

Abstract
Duchenne muscular dystrophy (DMD) and its milder allelic variant, Becker muscular dystrophy (BMD), result from mutations of the dystrophin gene and lead to progressive muscle deterioration. Enhanced activation of proteasomal degradation underlies critical steps in the pathogenesis of the DMD/BMD dystrophic process. Previously, we demonstrated that treatment with the proteasome inhibitor MG-132 rescues the cell membrane localization of dystrophin and the dystrophin glycoprotein complex in mdx mice, a natural genetic mouse model of DMD. The current work aims to thoroughly define the therapeutic potential in dystrophinopathies of Velcade, a drug that selectively blocks the ubiquitin-proteasome pathway. Velcade is particularly intriguing since it has been approved for the treatment of multiple myeloma. Therefore, its side effects in humans have been explored. Velcade effects were analyzed through two independent methodological approaches. First, we administered the drug systemically in mdx mice over a 2-week period. In this system, Velcade restores the membrane expression of dystrophin and dystrophin glycoprotein complex members and improves the dystrophic phenotype. In a second approach, we treated with the compound explants from muscle biopsies of DMD or BMD patients. We show that the inhibition of the proteasome pathway up-regulates dystrophin, α-sarcoglycan, and β-dystroglycan protein levels in explants from BMD patients, whereas it increases the proteins of the dystrophin glycoprotein complex in DMD cases.
Duchenne muscular dystrophy (DMD) is an incurable inherited disease, characterized by progressive muscle degeneration and weakness. The pathology results from the mutation of the DMD gene, which leads to deficiency of dystrophin, a 427-kDa protein found throughout the cytoplasmic face of the plasma membrane in both skeletal and cardiac muscle. Becker muscular dystrophy (BMD), a much milder allelic form of the disease, is caused by a reduction in the amount, or an alteration in the size, of dystrophin protein. In muscle cells, dystrophin binds through its amino-terminus to cytoskeletal F-actin and through the carboxyl-terminus to β-dystroglycan, a transmembrane component of a multimolecular complex [the dystrophin-glycoprotein complex (DGC)], which includes α, β, γ, δ-sarcoglycan, α, β-dystroglycan, the syntrophins, and dystrobrevin. In the DGC, dystroglycan appears to play a critical role, since it connects dystrophin with the DGC and it also binds laminin in the extracellular space, thus creating a mechanical linkage between cytoskeleton and extracellular matrix.1
Dystrophin deficiency is associated with a severe decrease or absence in the expression and membrane localization of the members of the DGC.2 It is feasible that dystrophin primary genetic loss leads to plasma membrane instability, thus causing a disarray of the multimeric complex and addressing its components to intracellular proteolysis. Indeed, different reports indicated that activation of protein degradation through the ubiquitin-proteasome system occurs in the pathogenesis of the DMD/BMD degenerative process.3
Consistently, our initial studies showed that local and systemic treatment with the proteasome inhibitor MG-132 rescues the expression of the DGC in mdx mice, a natural genetic mouse model of DMD.4 The effects of MG-132 were confirmed in freshly isolated skeletal muscle biopsies from patients affected by DMD or BMD, thus suggesting a possible therapeutic implication for these agents.5
In the present study we investigated the effects of the drug Velcade (bortezomib, PS-341), a proteasome inhibitor that belongs to the class of peptide boronates. Velcade is particularly appealing to us for different reasons.
First, while MG-132 exerts an inhibitory action on both the proteasome and the calpain system, Velcade displays a selective and high affinity to the proteasome. Therefore, this drug is an optimal probe to test the therapeutic potential of the inhibition of the proteasome pathway in muscular dystrophies. Secondarily, Velcade has been already approved by the Food and Drug Administration and the European Medicines Agency for the treatment of multiple myeloma. Thus, its side effects have been already explored and managed.6,7 Finally, Velcade is able to reduce the activity of nuclear factor-kappa B (NF-κB).8 Because NF-κB pathway has been shown to be involved in inflammation responses in myopathies and DMD, Velcade effects on this signaling molecule may have important clinical implications.9
Our previous results had indicated that Velcade, once injected locally into the gastrocnemius muscles of mdx mice, could up-regulate the expression and membrane localization of dystrophin and members of the DGC.10 However, the long-term actions of this proteasome inhibitor and its possible side effects in dystrophic animals are not known yet. To thoroughly evaluate Velcade’s therapeutic potential in dystrophinopathies, we adopted an integrated approach. First, we analyzed its efficacy using a systemic treatment and assessed the rescue of the dystrophic phenotype of mdx mice.
The mdx mouse model is characterized by a primary genetic defect of dystrophin due to a missense mutation in exon 23, which leads to a premature stop codon. Skeletal muscle tissue from mdx mutants is histologically normal early in postnatal development, but starting around 3 to 4 weeks of age, muscle necrosis develops, with some visible muscle weakness. Although skeletal limb muscles are characterized by a persistent and progressive degeneration and necrosis, at approximately 6 to 8 weeks of age, the muscle damage appears to plateau and it is possible to observe the beginning of a regenerative response activated by satellite cells, which continues until 12 weeks of age.11
Concurrently, we investigated the effects of the drug in human muscle explants of fresh biopsies from patients with DMD and BMD.
Materials and Methods
In Vivo Treatment with Velcade
C57BL/10ScSn-Dmdmdx/J mice (males, 7 weeks; Charles River Laboratories) were treated with 0.8 mg/kg of Velcade (or saline control) intravenously (i.v.) every 72 hours for two consecutive weeks. The mice were weighed every 72 hours before the injections.
The animals were sacrificed by CO2 inhalation at 2 weeks of treatment. The gastrocnemius muscles were removed bilaterally, immediately frozen in liquid nitrogen-cooled isopentane and stored at −80°C for morphological and immunohistochemical evaluations. The diaphragms were fixed in 10% formalin buffered with phosphate overnight (ON), and stored in 70% EtOH until processing. Processed muscles were cut longitudinally at the middle of the muscle and embedded into paraffin.
Serum was collected by intracardiac puncture at the time of sacrifice, and creatine kinase (CK) levels were measured using the CK-NAC quantitative system as per manufacturer’s instructions (BPC BioSED, Rome, Italy). CK values are reported as means ± SE.
All in vivo experiments were performed using four to six mice per group and were repeated five times. A group of age-matched C57BL/10ScSn (n = 4) mice was sacrificed as wild-type untreated controls for histopathological analyses and proteasome inhibition assay. Animal care, experiments, and protocols were approved by the Institutional Animal Care and Use Committee of the National Cancer Institute (Genoa, Italy).
Vital Staining with Evans Blue Dye
To detect damaged muscle fibers, 20 hours before the sacrifice Evans blue dye (EBD; stock solution: 10 mg/ml in phosphate buffered saline [PBS]) was injected (0.1 ml/10 g body weight) into the peritoneal cavity of each mouse.10
Following sacrifice, gastrocnemius muscle was excised, immediately frozen in nitrogen-cooled isopentane, and stored for further analyses. Gastrocnemius frozen sections, obtained by cutting the muscle on a cryostat, were observed under a Leica Diaplan inverted microscope to detect the EBD red autofluorescence signal. EBD-positive fibers were then counted in two sections for each muscle (n = four to seven mice for each experimental group, n = 400 to 600 myofibers) and expressed as percentage of the total fiber number.
Muscle Explants
All human samples were obtained after patients had signed informed consent forms in accordance with the guidelines of the G. Gaslini Institute Ethical Committee, including experimental protocols for muscle biopsies. Human explants were isolated from quadriceps muscle biopsies obtained for diagnostic purposes from eight patients with DMD and eight patients with BMD. Biopsies were cleaned from fat and connective tissue and divided into small pieces. These fragments were placed into a 60-mm-diameter dish containing F-14 medium (Dulbecco’s modified Eagle’s minimal essential medium (DMEM) containing 6 g/L glucose, 15% fetal bovine serum, 50 g/L fibroblast growth factor, 10 g/L epidermal growth factor, 2 mmol/L glutamine, 10 mg/L insulin, 1% penicillin-streptomycin-amphotericin B mixture, and 1 mg/L ATP) at 37°C in a humidified 95% air-5% CO2 atmosphere in the presence or absence of 1 and 10 μmol/L Velcade for 16 hours.5 At the end of the incubation period, explants were frozen in liquid nitrogen-cooled isopentane and stored in liquid nitrogen.
Proteasome Activity
Proteasome activity in skeletal muscle tissue from C57BL/10ScSn wild-types, untreated and Velcade-treated mdx mice was determined by cleavage of specific fluorogenic substrates.12 Briefly, the tissue was homogenized in 20 mmol/L Tris–HCl, pH 7.2, containing 0.1 mmol/L EDTA, 1 mmol/L 2-mercaptoethanol, 5 mmol/L ATP, 20% glycerol, and 0.04% (v/v) Nonidet P-40. Muscle homogenates were then centrifuged at 13,000 × g for 15 minutes at 4°C. The supernatant was collected and protein concentration determined. Aliquots of 100 μg protein were then incubated for 60 minutes at 37°C in the presence of a specific fluorogenic substrate (Suc-Leu-Leu-Val-Tyr-7-AMC; Sigma-Aldrich, Milan, Italy) in 50 mmol/L Hepes, pH 8.0, 5 mmol/L EGTA buffer. Fluorescence was read with a spectrofluorometer (380 nm excitation, 460 nm emission; Perkin–Elmer, Norwalk, CT). Negative and positive controls were performed as well. The activity, obtained by evaluating the release of free AMC, is expressed as relative fluorescence units and reported as means ± SE.
Histological Studies
Seven-micrometer (7 μm) sections from gastrocnemius muscle were cut on a cryostat and later stained with Hematoxylin and Eosin (H&E). Cross-sectional area (CSA) was quantified in a blinded manner in two sections for each muscle (n = four to seven mice for each experimental group, n = 400 to 600 myofibers) with ImageJ software (http://rsb.info.nih.gov/ij).13 Fiber size variability was calculated according to the formula: Standard Deviation × 1000/mean fiber diameter.14 In the same sections, central nucleation, and myofiber necrosis were also evaluated. The following three cytotypes were identified: 1) normal fibers, characterized by peripheral nuclei; 2) centrally nucleated fibers, identified by normal size but central nuclei; and 3) necrotic fibers, recognized by pale cytoplasm and peripheral macrophage invasion.15
