

Abstract
Although male sex is a well-recognized risk factor for stroke, the role of androgens in cerebral ischemia remains unclear. Therefore, we evaluated effects of testosterone on infarct size in both young adult and middle-aged rats (Wistar, 3-month versus 14-month old) and mice (C57/BL6, 3-month versus 12-month old) subjected to middle cerebral artery occlusion. In young adult groups, castrates displayed less ischemic damage as compared with intact males and castrates with testosterone replacement (Cortex: 24% in castrates versus 42% in intact versus 40% with testosterone; Striatum: 45% versus 73% versus 70%) at 22 h reperfusion. Surprisingly, supplementing testosterone in middle-aged rats to the physiologic levels ordinarily seen in young males reduced infarction (Cortex: 2% with testosterone versus 31%; Striatum: 38% with testosterone versus 68%). Testosterone effects on infarct size were blocked by the androgen receptor (AR) antagonist flutamide and further confirmed in young versus middle-aged mice. Baseline cerebral aromatase mRNA levels and activity were not different between young and middle-aged rats. Aromatase activity increased in ischemic tissue, but only in young males. Lastly, stroke damage was not different in aging aromatase knockout mice versus wild-type controls. Our findings indicate that testosterone's effects in experimental stroke are age dependent, mediated via AR, but not cerebral aromatase.
Keywords: testosterone, aromatase, androgen receptor, cerebral ischemia, stroke, aging
Introduction
One well-recognized risk factor for clinical stoke is male sex. Nevertheless, surprisingly little attention has been directed toward the role of male sex or androgenic steroids in outcome from cerebral ischemia. The role of androgens in men's susceptibility to stroke remains largely unknown. Low plasma testosterone is inversely associated with stroke severity, infarct size, and 6-month mortality (Jeppesen et al, 1996), and stroke incidence increases in men during middle age when androgen levels decline (Hollander et al, 2003). Available data from experimental stroke studies that evaluated androgens are exclusively obtained in young adult animals. Testosterone and its potent metabolite dihydrotestosterone increase stroke damage after cerebral ischemia in young adult rodents (Hawk et al, 1998; Yang et al, 2002; Cheng et al, 2007). However, another report suggests that testosterone exerts beneficial effects such as accelerating functional recovery after cerebral ischemia (Pan et al, 2005).
One explanation for the lack of agreement amongst these studies is that testosterone may shift brain susceptibility to ischemic damage in an age-dependent manner, one that reflects the duration of exposure to low gonadal androgen production. Male sex steroid production is not constant and declines throughout the middle years of life in many individuals. In some reports, neuroendocrine aging in men becomes significant by the fourth decade of life (for reviews see Anawalt and Merriam, 2001; Vermeulen, 2000). Unlike the rapid and overt loss of estrogen in women, loss of testosterone and 5α-dihydrotestosterone is gradual in men and can be clinically subtle. In this study, we hypothesized that loss of androgens in the middle years could change the brain's hormonal landscape and yield different stroke outcomes than those observed in a young male.
Testosterone can act directly on its cognate androgen receptors (AR) or after aromatization to protective 17β-estradiol by the cytochrome P450 aromatase. The latter metabolic step is essential in androgenic signaling under physiologic conditions, (Simpson et al, 2002; Roselli, 2007) and is a mechanism that has been increasingly recognized as one means by which brain defends itself against ischemic or traumatic injury (Azcoitia et al, 2001; McCullough et al, 2003; Carswell et al, 2005; Liu et al, 2007; Roselli, 2007). However, previous studies have not determined if testosterone's effects on cerebral ischemic outcome are mediated via AR signaling or via cerebral aromatase.
Accordingly, we used young adult and reproductively senescent, middle-aged male rats and mice to determine if androgens confer protection or exacerbate damage after cerebral ischemia in the male. We chose 12- to 14-month-old rodents of both species to model the effects of natural male sex steroid loss during midlife. To test the hypothesis that testosterone signals through the AR and impacts postischemic tissue outcomes, we determined if flutamide, a selective AR antagonist, blocks testosterone effects on infarct size in either age. Alternatively, we evaluated the possibility that aromatization of testosterone contributes to ischemic outcomes more greatly in reproductively senescent males versus young adults. To test this hypothesis, cerebral aromatase mRNA levels and enzymatic activity were measured under basal and postischemic conditions in both age groups. To further test the hypothesis, we used middle-aged mice genetically deficient in the aromatase and compared stroke outcome to that of age-matched, wild-type mice.
Materials and methods
Experimental Animals
This study was conducted in accordance with NIH guidelines, and all protocols were approved by the Animal Care and Use Committee of Oregon Health & Science University. Young adult male animals (3-month-old Wistar rats and C57/BL6 mice) were castrated under halothane anesthesia 1 week before middle cerebral artery occlusion (MCAO), as described previously (Toung et al, 1998). Reproductively senescent, mid-life male animals (14-month-old Wistar rats and 12-month-old C57/BL6 mice) were also studied under identical conditions of ischemia and reperfusion as the young adult males. Animals randomized to receive testosterone were implanted with subcutaneous pellets (25 mg/pellet for rat; 5 mg/pellet for mouse) with or without AR antagonist flutamide implants (25 mg/pellet for rat) 1 week before MCAO. Young adult rat experimental groups included intact males, castrated males, castrated males with 25-mg testosterone implantation, and castrated males with 25-mg testosterone plus 25-mg flutamide implantation. Middle-aged rat experimental groups included intact males, intact males with 25-mg testosterone implantation, and intact males with 25-mg testosterone plus 25-mg flutamide implantation. As gonadally intact males experience natural loss of testosterone production by middle age and hence very low androgen levels, these cohorts were not surgically castrated. Young adult mouse experimental groups included intact males, castrated males, and castrated males with 5-mg testosterone implantation. Middle-aged mouse experimental group included intact males and intact males with 5-mg testosterone implantation. Baseline aromatase mRNA and enzyme activity were measured on naive animals, and postischemic levels were assayed at 3 and 6 h reperfusion on intact young adult male rats and intact middle-aged male rats. Aromatase knockout (ArKO) mice were generated as described previously (McCullough et al, 2003), and genotype was confirmed by PCR using tail DNA (Robertson et al, 1999).
