, Minatoguchi S, Fujiwara T, Fujiwara H. Importance of recruitment of bone marrow-derived CXCR4+ cells in post-infarct cardiac repair mediated by G-CSF. Cardiovasc Res. 2006;71:455–465. doi: 10.1016/j.cardiores.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Willerson JT, Buja LM. Protection of the myocardium during myocardial infarction: pharmacologic protection during thrombolytic therapy. Am J Cardiol. 1990;65:35I–41I. doi: 10.1016/0002-9149(90)90124-j. [DOI] [PubMed] [Google Scholar]
- Schmidt U, del Monte F, Miyamoto MI, Matsui T, Gwathmey JK, Rosenzweig A, Hajjar RJ. Restoration of diastolic function in senescent rat hearts through adenoviral gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase. Circulation. 2000;101:790–796. doi: 10.1161/01.cir.101.7.790. [DOI] [PubMed] [Google Scholar]
- Hu X, Dai S, Wu WJ, Tan W, Zhu X, Mu J, Guo Y, Bolli R, Rokosh G. Stromal cell derived factor-1 alpha confers protection against myocardial ischemia/reperfusion injury: role of the cardiac stromal cell derived factor-1 alpha CXCR4 axis. Circulation. 2007;116:654–663. doi: 10.1161/CIRCULATIONAHA.106.672451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan JE, Zhao ZQ, Vinten-Johansen J. The role of neutrophils in myocardial ischemia-reperfusion injury. Cardiovasc Res. 1999;43:860–878. doi: 10.1016/s0008-6363(99)00187-x. [DOI] [PubMed] [Google Scholar]
- McQuibban GA, Butler GS, Gong JH, Bendall L, Power C, Clark-Lewis I, Overall CM. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem. 2001;276:43503–43508. doi: 10.1074/jbc.M107736200. [DOI] [PubMed] [Google Scholar]
- Zwacka RM, Zhang Y, Halldorson J, Schlossberg H, Dudus L, Engelhardt JF. CD4(+) T-lymphocytes mediate ischemia/reperfusion-induced inflammatory responses in mouse liver. J Clin Invest. 1997;100:279–289. doi: 10.1172/JCI119533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling M, Strecker JK, Schabitz WR, Ringelstein EB, Kiefer R. Effects of monocyte chemoattractant protein 1 on blood-borne cell recruitment after transient focal cerebral ischemia in mice. Neuroscience. 2009;161:806–812. doi: 10.1016/j.neuroscience.2009.04.025. [DOI] [PubMed] [Google Scholar]
- Scarabelli TM, Knight R, Stephanou A, Townsend P, Chen-Scarabelli C, Lawrence K, Gottlieb R, Latchman D, Narula J. Clinical implications of apoptosis in ischemic myocardium. Curr Probl Cardiol. 2006;31:181–264. doi: 10.1016/j.cpcardiol.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Colamussi ML, Secchiero P, Zella D, Curreli S, Mirandola P, Capitani S, Zauli G. Stromal derived factor-1alpha induces apoptosis in activated primary CD4+ T cells. Aids. 2000;14:748–750. doi: 10.1097/00002030-200004140-00017. [DOI] [PubMed] [Google Scholar]
- Wu Y, Tu X, Lin G, Xia H, Huang H, Wan J, Cheng Z, Liu M, Chen G, Zhang H, Fu J, Liu Q, Liu DX. Emodin-mediated protection from acute myocardial infarction via inhibition of inflammation and apoptosis in local ischemic myocardium. Life Sci. 2007;81:1332–1338. doi: 10.1016/j.lfs.2007.08.040. [DOI] [PubMed] [Google Scholar]
- Han Y, He T, Huang DR, Pardo CA, Ransohoff RM. TNF-alpha mediates SDF-1 alpha-induced NF-kappa B activation and cytotoxic effects in primary astrocytes. J Clin Invest. 2001;108:425–435. doi: 10.1172/JCI12629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Guo R, Schwarz EM, Boyce BF, Xing L. TNF inhibits production of stromal cell-derived factor 1 by bone stromal cells and increases osteoclast precursor mobilization from bone marrow to peripheral blood. Arthritis Res Ther. 2008;10:R37. doi: 10.1186/ar2391. [DOI] [PMC free article] [PubMed] [Google Scholar]