Immunohistochemical Analyses
Unfixed 5-μm-thick cryosections of mdx gastrocnemius muscles, or of patients’ muscle explants were incubated with primary antibodies ON at 4°C. After three washes with PBS (5 minutes each), sections were incubated with fluorescent secondary antibodies in common antibody diluent (BioGenex, San Ramon, CA) for 1 hour at room temperature in the dark. After being washed three times with PBS (5 minutes each), sections were mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA).
Quantification of fluorescence intensity of fibers stained for all of the studied proteins was performed using a Leica TCS SL confocal microscope equipped with Ar/He-Ne laser sources and a HCX PL APO CS 40.0 × 1.25 oil objective. An energy laser of 50% was applied to the specimens during acquisition. Data were acquired in an emission range of 590 to 670 nm, while the photomultiplier voltage gain and offset were set to eliminate both autofluorescence and overflow fluorescence of samples.
Single plane images, centered at the middle of fiber thickness of the specimen, were acquired and processed using the Leica LCS quantification software to obtain area and mean circumference fluorescence for each fiber.
Paraffin-embedded diaphragm muscle tissue was cut in cross section at the mid-belly of the muscle (7 μm thickness). Deparaffinization of the slides was done through a series of incubations in xylene, 100% EtOH, 95% EtOH, 70% EtOH, and water. Endogenous peroxidase activity was blocked by a 20-minute incubation in 10% H2O2/methanol solution. Antigen retrieval was done by treatment in 10 mmol/L citrate buffer solution for 1 hour at 98°C. Sections were subsequently blocked in 5% bovine serum albumin/PBS for 20 minutes at room temperature.
Primary antibodies were added and incubated ON in a humidified box at 4°C. Sections were then incubated with the Dako Real TM Envision TM Detection System peroxidase DAB+ reagent (DAKO, Milan, Italy) for 1 hour at room temperature. The reaction was visualized with the Dako real TM DAB+ chromogen according to manufacturer’s instructions. Slides were counterstained with hematoxylin, dehydrated with incubations in 70% EtOH, 95% EtOH, 100% EtOH, and xylene, and mounted with Vectashield mounting medium. Slides were observed under a Leica Diaplan microscope.
Western Blot Analysis
Tissue samples were prepared on ice as follows: five to eight 10-μm-thick cryosections of gastrocnemius muscles or of patients’ muscle explants were quickly dissolved by pipetting in 40 μl of radioimmunoprecipitation assay lysis buffer (Sigma-Aldrich) with 1:200 protease inhibitor cocktail (Sigma-Aldrich). Tissue lysates were then sonicated for 10 seconds on ice and centrifuged at 10,000 rpm for 15 minutes at 4°C. Supernatants were collected, and the protein concentration was determined using the Bradford protein assay method (Biorad, Milan, Italy).
To prepare nuclear extracts, muscle (about 50 mg) was homogenized in ice cold 10 mmol/L HEPES, pH 7.5, containing 10 mmol/L MgCl2, 5 mmol/L KCl, 0.1 mmol/L EDTA pH 8.0, 0.1% Triton X-100, 0.1 mmol/L phenylmethanesulfonyl fluoride [PMSF], 1 mmol/L DTT, 2 μg/ml aprotinin, and 2 μg/ml leupeptin. Samples were then centrifuged (5 minutes, 3000 × g), pellets resuspended in ice cold 20 mmol/L HEPES, pH 7.9, containing 25% glycerol, 500 mmol/L NaCl, 1.5 mmol/L MgCl2, 0.2 mmol/L EDTA, pH 8.0, 0.2 mmol/L phenylmethylsulfonyl fluoride, 0.5 mmol/L dithiothreitol, 2 μg/ml aprotinin, and 2 μg/ml leupeptin, and incubated on ice for 30 minutes. Cell debris was removed by centrifugation (5 minutes, 3000 × g) and the supernatant was collected and stored at −80°C. Protein concentration was determined using the Bradford protein assay method.
Equal amounts of protein (40 to 120 μg) were mixed with 4× SDS gel-loading buffer (100 mmol/L Tris-HCl, pH 6.8, 200 mmol/L dithiothreitol, 4% SDS, 0.2% bromophenol blue, and 20% glycerol), resolved on 6% to 15% SDS-polyacrylamide electrophoresis gel, and run at 80 V for 2 to 2.30 hours in electrophoresis buffer (25 mmol/L Tris, 250 mmol/L glycine, and 0.1% SDS). Proteins were then transferred onto polyvinylidene difluoride membranes (Immobilon PVDF; Millipore, Milan, Italy). Membranes were blocked with 5% nonfat dried milk in PBS/0.1% Tween 20 (PBST). Blots were then incubated for 2 hours at room temperature with primary antibodies all diluted in PBS/1% bovine serum albumin. Horseradish peroxidase anti-IgG were used to visualize bound primary antibodies with the enhanced chemiluminescence system (GE Health care, Milan, Italy). To normalize sample loading, membranes were then stripped by incubation in 62.5 mmol/L Tris-HCl, pH 6.7, containing 100 mmol/L 2-mercaptoethanol and 2% SDS for 30 minutes at 50°C, and reprobed with antibodies directed toward reference proteins (either spectrin or glyceraldehyde-3-phosphate dehydrogenase [GAPDH]). Band intensities were evaluated by densitometry using the ImageQuant 350 system (GE Health care).
Reagents and Antibodies
Velcade was provided by Janssen-Cilag (Milan, Italy) as a lyophilized white cake in 10-ml glass vials. Each sterile single-use vial contained 3.5 mg Velcade and 35 mg mannitol. Vials of Velcade were stored at 25°C. For in vitro experiments, the contents of each vial were dissolved in 910 μl dimethyl sulfoxide so that the final concentration of Velcade was 10 mmol/L, and the reconstituted drug was stored at −20°C; reconstituted drug was thawed and diluted in culture medium (the final dimethyl sulfoxide concentration was less than 0.1%) immediately before use. For in vivo experiments, the content of each vial was reconstituted in 3.5 ml of normal (0.9%) saline so that the reconstituted solution contained Velcade at a concentration of 1 mg/ml. Complete dissolution of the drug in 0.9% saline took approximately 10 seconds. The reconstituted solution was clear and colorless, had a final pH that ranged from 5 to 6, and was used immediately for injection into mice. EBD was purchased by Sigma-Aldrich.
The antibodies and dilutions used in the study include the following: monoclonal antibodies to β-dystroglycan (NCL-β-DG) (undiluted for immunohistochemistry [IHC-FR]; 1:50 for Western blot [WB]; α-sarcoglycan (NCL-α-sarco) (undiluted for IHC-FR; 1:50 for WB); human dystrophin (NCL-DYS1) (undiluted for IHC-FR; 1:200 for WB); human vinculin (1:100 for WB) were purchased from Novocastra (Newcastle on Tyne, UK). Polyclonal antibodies raised to mouse dystrophin (1:100 for IHC-FR; 1:25 for WB); mouse spectrin α II (1:250 for IHC-FR; 1:500 for WB); MyoD (1:200 for WB); Myf-5 (1:200 for WB); F4/80 (1:200 for WB, 1:50 for IHC-P); E-MYH1/2/3 (1:50 for IHC-P); GAPDH (1:200 for WB) were from Santa Cruz Biotechnology (Santa Cruz, CA). The monoclonal antibody to caveolin-3 (1:200 for IHC-FR; 1:10.000 for WB) was from BD Transduction Laboratories (San Jose, CA), the polyclonal antibody to α−1 syntrophin (1:200 for IHC-FR; 1:1000 for WB) was from Sigma-Aldrich, the monoclonal antibody to NF-κB p65 (1:1000 for WB) was from Cell Signaling Technology (Danvers, MA), and the polyclonal antibody to interleukin (IL)-6 (1:400 for WB) was from Abcam (Cambridge, UK).
Goat anti-mouse or anti-rabbit IgG antibodies Alexa Fluor 594 (1:500 for IHC-FR) were purchased from Invitrogen (Milan, Italy). Rabbit anti-mouse IgG horseradish peroxidase-conjugated (1:700 for WB) was from DAKO (Glostrup, Denmark), whereas donkey anti-rabbit IgG (1:5000 for WB) horseradish peroxidase-conjugated was from GE Health care.
Statistical Analysis
Differences in the groups were analyzed by a two-tailed Student’s t-test or by the nonparametric Mann-Whitney U-test in case of skewed distributions. Correlations between quantitative variables were analyzed by the Pearson’s correlation coefficient (r).
Results
Velcade Systemic Treatment in mdx Mice
To determine Velcade’s in vivo efficacy on the rescue of the dystrophic phenotype due to dystrophin deficiency, we chose to treat mdx male mice systemically at 7 weeks of age. Previous studies from oncologic research had shown in different mouse models that Velcade’s therapeutic window is quite narrow and is comprised between 0.7 and 1.3 mg/kg. A substantial inhibition of tumor growth is obtained at 1 mg/kg. This compound is injected i.v. every 72 hours, because it has been shown that it takes this long for the 26S proteasome activity to recover to control levels after being inhibited.16,17
On the basis of these data, in each of our experiments, we administered Velcade i.v. at 0.8 mg/kg, every 72 hours for two consecutive weeks. Age-matched mdx male mice injected with vehicle alone (saline solution) were used as controls. Experimental and control mice were sacrificed 24 hours following the last injection and gastrocnemius and diaphragm muscles were surgically isolated.
As it concerns drug side-effects, after 3 days of treatment, Velcade-treated mdx mice displayed a loss of body weight equivalent to 8 ± 1.5% (n = 10). However, the body weight remained stable during the following days of the experiment. No other significant adverse reactions were observed. The gastrocnemius muscles were weighed at the sacrifice, but no changes were observed between the untreated and Velcade-treated groups (data not shown). The experiments were repeated at least five times.
Among the limb skeletal muscles, the gastrocnemius shows a mixed fiber-type composition, displays the pseudo- hypertrophic pathological changes observed in DMD patients, is easily accessible and is invariably included in the research studies aimed to analyze the therapeutic effects of test agents in mdx mice. For these reasons, the majority of the experiments described in the paper were conducted on this muscle.
However, since based on phenotypic severity, the mdx diaphragm appears to show the closest homology to human DMD disease, the effects of Velcade on dystrophin up-regulation and on histopathological features of muscular dystrophy were confirmed in this muscle as well.
At first, we determined the percentage of inhibition of proteasome activity in mdx muscle tissue following Velcade exposure. For this purpose, gastrocnemius muscle lysates from Velcade-treated, untreated mdx, and age-matched wild-type C57Bl/10ScSn mice were analyzed via a standard proteasome activity assay. Notably, in accordance with our primary hypothesis, when compared with the wild-type group, proteasome activity was increased by 16-fold in the mdx untreated mice. At 24 hours following Velcade injection, proteasome activation was reduced by approximately 50% (Figure 1). This result is in accordance with the drug pharmacodynamic profile measured in human patients.17,18
Figure 1.