Rat Middle Cerebral Artery Occlusion
Middle cerebral artery occlusion (2 h) was induced by the intraluminal filament technique as previously described (Alkayed et al, 1998). Briefly, animals were anesthetized under halothane (5% for induction and 1% to 2% for the duration of the surgery and MCAO) via facemask in O2-enriched air. A femoral artery catheter was placed to monitor mean artery blood pressure and arterial blood pH and PaCO2. Rectal and temporalis temperatures were continuously monitored (Mon-a-therm; Mallinckrodt Medical Inc., St Louis, MO, USA) and maintained at 37 ± 0.5°C by heat lamps and water pads. Adequacy of vessel occlusion and reperfusion was assessed by laser-doppler flow signal (LDF, Moor Instruments Ltd, Oxford, England) via a probe placed 6 mm lateral and 2 mm posterior to the bregma. Occlusion was accomplished by introducing a 4-0 nylon monofilament surgical suture with a heat-rounded tip through the right common carotid artery and internal carotid artery to the origin of the middle cerebral artery as indicated by abrupt and significant reduction in LDF. Laser-doppler flow signal was continuously monitored during occlusion and 30 mins after reperfusion to confirm a uniform insult among the animals. Rats were decapitated at 22 h reperfusion, plasma was collected. Free and total testosterone levels were measured using commercial radioimmunoassay kit, per manufacturer's instruction (Diagnostic Products Corp., Los Angeles, CA, USA).
Mouse Middle Cerebral Artery Occlusion
Cerebral ischemia was induced by MCAO as previously described (Sampei et al, 2000a, b). Briefly, mice were anesthetized with 1% to 1.2% halothane in O2-enriched air by facemask. Rectal and temporalis muscle temperatures were controlled at 37 ± 0.5°C throughout the experiment with heat lamps and water pads. Unilateral MCAO was achieved by inserting a 6-0 nylon monofilament into the right internal carotid artery via an external carotid artery stump and then positioning the filament tip at a distance of 6 mm beyond the internal carotid/pterygopalatine bifurcation. Adequacy of artery occlusion was confirmed by monitoring cortical blood flow at the onset of the occlusion with a LDF probe affixed to the skull. After securing the filament in place, the surgical site was sutured closed. Then each animal was awakened during occlusion, and examined for gross neurologic deficits as follows: 0 = no deficit; 1 = failure to extend forelimb; 2 = circling; 3 = unilateral weakness; 4 = no spontaneous motor activity. Mice with clear neurologic deficits were reanesthetized with halothane for filament removal at 2 h occlusion. Mice were decapitated at 22 h reperfusion, and brains were collected for infarct volume analysis.
Infarct Volume Analysis
Brains were collected at 22 h reperfusion for infarct volume analysis. The rat brain was cut into seven 2-mm-thick coronal sections, and the mouse brain was cut into five 2-mm-thick coronal sections. Slices were then incubated in 2% 2,3,5-triphenyltetrazolium chloride (Sigma, St Louis, MO, USA) for 10 mins for each side, fixed in 10% formalin overnight, and then photographed. Images were analyzed in a masked fashion with Image Analysis Software (MCID; Imaging Research). Infarction sizes were expressed as a percentage of the ipsilateral structure.
Aromatase Activity Assay
Gonadally intact young adult or middle-aged male rats were subjected to 2 h MCAO, and tissue was subdissected from the ipsilateral and contralateral sides of the cortex, striatum, and hippocampus at two time points, 3 or 6 h reperfusion. Tissue was also collected from the same brain regions of naive animals to measure baseline aromatase activity. Collected samples were immediately frozen in 2-methylbutane at −30°C and stored at −80°C until analyzed. Aromatase activity was quantified with a previously validated radiometric assay that measures stereospecific loss of tritium from the C-1β position of [1β-3H]androstenedione; tritium is produced in proportion to the amount of estrogen formed by aromatase (Roselli and Resko, 1984). Briefly, brain tissues from individual rats were homogenized in 30 volumes of phosphate buffer (10 mmol/L KH2PO4, 100 mmol/L KCl, 1 mmol/L EDTA, and 1 mmol/L dithiothreitiol, pH 7.4). Protein concentrations were determined by the Bradford method. Aliquots of these homogenates (100 μL) were then incubated for 1 h at 37°C with 0.2 μmol/L [1β-3H]androstenedione (PerkinElmer, Boston, MA, USA; specific activity = 25.3 Ci/mmol) in the presence of an NADPH generating system. The reactions were stopped with 10% trichloroacetic acid (wt/vol) containing 20 mg/mL charcoal. The 3H2O generated was purified on Dowex minicolumns and quantified by liquid scintillation spectroscopy. Aromatase activity was expressed as fmol/h/mg protein. We validated this method in our preliminary studies, in which aromatase activity in striatum and hippocampus exhibited Michaelis–Menton enzyme kinetics and activity was inhibited by ∼80% when tissue homogenates were incubated in the presence of a 100-fold excess (20 μmol/L) of the aromatase inhibitor Fadrozole.