Inhibition of proteasome activation in muscles from 7-week-old mdx mice treated systemically with the proteasome inhibitor Velcade for 2 weeks. Proteasome activity was evaluated in gastrocnemius muscle lysates by means of specific fluorogenic substrates. Activity was assayed in age-matched wild-type C57BL/10 Sc/Sn (gray bar), untreated (white bar), and Velcade-treated (black bar) mdx mice. Notably, when compared with wild-type animals, proteasome degradation is markedly induced in untreated mdx mice. This effect is decreased by approximately 50% following Velcade exposure. Each column represents the mean ± SE from four to seven animals; *P < 0.05 Wild-type (Wt) versus Velcade-treated mdx; **P < 0.01 wild-type versus untreated mdx; §P < 0.05 untreated versus Velcade-treated mdx mice.
Hence, frozen skeletal muscle sections from untreated and Velcade-treated mdx were examined by IHC-FR analysis with antibodies directed against dystrophin, β-dystroglycan, and α-sarcoglycan. As expected, all these proteins were expressed at very low levels in the skeletal muscle fibers from untreated mdx muscles, whereas they were up-regulated and clearly detectable on the muscle plasma membrane of the mice treated with the proteasome inhibitor (Figure 2).
Figure 2.

Inhibition of the proteasome pathway up-regulates the membrane localization of dystrophin and the dystrophin-glycoprotein complex in 7-week-old mdx mice. Frozen gastrocnemius muscle sections from untreated (saline only) and Velcade-treated mdx mice were immunostained with specific antibodies directed against dystrophin (DYS), β-dystroglycan (β-DG), α-sarcoglycan (α-SG), and spectrin α II (Spectrin). Note that DYS, β-DG, and α-SG, were expressed at low levels in the skeletal muscle fibers from untreated mdx muscles, whereas they were up-regulated on the muscle plasma membrane of the mice treated with the proteasome inhibitor. Spectrin α II was used as a membrane protein control. Final magnification, ×25. Representative images are shown.
To further investigate the effects of Velcade on the formation and stability of the dystrophin complex, we analyzed the expression of α−1 syntrophin, a subsarcolemmal protein that is connected to dystrophin through the molecule dystrobrevin. α-1 Syntrophin is decreased in skeletal muscle biopsies from DMD patients and in mdx mice.19 Accordingly, α−1-syntrophin immunostaining was extremely weak in the mdx untreated group, while it was up-regulated in the Velcade-treated cohort (Figure 3).
Figure 3.

Inhibition of the proteasome pathway on caveolin 3 and α-1syntrophin expression in 7-week-old mdx mice. Frozen gastrocnemius muscle sections from untreated (saline only) and Velcade-treated mdx mice were immunostained with specific antibodies directed against caveolin 3 (Cav-3), α−1 synthophin (α−1 Syn) and spectrin α II (Spectrin). Note that Cav-3 is highly expressed in the untreated mdx group, while α−1 Syn staining is absent or just weakly positive. The proteasome inhibitor induced only a slight reduction of Cav-3 expression, while it enhanced α−1 Syn. Spectrin α II was used as a membrane protein control. Final magnification, ×25. Representative images are shown.
The intensity of each protein membrane fluorescent signal was quantified in the untreated and Velcade-treated mice by confocal microscopy (a range of 150 to 200 myofibers was counted for each experimental group, n = 3 mice). The data were hence evaluated through the Mann-Whitney U-test. The statistical analysis confirms that pharmacological inhibition of the proteasome pathway significantly increases the intensity of the membrane fluorescence of all of the protein evaluated (Supplemental data, Figure S1A, see http://ajp.amjpathol.org). Notably, the ratio intensity/cross-sectional myofiber area was also augmented (Supplemental Figure S1B, see http://ajp.amjpathol.org). In both the untreated or in Velcade-treated mdx mice, a weak inverse correlation between the cross-sectional myofiber area (CSA) and the intensity of the membrane fluorescence was observed (the smaller was the myofiber area (CSA), the higher was the intensity of the membrane fluorescence). This inverse correlation was identified in the IHC-FR for β-dystroglycan (r = 0.34), α-sarcoglycan (r = 0.24), and α−1-syntrophin (r = 0.25), but was absent in dystrophin fibers (r = 0.03).
Skeletal muscle biopsies from DMD patients display an increased number of caveolae and overexpression of the caveolin-3 protein product.20 Caveolin-3 membrane localization is elevated by two folds in dystrophin deficient skeletal muscle samples from mdx mice.21,22 In our experiments, the dystrophin increase observed in Velcade-treated mice is associated to an up-regulation of β-dystroglycan signal, but not to a significant reduction in caveolin-3 immunostaining. The molecule spectrin was used as a membrane protein control (Figures 2 and 3).
Next, skeletal muscle lysates from untreated (saline only) and from Velcade-treated mdx mice were subjected to Western Blot (WB) analysis with specific antibodies directed against β-dystroglycan, α-sarcoglycan, dystrophin, α−1 syntrophin, and caveolin-3. Figure 4A and B shows that β-dystroglycan, α-sarcoglycan, dystrophin, and α−1 syntrophin were increased in skeletal muscle lysates from Velcade-treated mice, as compared with untreated controls. Notably, in the Velcade-treated samples dystrophin migrates at a molecular weight equal to 120 kDa. This is in accordance with the predicted size of the protein on the basis of the mdx mutation (premature stop codon in exon 23). Consistent with the increase observed in dystrophin protein expression and membrane localization, caveolin-3 protein levels were reduced following proteasome treatment. The discrepancy observed between the IHC-FR and WB caveolin-3 experiments might be accounted for by the different sensitivity of the two assays.
Figure 4.

Inhibition of the proteasome pathway increases the protein levels of dystrophin and the dystrophin-glycoprotein complex in 7-week-old mdx mice. A: Gastrocnemius muscle lysates form untreated (saline only) and Velcade-treated mdx mice were separated by SDS-polyacrylamide gel electrophoresis, transferred onto nitrocellulose membrane, and subjected to WB analysis with specific antibodies against dystrophin (DYS), α-sarcoglycan (α-SG), β-dystroglycan (β-DG), caveolin 3 (Cav-3), α−1 synthophin (α−1-Syn), and Spectrin α II (Spectrin). Note that proteasome inhibition induced an increase of DYS and its associated proteins. Consistently with DYS up-regulation, Cav-3 protein levels were reduced in muscles from Velcade-treated mice, as compared with untreated controls. Notably, in the Velcade-treated mice, DYS migrates at molecular weight equal to 120 kDa. This is in accordance with the predicted size of the protein on the basis of the mdx mutation (premature stop codon in exon 23). Spectrin α II was used as a loading control. B: Bands quantification was performed by densitometric analysis. Data (mean ± SE from four to seven animals) are expressed as percentage of Velcade-treated versus untreated mdx mice. *P < 0.05 untreated (black bar) versus Velcade-treated (white bar) mdx mice.
We next evaluated Velcade’s actions on the dystrophic phenotype displayed by the mdx mice. To analyze the extent of muscle fiber degeneration, we used the EBD agent. This compound is a membrane-impermeant molecule that binds to serum albumin and is physically restricted from fibers unless the skeletal muscle membrane is damaged.4 When analyzed under the microscope, EBD emits a red autofluorescent signal. Hence, we injected EBD intraperitoneally in Velcade-treated and untreated mdx mice at 20 hours before the sacrifice. As shown in Figure 5, A and B, frozen sections of gastrocnemius muscles from untreated mdx mice showed a strong red fluorescent signal (about 60% of total fibers), whereas muscle fibers from the Velcade group were almost negative (less than 10% of fibers in the whole section), thus suggesting that the proteasome inhibitor improves membrane integrity. Consistent with these results, Velcade seems to improve the histopathology of mdx muscle lesions.
Figure 5.

Inhibition of the proteasome pathway reduces muscle degeneration and necrotic features in skeletal muscle fibers of 7-week-old mdx mice. A: To evaluate muscle fiber damage, Evans blue dye (EBD) agent was injected intraperitoneally in untreated (saline only) and Velcade-treated mdx mice 20 hours before the sacrifice. Frozen gastrocnemius muscle sections were examined by fluorescence microscopy. Sections from untreated mice displayed a strong red autofluorescent signal, whereas muscle fibers from the Velcade-treated group were negative. Representative images are shown. Final magnification, ×10. B: EBD-positive muscle fibers in the gastrocnemius from mdx mice, both untreated (white bar) and Velcade-treated (black bar), were evaluated and expressed as percentage of total fibers (two sections were counted for each muscle; n = 4 mice for each experimental group; n myofibers = 400 to 600). §§P < 0.01 untreated versus Velcade-treated mdx mice.
Frozen sections from the gastrocnemius muscles of Velcade-treated and untreated mdx mice were stained with H&E. Skeletal muscle biopsies from the untreated group showed clear signs of myopathic changes, including muscle fiber degeneration with many central nuclei, extreme variability in fiber diameter, inflammatory reaction, and connective tissue infiltration (Figure 6A). These histopathological features were significantly reduced by proteasome inhibition. In details, Velcade induced a 25% increase of myofibers CSA and a 36% reduction of the size variability index. Moreover, the drug led to a 20% decrease of the number of centrally nucleated and 50% reduction of degenerative/necrotic fibers (Figure 6B).
Figure 6.

Inhibition of the proteasome pathway improves histopathological features in skeletal muscle fibers of 7-week-old mdx mice. A: Frozen gastrocnemius muscle tissue sections from untreated (saline only) and Velcade-treated mdx mice were stained with standard H&E technique. Skeletal muscle from untreated mdx mice showed clear signs of myopathic changes, including muscle fiber degeneration characterized by central nuclei, variability in fibers size, lymphocytic, and connective tissue infiltration. Note that proteasome inhibition increased the diameter of the myofibers and decreased the signs of the inflammatory and fibrogenic response compared with the untreated mdx mice. Representative images are shown. Final magnification, ×10. B: Cross-sectional area (CSA) in age-matched wild-type C57BL/10 Sc/Sn (gray bar), untreated mdx (white bar), and Velcade-treated (black bar) mdx mice was determined by the Image J software (two sections were counted for each muscle (n = four to seven mice for each experimental group; n myofibers = 400 to 600). CSA data (mean ± SE from four to seven animals) are expressed as percentage of untreated and Velcade-treated mdx versus wild-type mice column. Both central nucleated and necrotic fibers are expressed as percentage of total fibers; **P < 0.001 wild-type versus untreated mdx; §P < 0.05 untreated versus Velcade-treated mdx mice; §§P < 0.01 untreated versus Velcade-treated mdx mice.
In accordance with the results determined in the gastrocnemius muscles, in the diaphragm tissue, Velcade up-regulated sarcolemmal dystrophin expression and significantly improved histopathological features of muscular dystrophy. In details, the drug reduced the number of degenerative/necrotic and centronucleated muscle cells by respectively 87 and 50%. As concerns the size variability index, this was decreased by 5% in the Velcade-treated mdx group (Figure 7, A−B). No differences in the CSA were observed.
Figure 7.

Inhibition of the proteasome pathway up-regulates dystrophin membrane levels and improves histopathological features in diaphragm muscle fibers of 7-week-old mdx mice. A: Paraffin diaphragm muscle sections from untreated (saline only) and Velcade-treated mdx mice were immunostained with a specific antibody directed against dystrophin (DYS) and hence counterstained with hematoxylin. Note that DYS was almost not detectable on the sarcolemma of diaphragm muscle fibers from untreated mdx muscles, whereas it was up-regulated on the muscle plasma membrane of the mice treated with the proteasome inhibitor (white arrows). Final magnification, ×25. Representative images are shown. B: Paraffin-embedded diaphragm muscle tissue sections from untreated (saline only) and Velcade-treated mdx mice were stained with standard H&E technique. Skeletal muscle from untreated mdx mice showed clear signs of myopathic changes, including muscle fiber degeneration characterized by central nuclei, variability in fiber size, and lymphocytic and connective tissue infiltration. Note that proteasome inhibition decreased the signs of the inflammatory and degenerative response. Representative images are shown. Final magnification, ×10. C: Cross-sectional area (CSA) in both untreated (white bar) and Velcade-treated (black bar) mdx mice was determined by the Image J software (two sections were counted for each muscle; n = four to seven mice for each experimental group; n myofibers = 400 to 600). CSA data (mean ± SE from four to seven animals) are expressed as percentage of Velcade-treated versus untreated mdx mice column, while both central nucleated and necrotic fibers are expressed as percentage of total fibers; §P < 0.05 untreated versus Velcade-treated mdx mice; §§P < 0.001 untreated versus Velcade-treated mdx mice.
To evaluate whether proteasome inhibition might have a role in skeletal muscle cell regeneration, gastrocnemius muscle sections from untreated and Velcade-treated mdx were immunostained with an antibody that recognizes embryonal (developmental) myosin heavy chain (E-MHC).23 The number of E-MHC positive myofibers were hence calculated in two sections for each mouse (n = 4 for each experimental group) and were found to be increased in Velcade-treated mice compared with untreated controls (Figure 8, A−B). Moreover, as shown in Figure 8, C−D, Velcade increased the protein levels of the two myogenic regulatory factors MyoD and Myf5.24
Figure 8.

Inhibition of the proteasome pathway enhances muscle cell regeneration in 7-week-old mdx mice. A: Frozen skeletal muscle sections from untreated (saline only) and Velcade-treated mdx mice were immunostained with a primary antibody directed toward the native form of E-MHC. Note that proteasome inhibition increases the number of E-MHC positive muscle fibers. Representative images are shown. Final magnification, ×10. B: E-MHC positive fibers in the gastrocnemius from mdx mice, both untreated (white bar) and treated (black bar), were quantified (two sections were counted for each muscle; n = four to seven mice for each experimental group; n myofibers = 400 to 600). Each column represents the mean ± SE from four to seven animals; §P < 0.05 untreated versus Velcade-treated mdx mice. C: Skeletal muscle nuclear lysates form untreated (saline only) and Velcade-treated mdx mice were subjected to immunoblot analysis with specific antibodies against MyoD and Myf-5, two common markers of regeneration. GAPDH was used to normalize sample loading. Note that Velcade administration to mdx mice increases both MyoD and Myf-5 levels, suggesting the hypothesis that muscle regeneration in treated animals is enhanced. D: Band quantification was performed by densitometric analysis. Data (mean ± SE from four to seven animals) are expressed as percentage of Velcade-treated versus untreated mdx mice. §P < 0.05, §§P < 0.01 untreated (white bar) versus Velcade-treated (black bar) mdx mice.
The H&E analysis of gastrocnemius and diaphragm muscle indicates that Velcade reduces the inflammatory reaction observed in the untreated mdx mice. To quantify this effect, gastrocnemius muscle sections from untreated and Velcade-treated mice were immunostained with an antibody directed to F4/80 a marker of activated macrophages.23 The number of labeled cells (brown colored) within two entire sections for each muscle was counted and the total area of each section was assessed using a calibrated square grid. The volume of muscle sample was calculated as the product of the section area and thickness (5 μm). Concentrations of macrophages were expressed as number of cells per cubic millimeter (mm3).23
Notably, the concentration of F4/80 positive cells was reduced in gastrocnemius muscle of mdx mice treated with the proteasome inhibitor when compared with untreated mice (Figure 9, A−B). Accordingly, IL-6 protein levels, measured by WB analysis in total muscle lysates from untreated and Velcade-treated mdx mice, were decreased following treatment with the drug. (Figure 9, C−D).25
Figure 9.

Inhibition of the proteasome pathway reduces signs of inflammation in the skeletal muscle of 7-week-old mdx mice. A: Frozen skeletal muscle sections from untreated (saline only) and Velcade-treated mdx mice were immunostained with a primary antibody directed toward the macrophage marker F4/80. Representative images are shown. Final magnification, ×10. B: In untreated (white bar) and Velcade-treated (black bar) mdx mice, the number of labeled cells (brown colored) was counted within two entire sections for each muscle (n = four to seven mice for each experimental group), and the total area of each section was assessed using a calibrated square grid. The volume of muscle sample was calculated as the product of the section area and thickness (5 μm). Concentrations of macrophages were expressed as number of cells per cubic millimeter (mm3). Each column represents the mean ± SE; §§P < 0.01 untreated versus Velcade-treated mdx mice. C: Skeletal muscle lysates form untreated (saline only) and Velcade-treated mdx mice were subjected to Immunoblot analysis with specific antibodies against IL-6. Determination of GAPDH expression was used to normalize sample loading. Please note that Velcade administration to mdx mice reduces the level of IL-6, thus further suggesting that proteasome inhibition is associated with a reduction of the inflammatory state. D: Quantification of IL-6 levels in untreated (white bar) and Velcade-treated (black bar) mdx mice by densitometric analysis. Data (mean ± SE from four to seven animals) are expressed as percentage of Velcade-treated versus untreated mdx mice. §P < 0.05 untreated versus Velcade-treated mdx mice.
In DMD, increased serum CK levels reflect and correlate with active muscle degeneration and CK measurement is used in the clinical setting as a rapid and useful tool to monitor the state and the progression of the disease. Mdx mice display CK levels increased by approximately 10- to 16-fold when compared with age-matched C57Bl/10 mice.26 To further evaluate the beneficial effects of proteasome inhibition on the process of muscle degeneration, we hence evaluated serum CK levels in Velcade-treated and untreated mdx controls. Notably, at 2 weeks of treatment, Velcade was able to decrease CK levels by 90% (Untreated mice: 1981.7 ± 795.4-n = 4; Velcade-treated mice: 202 ± 34.4-n = 7; means ± SE, P < 0.05) (Figure 10).
Figure 10.