Quantitative PCR Measurements of Aromatase mRNA
Gonadally intact young adult or middle-aged male rats were subjected to 2 h MCAO, and tissue samples were collected as described for aromatase activity assay. Total RNA was isolated using the RNeasy Mini kit (Qiagen, Valencia, CA, USA) and further treated with DNase I, per manufacturer's instructions. First strand cDNA was reverse transcribed from 500 ng total RNA with a High Capacity cDNA archive Kit (Applied Biosystems, Foster City, CA, USA), and quantitative PCR (qPCR) reactions were performed in triplicate on an ABI Prism 7000 sequence detection system using 50 ng cDNA. Parameters for qPCR were incubation at 50°C for 2 mins, denaturing at 95°C for 10 mins followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. The qPCR primers and probe were designed from the known sequence (GenBank accession no. NM_017085) using Primer Express 2.0 (Applied Biosystems): 5′-CCGGAAACTGTGCCTGTC AT-3′ (forward primer), 5′-GAGCTCTCACAATTCCG AATCA-3′ (reverse primer), and 5′-FAM-TGCCACT TCTGCTGATCATGGGCC-TAMRA (Probe), targeting the second exon of the aromatase gene, which have been reported to be specifically absent in rat cortical aromatase mRNA but present in the full-length mRNA (Yamada-Mouri et al, 1997). 18S RNA was also assayed for each sample using 5 ng total cDNA (Eurogentec North America Inc., San Diego, CA, USA). Final results were normalized to 18S RNA and expressed as the ratio of target gene to 18S.
Statistical Analysis
Values are expressed as mean ± s.e.m. Comparisons were subjected to t-test (for two-group comparisons), one-way analysis of variance (for multi-group comparisons), or two-way analysis of variance (for physiologic variables) with post hoc Newman–Keuls test. Statistical significance was set at P < 0.05. All statistical analyses were performed with SigmaStat Statistical Software Version 2.0 (SPSS Inc., Chicago, IL, USA).
Results
To test our hypothesis that testosterone exerts age-dependent effects on stroke outcome, we assessed the infarct sizes after MCAO in both young adult and middle-aged rats and mice with or without physiologic levels of testosterone. During MCAO, there was no difference in physiologic parameters and LDF reduction among experimental groups (Table 1). No rats or mice died before 22 h reperfusion, and no mice were excluded because of the absence of neurologic deficits after occlusion.
Table 1.
Intraischemic physiological parameters and reduction in LDF
| Groups | pH | PaCO2 (mmHg) | MAP (mmHg) | Reduction in LDF (% of baseline) |
|---|---|---|---|---|
| Young adult rats | ||||
| Intact (n = 11) | 7.39 ± 0.01 | 40 ± 2 | 99 ± 3 | 30 ± 2 |
| Cast (n = 11) | 7.40 ± 0.01 | 37 ± 1 | 109 ± 3 | 30 ± 3 |
| Cast+T25 (n = 11) | 7.40 ± 0.01 | 40 ± 2 | 105 ± 3 | 32 ± 3 |
| Cast+T25 +Flu (n = 9) | 7.43 ± 0.01 | 37 ± 2 | 101 ± 3 | 31 ± 3 |
| Middle-aged rats | ||||
| Intact (n = 9) | 7.39 ± 0.01 | 44 ± 2 | 102 ± 6 | 31 ± 4 |
| Intact+T25 (n = 7) | 7.36 ± 0.02 | 51 ± 4 | 89 ± 4 | 42 ± 1 |
| Intact+T25+Flu (n = 9) | 7.37 ± 0.02 | 51 ± 2 | 104 ± 5 | 35 ± 3 |
| Young adult mice | ||||
| Intact (n = 10) | 7.39 ± 0.01 | 43 ± 2 | 74 ± 2 | 18 ± 1 |
| Cast (n = 10) | 7.39 ± 0.01 | 44 ± 2 | 77 ± 2 | 18 ± 2 |
| Cast+T5 (n = 10) | 7.38 ± 0.02 | 44 ± 2 | 84 ± 5 | 19 ± 1 |
| Middle-aged mice | ||||
| Intact (n = 10) | 7.38 ± 0.01 | 42 ± 1 | 78 ± 3 | 17 ± 2 |
| Intact+T5 (n =10) | 7.36 ± 0.02 | 42 ± 2 | 73 ± 2 | 19 ± 2 |
Compared with naive young adult male animals, middle-aged male animals displayed much lower plasma total testosterone levels: 8 ± 4 ng/mL in young adult rats (n = 10) versus 3 ± 2 ng/mL in middle-aged rats (n = 10); and 5 ± 2 ng/mL in young adult mice (n = 8) versus 1 ± 1 ng/mL in middle-aged mice (n = 7). Similarly, free testosterone levels were also lower in middle-aged animals: 9 ± 3 pg/mL in young adult rats versus 0.4 ± 1 pg/mL in middle-aged rats; and 11 ± 3 pg/mL in young adult mice versus 4 ± 2 pg/mL in middle-aged mice. After castration, total and free testosterone levels were undetectable in all young adult animals. Testosterone implantation restored free testosterone levels in castrated young adult rats (15 ± 8 pg/mL, n = 11) and mice (30 ± 11 pg/mL n = 9) and intact middle-aged animals (16 ± 3 pg/mL in rats, n = 7; 30 ± 7 pg/mL in mice, n = 7) to the physiologically relevant levels seen in reproductively active male animals.