Inhibition of the proteasome pathway dramatically decreases serum CK levels in 7-week-old mdx mice. Blood samples from untreated (saline only) and Velcade-treated mdx mice were obtained by intracardiac punctures at 2 weeks of treatment. Each column represents the mean ± SE from four to seven animals. *P < 0.05 untreated (white bar) versus Velcade-treated (black bar) mdx mice.
Part of Velcade’s mechanisms of action is the inhibition of the activation of the transcription factor NF-κB.8 NF-κB pathway is activated in DMD, and is involved in muscle degeneration and regeneration in dystrophin-deficient fibers. Interestingly, lysates from skeletal muscle fibers of Velcade-treated mdx mice, display a decrease of the nuclear p65 active form of NFκB when compared with the untreated groups. This result may be due to the block of proteasomal degradation of the inhibitory protein IκB, which increases the stability of the cytoplasmatic inactive form of NFκB (Figure 11, A and B).
Figure 11.
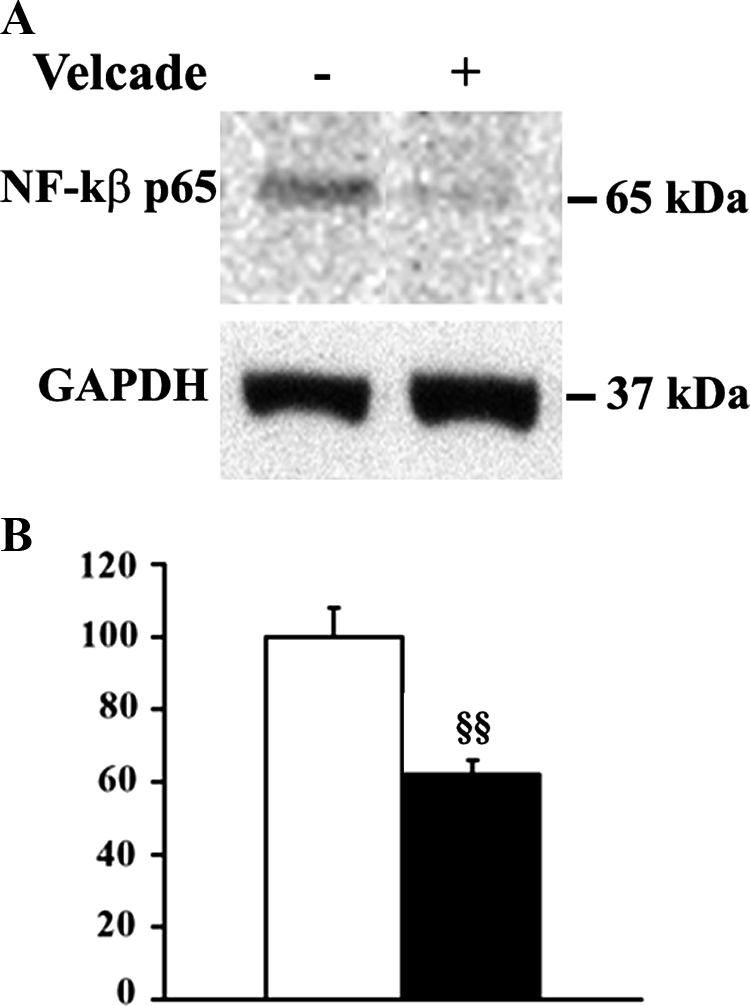
Inhibition of the proteasome pathway decreases NF-κB activation in 7-week-old mdx mice. A: Skeletal muscle nuclear lysates from untreated (saline only) and Velcade-treated mdx mice were subjected to immunoblot analysis with specific antibodies against total NF-κB (p65). The membrane was then stripped and reprobed with GAPDH antibody to normalize sample loading. Please note that Velcade administration to mdx mice reduces the level of p65 NF-κB allowed to translocate at nuclear level, thus suggesting that NF-κB activation is inhibited. B: Bands quantification was performed by densitometric analysis. Data (mean ± SE from four to seven animals) are expressed as percentage of Velcade-treated versus untreated mdx mice. §§P < 0.01 untreated (white bar) versus Velcade-treated (black bar) mdx mice.
Velcade Treatment in Muscle Explants from DMD and BMD Patients
In a complementary set of experiments, we tested the effects of Velcade in explants from fresh human skeletal muscle biopsies from patients with DMD and BMD. More specifically, the actions of the drug were analyzed in muscle explants from eight DMD and eight BMD patients. All of the patients were genetically characterized through multiplex quantitative real-time PCR assays for the detection of gene deletions or duplications, and DNA sequencing for the analysis of missense mutations (Table 1).27
Table 1.
Genetic, IHC-FR, and WB Data from Skeletal Muscle Explants Extracted from DMD and BMD Patients and Treated with Velcade at 1 and 10 μmol/L
| Disease | Patient | Mutation | Velcade
|
Notes | ||
|---|---|---|---|---|---|---|
| α-sarcoglycan | β-dystroglycan | Dystrophin | ||||
| DMD | D1 | d 46–52 | ++ | + | + IHC only | |
| DMD | D2 | d 49–52 | + | ++ | + IHC only | |
| DMD | D3 | d 17–44 | ∼ | ∼ | ∼ | + connective tissue |
| DMD | D4 | d 45 | + | + | + IHC only | |
| DMD | D5 | d 45 | ∼ | ∼ | ∼ | + connective tissue |
| DMD | D6 | d 49–52 | ++ | ++ | + IHC only | |
| DMD | D7 | d 57–52 | + | + | + IHC only | |
| DMD | D8 | d 45–52 | ++ | ++ | + IHC only | |
| BMD | B1 | d 42–51 | + | + | + | |
| BMD | B2 | d 45–51 | + | + | + | |
| BMD | B3 | d 48–55 | + | ++ | + | |
| BMD | B4 | d 45–49 | + | + | + | |
| BMD | B5 | c.7098 + 1G>A | + | + | + | |
| BMD | B6 | DD 3–4 | + | + | + | |
| BMD | B7 | DD 13–17 | + | ++ | + | |
| BMD | B8 | d 48 | + | + | + | |
d = Deletion; DD = Duplication; IHC = Immunohistochemistry; ∼ = No effect; + = Increase lower than 50% at densitometric analysis of Western Blot results; ++ = Increase higher than 50% at densitometric analysis of Western Blot results.
Briefly, muscle explants were isolated from the biopsy, divided into small pieces and incubated in culture medium supplemented with 15% fetal bovine serum at 37°C in the presence or absence of Velcade. Time-course experiments defined that the best length of treatment was 16 hours (data not shown). Initially, Velcade was administered at doses from 100 nmol/L to 50 μmol/L. When the drug was used at the concentration of 100 nmol/L we did not observe any effect, while the doses of 1, 10, and 50 μmol/L were equally effective. Because of the small amount of tissue available from the biopsies, we focused in the following experiments on the 1 and 10 μmol/L concentrations. After treatment, explants were snap-frozen in liquid nitrogen and subsequently used for IHC-FR and WB analysis for dystrophin, β-dystroglycan, and α-sarcoglycan.
As concerns the BMD cohort, Velcade significantly up-regulated the expression of dystrophin and members of the DGC, in all of the patients examined (Table 1). In IHC-FR, as expected, α-sarcoglycan, β-dystroglycan, and dystrophin were reduced and exhibited a disorganized staining in the untreated samples, while they showed a more uniform and enhanced staining following proteasome inhibition (Figure 12). These results were confirmed by WB analysis, which demonstrated that dystrophin protein levels were increased on treatment. Dystrophin migrated at lower molecular weights according to each patient’s mutation (Figure 13). Notably, in most of our BMD patients, Velcade also up-regulated a lower protein band migrating at approximately 200 to 260 kDa. We hypothesize that this signal could relate to dystrophin isoform Dp260, whose transcripts originate from a promoter/sequence located in intron 29 of dystrophin gene.
Figure 12.

Inhibition of the proteasome pathway up-regulates the membrane expression of dystrophin and its associated proteins in muscle explants from BMD patients. Muscle explants were incubated in the absence or presence of Velcade at 1 and 10 μmol/L concentrations for 16 hours. Frozen skeletal muscle sections from untreated and Velcade-treated BMD explants were immunostained with specific antibodies directed against dystrophin (DYS), β-dystroglycan (β-DG), and α-sarcoglycan (α-SG). DYS, α-SG, and β-DG were reduced and showed an irregular membrane staining in the skeletal muscle fibers from untreated muscle explants, whereas they displayed an enhanced and uniform positivity on the muscle plasma membrane following Velcade exposure. Final magnification, ×25. Representative images are shown.
Figure 13.

Inhibition of the proteasome pathway up-regulates protein levels of members of the dystrophin-glycoprotein complex in human muscle explants from BMD patients. Muscle explants were incubated in the absence or presence of Velcade at 1 and 10 μmol/L concentrations for 16 hours. Total protein lysates from control, BMD untreated and Velcade-treated muscle explants were separated by SDS-polyacrylamide gel electrophoresis, transferred onto nitrocellulose membrane, and subjected to WB analysis with specific antibodies against dystrophin (DYS), α-sarcoglycan (α-SG), and β-dystroglycan (β-DG). In the Velcade-group DYS protein levels were significantly increased. Notably, DYS migrated at a molecular weight of 357 kDa that is expected according to the patient’s mutation (black arrow). A lower protein band (approximately 200 kDa) was detected (black arrowhead). This signal could relate to the dystrophin isoform Dp260, migrating at a lower molecular size because of the patient’s mutation. α-SG and β-DG were increased by 30% and 60%. Vinculin (Vinc) was used as a loading control.
To quantify Velcade’s effects on the protein levels of the members of the DGC, we included as controls muscle explants extracted from the biopsy of a patient affected by a mitochondrial or metabolic disorder. These samples were incubated in parallel and in the same conditions as the explants collected from BMD/DMD patients. When compared with the controls, BMD α-sarcoglycan levels were mildly decreased and they were up-regulated by 30% following Velcade exposure. Differently, β-dystroglycan was more severely affected in the BMD untreated group and in the presence of the proteasome inhibitor was up-regulated by 60% (Figure 13).
In the DMD group, Velcade increased the expression and membrane targeting of the dystrophin-associated proteins α-sarcoglycan and β-dystroglycan in six out of eight cases. In the DMD explants, we could detect positivity for dystrophin staining at the IHC-FR assays in six out of eight cases, but in no one at the WB analysis (Table 1). It is possible that the concentration of the dystrophin protein rescued by the drug is below the sensitivity of the WB assay used. Interestingly, the two DMD patients who did not respond at all to the treatment with the proteasome inhibitor displayed severe muscle damage, with the histological analysis of the biopsy showing a high replacement of muscle fibers with fibro-fatty tissue (Table 1).
Figure 14 shows IHC-FR data of Velcade treatment in one representative DMD responsive patient. In the untreated samples, α-sarcoglycan displays an irregular staining on the plasma membrane with some membrane areas totally negative. Similar data are shown for β-dystroglycan, which also seems to accumulate intracellularly. As expected, dystrophin is almost negative (the percentage of positive fibers is 1.2% ± 0.01, mean ± SE, n = 2 sections for each of the eight patients). In the 1 and 10 μmol/L Velcade-treated groups, α-sarcoglycan and β-dystroglycan display an enhanced and more uniform sarcolemmal staining. Notably, few fibers show also positivity for dystrophin (the percentage is 7.4 ± 0.5% and 5.8 ± 0.8% respectively for Velcade at 1 and 10 μmol/L; mean ± SE, n = 2 sections for each of the eight patients). To quantify these results, we next performed WB analysis from the total muscle lysates. When compared with a control sample, DMD α-sarcoglycan and β-dystroglycan protein levels were severely reduced and were increased respectively by 60% and 70% following proteasome inhibition (Figure 15). As mentioned above, we did not detect an effect on dystrophin.
Figure 14.

Inhibition of the proteasome pathway up-regulates the membrane expression of members of the dystrophin-glycoprotein complex in human muscle explants from DMD patients. Muscle explants were incubated in the absence or presence of Velcade at 1 and 10 μmol/L concentrations for 16 hours. Frozen skeletal muscle sections from untreated and Velcade-treated DMD explants were immunostained with specific antibodies directed against dystrophin (DYS), β-dystroglycan (β-DG), and α-sarcoglycan (α-SG). A-SG, β-DG were reduced in the skeletal muscle fibers from untreated muscle explants, whereas they displayed an enhanced and uniform membrane staining on the muscle plasma membrane following Velcade exposure. As expected in the untreated group, DYS is negative, while, in the Velcade samples, it displays positivity in few fibers. Final magnification, ×25. Representative images are shown.
Figure 15.