In young adult rats, infarct sizes in both cortex and striatum were significantly smaller in castrated versus intact males, whereas castrates supplemented with physiologic testosterone sustained similar infarct sizes as seen in intact males. Furthermore, infarct sizes in castrated young rats implanted with both testosterone and flutamide were equivalent to those in young castrated males and smaller than those in intact males or castrates supplemented only with testosterone (Figure 1A). Intact middle-aged rats supplemented with physiologic testosterone displayed significantly smaller infarctions compared with age-matched intact rats without testosterone supplementation. However, infarct sizes in rats treated with both testosterone and flutamide were comparable to those in un-supplemented intact middle-aged rats (Figure 1B).
Figure 1.
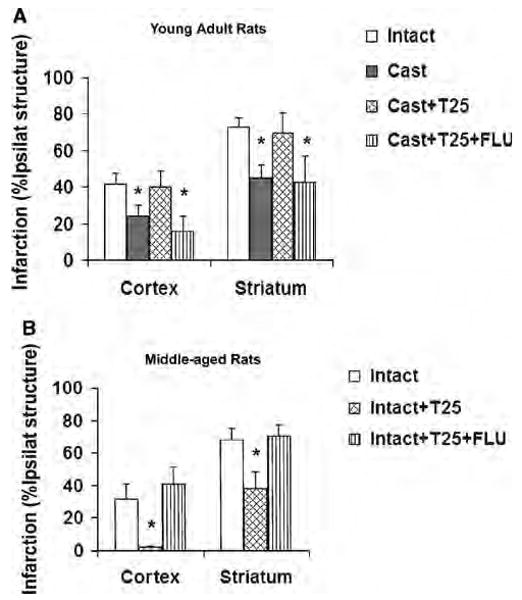
Cortical and striatal infarction (percentage of ipsilateral structure) at 22 h reperfusion in young adult and middle-aged rats. (A) Testosterone exacerbated cerebral ischemic damage in young adult rats, but AR antagonist flutamide blocked this deleterious effect. (Intact (n = 11): untreated, Cast (n = 11): castrated, Cast + T25 (n = 11): castrated and implanted with 25 mg testosterone, Cast + T25 + FLU (n = 9): castrated and implanted with 25 mg testosterone plus 25 mg flutamide.) *P < 0.05 from Intact and Cast + T25. (B) Supplemental testosterone protected middle-aged rats from ischemic damage, but AR antagonist flutamide blocked this neuroprotection. (Intact (n = 9): untreated, Intact + T25 (n = 7): intact and implanted with 25 mg testosterone, Intact + T25 + FLU (n = 9): intact and implanted with 25 mg testosterone plus 25 mg flutamide.) *P < 0.05 from Intact and from Intact + T25 + Flu.
Similar age-related differences in infarct size were confirmed in the mouse MCAO model. As shown in Figure 2A, both cortical and striatal infarct damage in castrated young adult mice was remarkably less than that in young intact male mice. Testosterone-supplemented castrated young mice sustained equivalent levels of cortical ischemic damage as seen in intact young mice. Figure 2B illustrates that intact middle-aged mice had significantly larger cortical infarct sizes than age-matched mice supplemented with testosterone. Striatal damage was not different between the two groups.
Figure 2.
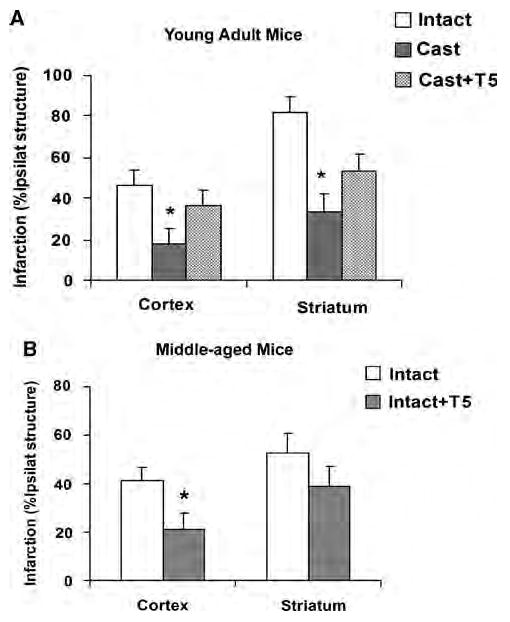
Cortical and striatal infarction (percentage of ipsilateral structure) at 22 h reperfusion in young adult and middle-aged mice. (A) Testosterone exacerbated cerebral ischemic damage in young adult mice. (Intact (n = 10): untreated, Cast (n = 10): castrated, Cast + T5 (n = 10): castrated and implanted with 5 mg testosterone.) *P < 0.05 from Intact and Cast + T5. (B) Supplemental testosterone decreased cortical infarction in intact middle-aged mice. (Intact (n = 10): untreated, Intact + T5 (n = 10): intact and implanted with 5 mg testosterone.) *P < 0.05 from Intact.
To test our hypothesis that the age-dependent effects of testosterone on ischemic damage can be attributed to higher cerebral testosterone aromatization in middle-aged animals versus lower aromatization in young adult animals, we measured both cerebral aromatase activity and mRNA expression in young and middle-aged intact rats before and after MCAO. Relatively high aromatase activity has been reported in the hippocampus (Roselli and Resko, 1993), which served as positive control in this study (12 to 21 fmol/h/mg protein, n = 13). As expected, the hippocampus had the highest aromatase activity among the three brain regions. In the cortex, both baseline (n = 13) and postischemic aromatase activities at 3 (n = 8) and 6 h reperfusion (n = 8) were barely detectable in both young and middle-aged animals (2.0 to 2.7 fmol/h/mg protein). In striatum, baseline aromatase activities were not different between intact young adult and middle-aged animals. Aromatase activities were not induced at 3 h reperfusion in either young adult (n = 8) or middle-aged rats (n = 8). However, a significant induction of striatal aromatase activity was detected at 6 h reperfusion in young adult rats (n = 8), but not in middle-aged rats (n = 7; Figure 3).
Figure 3.
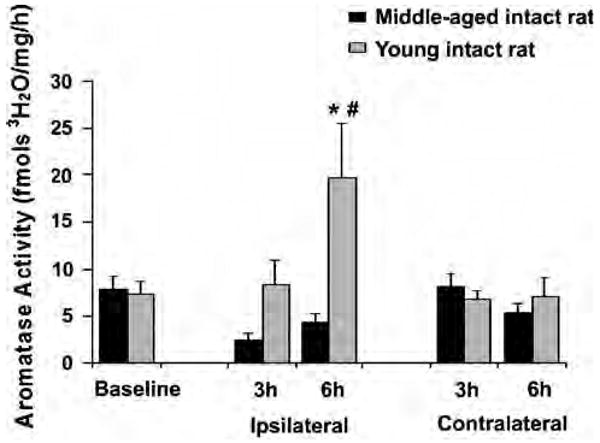
Striatal aromatase activity was significantly induced at 6 h reperfusion in young adult rats (n = 8). *P < 0.05 from baseline (n = 13); #P < 0.05 from ipsilateral aromatase activity at 3 h in young (n = 8) or middle-aged rats (n = 8) and from 6 h reperfusion in middle-aged rats (n = 7).
As aromatase activity was not detected in the cerebral cortex, we focused our measurements of aromatase mRNA to the striatum. Neither baseline nor postischemic aromatase mRNA levels at 3 and 6 h after reperfusion was different between young (n = 7) and middle-aged animals (n = 7). No postischemic induction of aromatase mRNA transcription was detected either in young adult or middle-aged rats at both time points (Figure 4).
Figure 4.
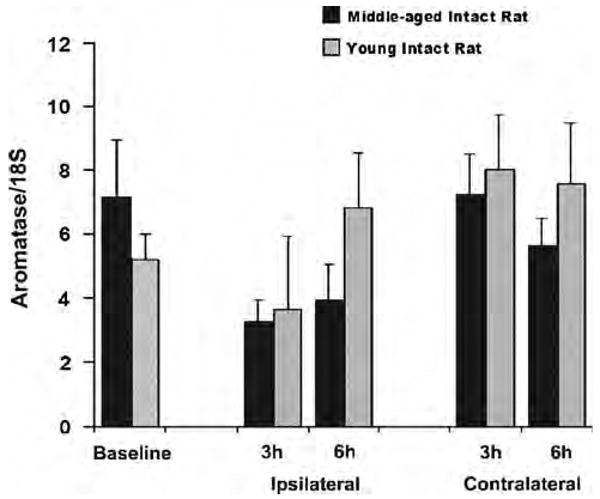
Neither baseline nor postischemic aromatase mRNA levels in striatum were different among experimental groups (n = 7 for each group). Total RNA was extracted from naive striatal tissue or ischemic striatal tissue at 3 or 6 h reperfusion. Aromatase mRNA levels were measured using Taqman quantitative RT-PCR technique with normalization to 18S for each sample. Final results were expressed as the ratios of aromatase/18S.
To further examine whether aromatase plays a role in mediating the age-related effects of testosterone on cerebral ischemia, we compared the infarct size in aging animals with aromatase gene deletion (ArKO) with that in age-matched wild-type male mice. Both cortical and striatal infarct sizes in 12-month-old male ArKO mice (n = 10) were not different from those in age-matched wild-type (n = 10) male mice at 22 h reperfusion after MCAO (Figure 5).
Figure 5.
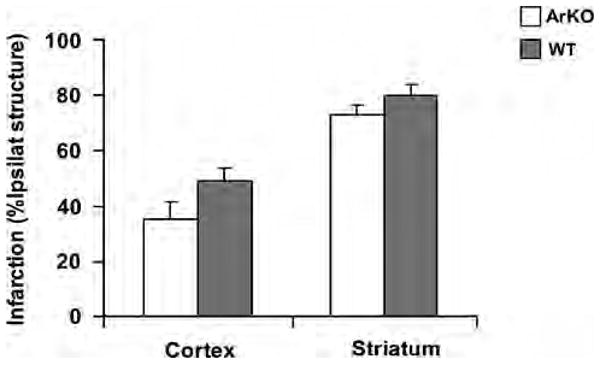
Infarct sizes at 22 h reperfusion were comparable in middle-aged male aromatase knockout (ArKO) mice (n = 10) versus age-matched wild-type (WT) mice (n = 10).
Discussion
This study shows three important findings. First, we found that testosterone's ability to worsen ischemic damage is age dependent and that steroid repletion in middle-aged brain is surprisingly beneficial. Second, testosterone acts via the AR during cerebral ischemia in both ages, i.e., both the neuroprotection and the enhancement of tissue damage are linked to testosterone–AR signaling. Lastly, our assays did not show higher cerebral aromatase activity and mRNA levels in middle-aged versus young adult males, suggesting that cerebral aromatization of testosterone to estradiol is not likely to contribute to the protection conferred by testosterone in the middle-aged stroke. Furthermore, infarct size in middle-aged males is not altered by aromatase deficiency, again suggesting that aromatization does not play an important role in mice of this age. We conclude that androgens have complex effects during cerebral ischemia that are likely linked to age and the duration of life in an androgen-deficient physiology.