Inhibition of the proteasome pathway up-regulates the protein levels of members the dystrophin-glycoprotein complex in muscle explants from DMD patients. Muscle explants were incubated in the absence or presence of Velcade at 1 and 10 μmol/L concentrations for 16 hours. Total protein lysates from control, DMD untreated, and Velcade-treated muscle explants were separated by SDS-PAGE, transferred onto nitrocellulose membrane, and subjected to immunoblot analysis with specific antibodies against α-sarcoglycan (α-SG) and β-dystroglycan (β-DG). Note that α-SG and β-DG are severely reduced in the untreated muscle explant from the DMD patient when compared with the control subject (C). Velcade increases α-SG and β-DG protein levels by 60% and 70%. Vinculin (Vinc) was used as a loading control.
Discussion
DMD is a severe progressive disease affecting one of every 3500 males born and causing progressive muscle degeneration and weakness. A successful treatment for these patients has not been defined, as of yet. The high incidence of sporadic cases (1 in 10,000 sperm or eggs) indicates that genetic screening will never eliminate this disease and emphasizes the need for an effective therapy. Studies in different animal models have explored possible therapeutic strategies, ranging from skipping of dystrophin mutated exon, utrophin overexpression or cell therapy with mesangioblast stem cells, to the use of deacetylase inhibitors or blockage of myostatin pathway. Although encouraging, these approaches still lack approval for use in patients and data on long-term efficacy, safety, and tolerability are being developed.28
The present paper aims to thoroughly analyze the effects, and therapeutic potential of Velcade, a low-molecular-weight dipeptil boronic acid that binds with high affinity to the proteasome and dissociates slowly, resulting in a stable but reversible inhibition of the chymotryptic enzyme activity.
In recent years, several reports have highlighted the importance of the activation of the ubiquitin/proteasome proteolytic system in muscular dystrophies. Skeletal muscle has at least three different pathways for protein degradation: 1) proteolysis by lysosomal proteases, such as cathepsins; 2) proteolysis by non-lysosomal Ca2+-dependent proteases, such as calpains; and 3) proteolysis by non-lysosomal ATP-ubiquitin-dependent proteases, ie, the multi catalytic protease complex (or proteasome).3,29,30 In this context, DMD/BMD necrotic and degenerative muscle fibers display an overactivation of the ubiquitin-proteasome system.3,30
In dystrophic muscle tissue, the ubiquitin pathway can be triggered by multiple mechanisms: (i) distinct dystrophin mutations affect dystrophin folding properties, thus impairing its correct targeting to the plasma membrane; these mutants can be therefore retained in a intracellular compartment (endoplasmic reticulum or Golgi) where they are rapidly degraded; (ii) the primary dystrophin defect impairs the assembly of the DGC, thus releasing the single subunits intracellularly and addressing them to proteolytic degradation; or (iii) the damage on the cell membrane increases subsarcolemmal Ca++ levels, which then overactivate proteolytic enzymatic cascades.31,32
Among the number of small molecules that are able to pharmacologically target the proteasome, Velcade is particularly intriguing since it is the first inhibitor proven to be clinically useful. This drug was approved by the Food and Drug Administration and the European Medicines Agency for the primary treatment of multiple myeloma. Moreover, this compound is currently tested in different phase II clinical trials as possible antitumor agent in ovarian, lung, prostate, and pancreatic cancers, and melanoma, and glioblastoma.7,8,33,34 Notably, two phase I studies involving Velcade were conducted in children affected by refractory leukemia and solid tumors.17,35
Our previous studies had indicated that Velcade up-regulates the expression of dystrophin and members of the DGC once injected locally into the gastrocnemius muscles of mdx mice.10
Here, through a 2-week period of systemic treatment, we confirm the efficiency of the drug on the rescue of dystrophin, α-sarcoglycan, and β-dystroglycan muscle membrane localization and provide new data on Velcade effects on histopathological and clinical aspects of the mdx phenotype and on the drug mechanisms of action.
In gastrocnemius muscles, the dystrophin/DGC displays a positive immunostaining on the sarcolemma of Velcade-treated mdx mice, whereas they are absent or greatly reduced in untreated mdx controls. These results are quantified by WB analysis and confocal microscopy, which shows that the drug trends to have a more pronounced effect in small diameter muscle cells. At the histological examination, skeletal muscle biopsies from untreated mdx mice show all the key histological features of muscular dystrophy, including variable fiber size, centralized nuclei, macrophagic infiltration, lipidic vesicles, and connective tissue replacement. These inflammatory and degenerative aspects are greatly improved in skeletal muscle biopsies from Velcade-treated mice. In details, inhibition of the proteasome pathway is associated to a decrease of the number of F4/80 positive macrophages and to a reduction of IL-6 protein levels.
Accordingly, untreated mdx muscle fibers display a strong red autofluorescent EBD signal, a marker of muscle injury, whereas Velcade-treated mdx muscle cells show little or no signal.
The effects of the proteasome inhibitor on dystrophin expression and on histological signs of muscular dystrophy were hence confirmed in diaphragm tissue, which in terms of phenotypic severity, most closely resembles the histopathological features described in DMD patients.
We next evaluated whether part of Velcade positive effects on mdx muscle lesions could be related to an enhancement of muscle cell regeneration. Indeed, after 2 weeks of treatment, the proteasome inhibitor increased the expression of embryonal-MHC, typically present in myofibers at an early phase of differentiation and up-regulated MyoD and Myf-5, two molecules expressed in activated muscle satellite cells and thus markers of regeneration.24 Notably, Velcade is known to enhance osteoblastogenesis when administered in vivo in a murine model of multiple myeloma. This effect is partially caused by the block of proteasomal degradation of the osteoblast differentiation factor Runx-2.36 It is thus possible that in muscle tissue the drug exerts a similar direct effect by acting on the cell turnover of transcription factors involved in the recruitment of undifferentiated myoblasts.
In cancer cells, Velcade’s mechanism of action includes the inhibition of NF-κB, a molecule involved in growth and survival of neoplastic cells, in inflammatory responses and in the processes of muscle degeneration/regeneration observed in DMD.8 NF-κB forms a dimer with the inhibitory protein IκB, which maintains it in the cytoplasm in an inactive form. Under a variety of signals, IkB is phosphorylated and is targeted to degradation by the proteasome. The released NF-κB complex hence translocates into the nucleus where it regulates the transcription of distinct growth/survival factor-associated genes. By blocking proteasomal activity, treatment with Velcade leads to an up-regulation of total inactive NF-κB, thus reducing anti-apoptotic factors and inflammatory molecules.37 Our results show here that Velcade treatment decreases the nuclear levels of p65 NF-κB, corroborating the idea that inhibition of NF-κB pathway represents a direct effect of the drug on the inflammatory component of the mdx dystrophic phenotype.
Although the mdx mouse model is used as the platform where to first assess the potential of new therapeutic approaches to DMD, there are many differences in the clinical phenotype and in the pathogenetic mechanisms when compared with the human pathology. These discrepancies constitute major caveats to this approach.38 For this reason, we evaluated the effects of proteasome inhibition also in explants from fresh human muscular biopsies of DMD and BMD patients. We believe that an ex vivo model can substantially integrate and validate the data obtained from the in vivo experiments. Small fragments from the bioptic tissue are immediately placed in culture medium enriched with growth factors, tested with compounds of interest, and hence analyzed.
Our results indicate that in explants from BMD patients, blocking of the proteasome pathway is able to promote the membrane localization of dystrophin mutants and, accordingly, of α-sarcoglycan and β-dystroglycan proteins. This effect is observed in all our BMD patients and appears to be independent from the type of mutation diagnosed. In our BMD cohort, six patients display a deletion or a missense mutation (one case) in the protein segments of repeats 16 to 21 of the central rod domain, one patient has a duplication in repeats 2 to 3 of the rod segment, and one patient has a duplication in the N-terminal dystrophin actin-binding region.
As concerns the DMD group, Velcade effects are straightforward on the members of the DGC, α-sarcoglycan and β-dystroglycan, which, following exposure to the drug, are increased and are targeted to the plasma membrane. In contrast, as concerns dystrophin, Velcade treatment induces only a modest increase of the protein level by IHC-FR analysis.
These results are only partially surprising. First, a number of dystrophin mutations may result in reduced dystrophin mRNA stability and in activation of the nonsense-mediated mRNA decay cell quality control mechanism, thus leading to nearly undetectable concentrations of truncated protein.39 These are most likely the cases where inhibition of the proteasome pathway might fail to up-regulate dystrophin protein levels.
Secondarily, according to our results in DMD patients, Velcade effects appear to inversely correlate with the severity of healthy muscle tissue replacement with connective tissue. This would suggest that treatment with proteasome inhibitors should be started at an early stage of disease.
Finally, the culture conditions of the explant methodology we adopted might still not be optimized to analyze highly unstable truncated proteins, which are extremely susceptible to aspecific culture-related proteolytic degradation.
After taking these issues into consideration, it is still important to emphasize that the aim of our work was to evaluate the consequences of the inhibition of the proteasome pathway on those proteins of the DGC, which are secondarily affected in DMD. Indeed, the role and biological relevance of a functional DGC in conditions of dystrophin deficiency is only partially clarified, and data in literature are controversial.40,41,42,43
Previous studies indicated that an intact sarcoglycan complex provides minimal protection in the presence of the shorter N-terminal domain-truncated dystrophin isoforms, such as Dp71 and Dp116.40 In contrast, recent data showed that subphysiological sarcoglycan expression plays a role in maintaining the mild dystrophic phenotype observed in mdx mice and the double δ-sarcoglycan/mdx mice exhibit a severe worsening of the muscular degeneration process.44
In DMD patients, up to now, no data have been developed on the correlation between degrees of expression or membrane localization of dystrophin-associated proteins and severity or prognosis of the clinical phenotype. This lack of information is probably related to the high degree of muscle tissue degeneration observed in most of the newly diagnosed DMD patients and to the limited number of clinical outcomes evaluated by the physicians. It is therefore possible to speculate that in DMD patients an efficient rescue of a functional membrane DGC might improve the progression of the disease.
In conclusion, inhibition of the proteasome pathway through the drug Velcade promotes the expression and membrane localization of dystrophin and dystrophin-associated proteins in mdx mice and in pathological models of BMD. In explants from DMD patients, Velcade increases the protein levels and the membrane targeting of members of the DGC, while its effect on dystrophin are modest and detectable only by IHC-FR.
Our hypothesis is that proteasome inhibitors could act on two aspects of the dystrophic process: on one hand, they prevent the proteolysis of dystrophin and/or of the proteins forming the dystrophin complex (sarcoglycans, dystroglycans), thus contributing to the stability of the muscle fiber; on the other hand, they modulate the inflammatory (NF-κB) and fibrogenic (transforming growth factor β) response.
Future studies should address the effects of pharmacological proteasomal inhibition on mdx muscle strength and functional motility. Moreover, as chronic exercise may accelerate the active cycle of degeneration/regeneration in mdx skeletal muscle, this model could further define the effects of Velcade on the expression of dystrophin and members of the DGC.