A large body of evidence suggests that stroke is a sexually dimorphic disease, and that sex steroids play essential mechanistic roles when stroke occurs (Hall et al, 1991; Alkayed et al, 1998; Toung et al, 1998, 2004; Alkayed et al, 2000; Hurn et al, 2005). Testosterone is the major male sex steroid in circulation; however, in contrast to the well-established neuroprotective role of the female sex steroid estrogen, the effects of testosterone after cerebral ischemia remain unclear. In this study, we found that the absence of testosterone in young castrated adult rodents resulted in decreased ischemic damage, whereas testosterone replacement to physiologically relevant levels restored ischemic vulnerability through AR dependent mechanisms. Our results are consistent with previous studies showing that testosterone and its metabolite dihydrotestosterone exacerbate ischemic damage from experimental stroke in young adult male animals (Hawk et al, 1998; Yang et al, 2002; Cheng et al, 2007). Therefore, androgens appear to be significant factors in the high susceptibility of young adult males to ischemic damage. Although published data in experimental stroke (Hawk et al, 1998; Yang et al, 2002; Cheng et al, 2007) and the present data show that androgens can play deleterious roles in cerebral ischemia, available clinical studies contradict this conclusion. First, stroke incidence aggressively increases in males during middle age when androgen levels begin to decline (Hollander et al, 2003); secondly, in elderly men, lower testosterone levels are associated with greater infarct size and poorer functional recovery (Jeppesen et al, 1996). To reconcile these differences, we now offer the explanation that any causative links between androgen loss and male sensitivity to ischemic injury will depend on age and duration of low gonadal androgen availability.
Our results showed that there is a natural ‘andro-pause’, a loss of serum testosterone, that is present by 12 to 14 months of age in male rats or mice. Testosterone, like other lipophilic sex steroids, circulates systemically as part of a complex with sex hormone binding globulin. The free steroid fraction is the small but biologically active component (Moffat et al, 2002; Yeap et al, 2008), hence we assessed both total and free steroid levels in these experiments. We used a standard subcutaneous pellet implantation technique to target restoration of the free steroid component to physiologically relevant serum levels in gondally intact middle-aged males. With this model, we uncovered the surprisingly beneficial effect of testosterone replacement in previously ‘testosterone deficient’ middle-aged males. Our results provide a line of evidence suggesting that testosterone decline is one mechanism that accounts for the male's increased sensitivity to ischemic damage after middle age. As we confirmed the age-dependent effects of testosterone in both rat and mouse MCAO models, these effects are unlikely to be species related.
Cerebral aromatase is thought to be an important mediator of testosterone signaling in brain, and several studies suggest the enzyme's involvement in stroke pathologic study (for review see Roselli, 2007). Aromatase expression and activity have been reported in multiple brain regions including hippocampus, hypothalamus, and cortex (Roselli and Resko, 1984); and cerebral aromatase protein levels are elevated in cortical penumbra after MCAO (Carswell et al, 2005). Increasing evidence suggests that aromatase-mediated cerebral estradiol synthesis is an endogenous neuroprotective mechanism under various neuron-degenerative conditions. For example, in an in vitro ischemia model, primary astrocyte cultures derived from neonatal female rat brain are less sensitive than male cells to oxygen-glucose deprivation, in part because of higher aromatase activity and estradiol synthesis in female astrocytes (Liu et al, 2007). Conversely, if cerebral aromatase activity is inhibited by direct administration of aromatase inhibitor fadrozole into brain, kainite-induced hilar neuronal death increases (Azcoitia et al, 2001). Finally, studies on ArKO female mice indicate that cerebral aromatase contributes to estrogen-mediated neuroprotection after MCAO (McCullough et al, 2003). These findings led us to hypothesize that the age-dependent effects of testosterone may be mediated by differential cerebral aromatase activity between young adult and middle-aged males.
However, the results of our aromatase activity assays do not support this hypothesis. As both basal and postischemic aromatase activity are almost undetectable in cortex of both young adult and middle-aged rats, we do not expect that aromatase contributes to the age-dependent effects of testosterone in cortex. These remarkably low levels of aromatase activity in cortex are unlikely attributed to the insensitivity of the method because (1) we detected relatively high aromatase activity in the hippocampus; (2) induction of aromatase activity was also detected in striatum; and (3) the results are consistent with previous studies demonstrating that aromatase activity is low or absent in cortex (Roselli et al, 1985). In striatum, baseline aromatase activity in young adult versus middle-aged rats was not significantly different. Interestingly, and in contrast to our speculation that aromatase activity is higher in middle-aged versus young adult rats, striatal aromatase activity was increased at 6 h reperfusion in young adult rats but was not increased from basal values at any timepoint in middle-aged rats. These findings indicate that aromatase/estradiol also does not appear to contribute to the age-dependent effects of testosterone in striatum. The observation that aromatase activity increased in young animals at 6 h of reperfusion is consistent with previous reports, however we did not identify the cell type with enhanced aromatase activity in striatum. Previous work suggests that aromatase expression (Carswell et al, 2005) and activity (Liu et al, 2007) are increased in astrocytes after in vivo or in vitro ischemia.
The aromatase mRNA results from our qPCR assay also strengthen the notion that cerebral aromatase does not contribute to testosterone's differential effects in middle-aged animals. We used qPCR assay to confirm aromatase activity because aromatase activity is regulated at the cellular level mainly by transcriptional control (Simpson et al, 2002). Several splicing variants of aromatase are present in the brain, and the cortex-specific variant, in particular, is reportedly nonfunctional (Yamada-Mouri et al, 1997). So, we designed qPCR primers and probe to target the exon that are specifically present in the full-length aromatase mRNA but absent in the cortex-specific variant. Unlike commercially available primers and probe that recognize common exons present in both the full-length and cortex-specific variants (Applied Biosystem: Rn0056722_g1), we found that the mRNA levels detected with our designed primers and probe coincided perfectly with the activity levels in these brain regions. Under basal conditions, the aromatase mRNA levels are highest in hippocampus, lower in striatum, and barely detectable in cortex (data not shown). In striatum, aromatase mRNA expression was not different in young adult versus middle-aged rats before or after MCAO. Therefore, the mRNA results are largely consistent with our aromatase activity assays and further support the notion that aromatase does not contribute to age-dependent effects of testosterone on ischemic damage. However, aromatase activity was induced in striatum at 6 h reperfusion in young adult rats, whereas mRNA level was not increased at either 3 or 6 h reperfusion. This may suggest that, in addition to transcriptional control, other mechanisms, such as ATP-dependent phosphorylation, contribute to the regulation of aromatase activity in striatum (Balthazart et al, 2001).
The mechanism by which testosterone–AR signaling protects in middle age and enhances damage in the young male remains to be identified. We have previously identified numerous genes expressed after MCAO in the young male rat with or without androgen (Cheng et al, 2007), and these candidate genes remain to be explored and contrasted in the middle-aged males. Although differences in aromatase expression/activity did not explain tesosterone's age dependent actions, the role of the 5α-reductase remains to be explored. The reductase converts testosterone to 5α-dihydrotestosterone, a more potent AR agonist. If the 5α-reductase is differentially active with age, then the ratio of testosterone to 5α-dihydrotestosterone signaling at the AR may be altered with opposing effects on ischemic tissue outcomes. We used laser Doppler flowmetry to confirm adequate vascular occlusion in all animals. Our finding that intra-ischemic perfusion, as assessed by this technique, was not different amongst young and middle-aged animals does not exclude the possibility that testosterone depletion or repletion alters baseline cerebral blood flow. If so, then vascular mechanisms may be involved in the differential effects of testosterone on infarct size. Measurement of absolute blood flow will be needed in future to address this possibility. Finally, it should also be noted that we examined testosterone effects on the outcomes from experimental stroke at relatively early reperfusion time point (22 h). Further histologic as well as behavioral studies are needed to confirm the long-term effects of testosterone on stroke outcomes.
It was not our intent to characterize ischemic damage relative to age per se, and our studies were designed to evaluate a treatment within a specific age cohort. However we observed unexpectedly that intact middle-aged animals displayed smaller infarct sizes than intact young adult males (Figures 1 and 2). In human stroke, age impacts infarct size (Engelter et al, 2003; Nakayama et al, 1994). In experimental stroke, effects of age on outcome remain controversial with reports of better (Shapira et al, 2002), worse (DiNapoli et al, 2008), or a lack of difference (Petcu et al, 2007) in ischemic outcomes in aged rodents. In part, discrepancies amongst findings may be explained by the concept that aged animals develop cytologic responses to tissue damage and functional deficits over a different time course than do young animals (Petcu et al, 2007; Popa-Wagner et al, 2007). Our data are taken from middle-aged animals, younger than in these previous studies, and we evaluated only histologic injury at an early point in infarct maturation. In any event, it is unlikely that the somewhat smaller infarcts observed in middle-aged versus young rodents influenced the age-specific actions of testosterone.
In conclusion, testosterone exerts opposite effects on early stroke outcome in young versus mid-life, reproductively senescent rats and mice. These age-dependent effects are not dependent on cerebral aromatase activity and involve testosterone–AR signaling. Further studies are needed to dissect the potentially divergent mechanisms of testosterone signaling in aging ischemic brain.
Acknowledgments
This research was funded by US Public Health Service grants NS049210 and NS33668 and American Heart Association Grant 0825526G.
Footnotes
Disclosure/conflict of interest: The authors declare there is no conflict of interest.