Finally, although our paper focused on the efficacy of Velcade as single agent in dystrophinopathies, we recognize the importance of combining this drug with other agents, such as corticosteroids. These experiments and the analysis of the drug mechanisms of action in dystrophin-deficient muscle tissue will be the object of our future research.
Acknowledgments
We acknowledge Dr. Paolo Broda for helpful technical advice.
Footnotes
Address reprint requests to Elisabetta Gazzerro, M.D., Muscular and Neurodegenerative Disease Unit, University of Genoa, G. Gaslini Institute, Largo G. Gaslini 5, I-16147 Genoa, Italy. E-mail: elisabettagazzerro@ospedale-gaslini.ge.it.
Supported by the Association Française contre les Myopathies (AFM) grant 2007 # 13036 and AFM grant 2008 # 13771 and from funds donated by Prof. R. Bulgarelli. M.P.L., F.S., and G.B. were supported by a grant from the Muscular Dystrophy Association.
E.G. and S.A. contributed equally to this work.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
A guest editor acted as editor-in-chief for this manuscript. No person at Thomas Jefferson University or Albert Einstein College of Medicine was involved in the peer review process or final disposition for this article.
References
- Nowak KJ, Davies KE. Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO Rep. 2004;5:872–876. doi: 10.1038/sj.embor.7400221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deconinck N, Dan B. Pathophysiology of Duchenne muscular dystrophy: current hypotheses. Pediatr Neurol. 2007;36:1–7. doi: 10.1016/j.pediatrneurol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Kumamoto T, Fujimoto S, Ito T, Horinouchi H, Ueyama H, Tsuda T. Proteasome expression in the skeletal muscles of patients with muscular dystrophy. Acta Neuropathol. 2000;100:595–602. doi: 10.1007/s004010000229. [DOI] [PubMed] [Google Scholar]
- Bonuccelli G, Sotgia F, Schubert W, Park DS, Frank PG, Woodman SE, Insabato L, Cammer M, Minetti C, Lisanti MP. Proteasome inhibitor (MG-132) treatment of mdx mice rescues the expression and membrane localization of dystrophin and dystrophin-associated proteins. Am J Pathol. 2003;163:1663–1675. doi: 10.1016/S0002-9440(10)63523-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assereto S, Stringara S, Sotgia F, Bonuccelli G, Broccolini A, Pedemonte M, Traverso M, Biancheri R, Zara F, Bruno C, Lisanti MP, Minetti C. Pharmacological rescue of the dystrophin-glycoprotein complex in Duchenne and Becker skeletal muscle explants by proteasome inhibitor treatment. Am J Physiol Cell Physiol. 2006;290:C577–C582. doi: 10.1152/ajpcell.00434.2005. [DOI] [PubMed] [Google Scholar]
- Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol. 2001;8:739–758. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- Armand JP, Burnett AK, Drach J, Harousseau JL, Lowenberg B, San Miguel J. The emerging role of targeted therapy for hematologic malignancies: update on bortezomib and tipifarnib. Oncologist. 2007;12:281–290. doi: 10.1634/theoncologist.12-3-281. [DOI] [PubMed] [Google Scholar]
- Nencioni A, Grunebach F, Patrone F, Ballestrero A, Brossart P. Proteasome inhibitors: antitumor effects and beyond. Leukemia. 2007;21:30–36. doi: 10.1038/sj.leu.2404444. [DOI] [PubMed] [Google Scholar]
- Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PM, Carathers M, Li ZW, Beg AA, Ghosh S, Sahenk Z, Weinstein M, Gardner KL, Rafael-Fortney JA, Karin M, Tidball JG, Baldwin AS, Guttridge DC. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. 2007;117:889–901. doi: 10.1172/JCI30556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonuccelli G, Sotgia F, Capozza F, Gazzerro E, Minetti C, Lisanti MP. Localized treatment with a novel FDA-approved proteasome inhibitor blocks the degradation of dystrophin and dystrophin-associated proteins in mdx mice. Cell Cycle. 2007;6:1242–1248. doi: 10.4161/cc.6.10.4182. [DOI] [PubMed] [Google Scholar]
- Vainzof M, Ayub-Guerrieri D, Onofre PC, Martins PC, Lopes VF, Zilberztajn D, Maia LS, Sell K, Yamamoto LU. Animal models for genetic neuromuscular diseases. J Mol Neurosci. 2008;34:241–248. doi: 10.1007/s12031-007-9023-9. [DOI] [PubMed] [Google Scholar]
- Costelli P, Bossola M, Muscaritoli M, Grieco G, Bonelli G, Bellantone R, Doglietto GB, Baccino FM, Rossi Fanelli F. Anticytokine treatment prevents the increase in the activity of ATP-ubiquitin- and Ca(2+)-dependent proteolytic systems in the muscle of tumour-bearing rats. Cytokine. 2002;19:1–5. doi: 10.1006/cyto.2002.1036. [DOI] [PubMed] [Google Scholar]
- Rasband WS. US National Institute of Health,: Bethesda MD,; ImageJ, 1997–2007 [Google Scholar]
- Dubowitz V, Sewry C. Muscle biopsy. Philadelphia: Saunders, M.J.H. and L.C. Elsevier,; 2007:p. 84. [Google Scholar]
- Messina S, Altavilla D, Aguennouz M, Seminara P, Minutoli L, Monici MC, Bitto A, Mazzeo A, Marini H, Squadrito F, Vita G. Lipid peroxidation inhibition blunts nuclear factor-kappaB activation, reduces skeletal muscle degeneration, and enhances muscle function in mdx mice. Am J Pathol. 2006;168:918–926. doi: 10.2353/ajpath.2006.050673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignole C, Marimpietri D, Pastorino F, Nico B, Di Paolo D, Cioni M, Piccardi F, Cilli M, Pezzolo A, Corrias MV, Pistoia V, Ribatti D, Pagnan G, Ponzoni M. Effect of bortezomib on human neuroblastoma cell growth, apoptosis, and angiogenesis. J Natl Cancer Inst. 2006;98:1142–1157. doi: 10.1093/jnci/djj309. [DOI] [PubMed] [Google Scholar]
- Blaney SM, Bernstein M, Neville K, Ginsberg J, Kitchen B, Horton T, Berg SL, Krailo M, Adamson PC. Phase I study of the proteasome inhibitor bortezomib in pediatric patients with refractory solid tumors: a Children’s Oncology Group study (ADVL0015). J Clin Oncol. 2004;22:4804–4809. doi: 10.1200/JCO.2004.12.185. [DOI] [PubMed] [Google Scholar]
- Houghton PJ, Morton CL, Kolb EA, Lock R, Carol H, Reynolds CP, Keshelava N, Maris JM, Keir ST, Wu J, Smith MA. Initial testing (stage 1) of the proteasome inhibitor bortezomib by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;50:37–45. doi: 10.1002/pbc.21214. [DOI] [PubMed] [Google Scholar]
- Wakayama Y, Inoue M, Kojima H, Jimi T, Yamashita S, Kumagai T, Shibuya S, Hara H, Oniki H. Altered alpha1-syntrophin expression in myofibers with Duchenne and Fukuyama muscular dystrophies. Histol Histopathol. 2006;21:23–34. doi: 10.14670/HH-21.23. [DOI] [PubMed] [Google Scholar]
- Repetto S, Bado M, Broda P, Lucania G, Masetti E, Sotgia F, Carbone I, Pavan A, Bonilla E, Cordone G, Lisanti MP, Minetti C. Increased number of caveolae and caveolin-3 overexpression in Duchenne muscular dystrophy. Biochem Biophys Res Commun. 1999;261:547–550. doi: 10.1006/bbrc.1999.1055. [DOI] [PubMed] [Google Scholar]
- Vaghy PL, Fang J, Wu W, Vaghy LP. Increased caveolin-3 levels in mdx mouse muscles. FEBS Lett. 1998;431:125–127. doi: 10.1016/s0014-5793(98)00738-8. [DOI] [PubMed] [Google Scholar]
- Sotgia F, Lee JK, Das K, Bedford M, Petrucci TC, Macioce P, Sargiacomo M, Bricarelli FD, Minetti C, Sudol M, Lisanti MP. Caveolin-3 directly interacts with the C-terminal tail of beta-dystroglycan. Identification of a central WW-like domain within caveolin family members. J Biol Chem. 2000;275:38048–38058. doi: 10.1074/jbc.M005321200. [DOI] [PubMed] [Google Scholar]
- Li H, Mittal A, Makonchuk DY, Bhatnagar S, Kumar A. Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum Mol Genet. 2009;18:2584–2598. doi: 10.1093/hmg/ddp191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straface G, Aprahamian T, Flex A, Gaetani E, Biscetti F, Smith RC, Pecorini G, Pola E, Angelini F, Stigliano E, Castellot JJ, Jr, Losordo DW, Pola R. Sonic hedgehog regulates angiogenesis and myogenesis during post-natal skeletal muscle regeneration. J Cell Mol Med. 2008;13:2424–2435. doi: 10.1111/j.1582-4934.2008.00440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidball JG. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum Mol Genet. 2009;18:482–496. doi: 10.1093/hmg/ddn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glesby MJ, Rosenmann E, Nylen EG, Wrogemann K. Serum CK, calcium, magnesium, and oxidative phosphorylation in mdx mouse muscular dystrophy. Muscle Nerve. 1988;11:852–856. doi: 10.1002/mus.880110809. [DOI] [PubMed] [Google Scholar]
- Traverso M, Malnati M, Minetti C, Regis S, Tedeschi S, Pedemonte M, Bruno C, Biassoni R, Zara F. Multiplex real-time PCR for detection of deletions and duplications in dystrophin gene. Biochem Biophys Res Commun. 2006;339:145–150. doi: 10.1016/j.bbrc.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Ludolph AC. Neuromuscular diseases: new hopes for alleviation and elimination. Lancet Neurol. 2009;8:16–17. doi: 10.1016/S1474-4422(08)70277-1. [DOI] [PubMed] [Google Scholar]
- Costelli P, Reffo P, Penna F, Autelli R, Bonelli G, Baccino FM. Ca(2+)-dependent proteolysis in muscle wasting. Int J Biochem Cell Biol. 2005;37:2134–2146. doi: 10.1016/j.biocel.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Briguet A, Erb M, Courdier-Fruh I, Barzaghi P, Santos G, Herzner H, Lescop C, Siendt H, Henneboehle M, Weyermann P, Magyar JP, Dubach-Powell J, Metz G, Meier T. Effect of calpain and proteasome inhibition on Ca2+-dependent proteolysis and muscle histopathology in the mdx mouse. FASEB J. 2008;22:4190–4200. doi: 10.1096/fj.07-099036. [DOI] [PubMed] [Google Scholar]
- Whitehead NP, Yeung EW, Allen DG. Muscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen species. Clin Exp Pharmacol Physiol. 2006;33:657–662. doi: 10.1111/j.1440-1681.2006.04394.x. [DOI] [PubMed] [Google Scholar]
- Galbiati F, Volonte D, Minetti C, Chu JB, Lisanti MP. Phenotypic behavior of caveolin-3 mutations that cause autosomal dominant limb girdle muscular dystrophy (LGMD-1C). Retention of LGMD-1C caveolin-3 mutants within the golgi complex. J Biol Chem. 1999;274:25632–25641. doi: 10.1074/jbc.274.36.25632. [DOI] [PubMed] [Google Scholar]
- Davis NB, Taber DA, Ansari RH, Ryan CW, George C, Vokes EE, Vogelzang NJ, Stadler WM. Phase II trial of PS-341 in patients with renal cell cancer: a University of Chicago phase II consortium study. J Clin Oncol. 2004;22:115–119. doi: 10.1200/JCO.2004.07.165. [DOI] [PubMed] [Google Scholar]
- Faderl S, Rai K, Gribben J, Byrd JC, Flinn IW, O'Brien S, Sheng S, Esseltine DL, Keating MJ. Phase II study of single-agent bortezomib for the treatment of patients with fludarabine-refractory B-cell chronic lymphocytic leukemia. Cancer. 2006;107:916–924. doi: 10.1002/cncr.22097. [DOI] [PubMed] [Google Scholar]
- Horton TM, Pati D, Plon SE, Thompson PA, Bomgaars LR, Adamson PC, Ingle AM, Wright J, Brockman AH, Paton M, Blaney SM. A phase 1 study of the proteasome inhibitor bortezomib in pediatric patients with refractory leukemia: a Children’s Oncology Group study. Clin Cancer Res. 2007;13:1516–1522. doi: 10.1158/1078-0432.CCR-06-2173. [DOI] [PubMed] [Google Scholar]
- Pennisi A, Li X, Ling W, Khan S, Zangari M, Yaccoby S. The proteasome inhibitor, bortezomib suppresses primary myeloma and stimulates bone formation in myelomatous and nonmyelomatous bones in vivo. Am J Hematol. 2009;84:6–14. doi: 10.1002/ajh.21310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani M, Crinelli R, Bianchi M, Antonelli A. The ubiquitin-dependent proteolytic system and other potential targets for the modulation of nuclear factor-kB (NF-kB). Curr Drug Targets. 2000;1:387–399. doi: 10.2174/1389450003349056. [DOI] [PubMed] [Google Scholar]
- Spurney CF, Gordish-Dressman H, Guerron AD, Sali A, Pandey GS, Rawat R, Van Der Meulen JH, Cha HJ, Pistilli EE, Partridge TA, Hoffman EP, Nagaraju K. Preclinical drug trials in the mdx mouse: assessment of reliable and sensitive outcome measures. Muscle Nerve. 2009;39:591–602. doi: 10.1002/mus.21211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2:731–740. doi: 10.1016/s1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- Cox GA, Sunada Y, Campbell KP, Chamberlain JS. Dp71 can restore the dystrophin-associated glycoprotein complex in muscle but fails to prevent dystrophy. Nat Genet. 1994;8:333–339. doi: 10.1038/ng1294-333. [DOI] [PubMed] [Google Scholar]
- Gardner KL, Kearney JA, Edwards JD, Rafael-Fortney JA. Restoration of all dystrophin protein interactions by functional domains in trans does not rescue dystrophy. Gene Ther. 2006;13:744–751. doi: 10.1038/sj.gt.3302686. [DOI] [PubMed] [Google Scholar]
- Li D, Yue Y, Duan D. Preservation of muscle force in Mdx3cv mice correlates with low-level expression of a near full-length dystrophin protein. Am J Pathol. 2008;172:1332–1341. doi: 10.2353/ajpath.2008.071042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge LM, Haraguchiln M, Chamberlain JS. Dissecting the signaling and mechanical functions of the dystrophin-glycoprotein complex. J Cell Sci. 2006;119:1537–1546. doi: 10.1242/jcs.02857. [DOI] [PubMed] [Google Scholar]
- Li D, Long C, Yue Y, Duan D. Sub-physiological sarcoglycan expression contributes to compensatory muscle protection in mdx mice. Hum Mol Genet. 2009;18:1209–1220. doi: 10.1093/hmg/ddp015. [DOI] [PMC free article] [PubMed] [Google Scholar]