References
- Alkayed NJ, Harukuni I, Kimes AS, London ED, Traystman RJ, Hurn PD. Gender-linked brain injury in experimental stroke. Stroke. 1998;29:159–65. doi: 10.1161/01.str.29.1.159. [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Murphy SJ, Traystman RJ, Hurn PD, Miller VM. Neuroprotective effects of female gonadal steroids in reproductively senescent female rats. Stroke. 2000;31:161–8. doi: 10.1161/01.str.31.1.161. [DOI] [PubMed] [Google Scholar]
- Anawalt BD, Merriam GR. Neuroendocrine aging in men. Andropause and somatopause. Neuroendocrinol Metab Clin North Am. 2001;30:647–69. doi: 10.1016/s0889-8529(05)70206-1. [DOI] [PubMed] [Google Scholar]
- Azcoitia I, Sierra A, Veiga S, Honda S, Harada N, Garcia-Segura LM. Brain aromatase is neuroprotective. J Neurobiol. 2001;47:318–29. doi: 10.1002/neu.1038. [DOI] [PubMed] [Google Scholar]
- Balthazart J, Baillien M, Ball GF. Phosphorylation processes mediate rapid changes of brain aromatase activity. J Steroid Biochem Mol Biol. 2001;79:261–77. doi: 10.1016/s0960-0760(01)00143-1. [DOI] [PubMed] [Google Scholar]
- Carswell HV, Dominiczak AF, Garcia-Segura LM, Harada N, Hutchison JB, Macrae IM. Brain aromatase expression after experimental stroke: topography and time course. J Steroid Biochem Mol Biol. 2005;96:89–91. doi: 10.1016/j.jsbmb.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Cheng J, Alkayed NJ, Hurn PD. Deleterious effects of dihydrotestosterone on cerebral ischemic injury. J Cereb Blood Flow Metab. 2007;27:1553–62. doi: 10.1038/sj.jcbfm.9600457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNapoli VA, Huber JD, Houser K, Li X, Rosen C. Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol Aging. 2008;29:753–64. doi: 10.1016/j.neurobiolaging.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelter ST, Provenzale JM, Petrella JR, DeLong DM, Alberts MJ. Infarct volume on apparent diffusion coefficient maps correlates with length of stay and outcome after middle cerebral artery stroke. Cerebrovasc Dis. 2003;15:188–91. doi: 10.1159/000068826. [DOI] [PubMed] [Google Scholar]
- Hall ED, Pazara KE, Linseman KL. Sex differences in postischemic neuronal necrosis in gerbils. J Cereb Blood Flow Metab. 1991;11:292–8. doi: 10.1038/jcbfm.1991.61. [DOI] [PubMed] [Google Scholar]
- Hawk T, Zhang YQ, Rajakumar G, Day AL, Simpkins JW. Testosterone increases and estradiol decreases middle cerebral artery occlusion lesion size in male rats. Brain Res. 1998;796:296–8. doi: 10.1016/s0006-8993(98)00327-8. [DOI] [PubMed] [Google Scholar]
- Hollander M, Koudstaal PJ, Bots ML, Grobbee DE, Hofman A, Breteler MM. Incidence, risk, and case fatality of first ever stroke in the elderly population. The Rotterdam Study. J Neurol Neurosurg Psychiatry. 2003;74:317–21. doi: 10.1136/jnnp.74.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurn PD, Vannucci SJ, Hagberg H. Adult or perinatal brain injury: does sex matter? Stroke. 2005;36:193–5. doi: 10.1161/01.STR.0000153064.41332.f6. [DOI] [PubMed] [Google Scholar]
- Jeppesen LL, Jorgensen HS, Nakayama H, Raaschou HO, Olsen TS, Winther K. Decreased serum testosterone in men with acute ischemic stroke. Arterioscler Thromb Vasc Biol. 1996;16:749–54. doi: 10.1161/01.atv.16.6.749. [DOI] [PubMed] [Google Scholar]
- Liu M, Hurn PD, Roselli CE, Alkayed NJ. Role of P450 aromatase in sex-specific astrocytic cell death. J Cereb Blood Flow Metab. 2007;27:135–41. doi: 10.1038/sj.jcbfm.9600331. [DOI] [PubMed] [Google Scholar]
- McCullough LD, Blizzard K, Simpson ER, Oz OK, Hurn PD. Aromatase cytochrome P450 and extragonadal estrogen play a role in ischemic neuroprotection. J Neurosci. 2003;23:8701–5. doi: 10.1523/JNEUROSCI.23-25-08701.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat SD, Zonderman AB, Metter EJ, Blackman MR, Harman SM, Resnick SM. Longitudinal assessment of serum free testosterone concentration predicts memory performance and cognitive status in elderly men. J Clin Endocrinol Metab. 2002;87:5001–7. doi: 10.1210/jc.2002-020419. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Jorgensen HS, Raaschou HO, Olsen TS. The influence of age on stroke outcome. The Copenhagen Stroke Study. Stroke. 1994;25:808–13. doi: 10.1161/01.str.25.4.808. [DOI] [PubMed] [Google Scholar]
- Pan Y, Zhang H, Acharya AB, Patrick PH, Oliver D, Morley JE. Effect of testosterone on functional recovery in a castrate male rat stroke model. Brain Res. 2005;1043:195–204. doi: 10.1016/j.brainres.2005.02.078. [DOI] [PubMed] [Google Scholar]
- Petcu EB, Sfredel V, Platt D, Herndon JG, Kessler C, Popa-Wagner A. Cellular and molecular events underlying the dysregulated reponse of the aged brain to stroke. Gerontology. 2007;54:6–17. doi: 10.1159/000112845. [DOI] [PubMed] [Google Scholar]
- Popa-Wagner A, Badan I, Walker L, Groppa S, Patrana N, Kessler C. Accelerated infarct development, cytogenesis and apoptosis following transient cerebral ischemia in aged rats. Acta Neuropathol. 2007;113:277–93. doi: 10.1007/s00401-006-0164-7. [DOI] [PubMed] [Google Scholar]
- Robertson KM, O'Donnell L, Jones ME, Meachem SJ, Boon WC, Fisher CR, Graves KH, McLachlan RI, Simpson ER. Impairment of spermatogenesis in mice lacking a functional aromatase (cyp 19) gene. Proc Natl Acad Sci. 1999;96:7986–91. doi: 10.1073/pnas.96.14.7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselli CE, Horton LE, Resko JA. Distribution and regulation of aromatase activity in the rat hypothalamus and limbic system. Endocrinology. 1985;117:2471–7. doi: 10.1210/endo-117-6-2471. [DOI] [PubMed] [Google Scholar]
- Roselli CE, Resko JA. Androgens regulate brain aromatase activity in adult male rats through a receptor mechanism. Endocrinology. 1984;114:2183–9. doi: 10.1210/endo-114-6-2183. [DOI] [PubMed] [Google Scholar]
- Roselli CE, Resko JA. Aromatase activity in the rat brain: hormonal regulation and sex differences. J Steroid Biochem Mol Biol. 1993;44:499–508. doi: 10.1016/0960-0760(93)90254-t. [DOI] [PubMed] [Google Scholar]
- Roselli CF. Brain aromatase: roles in reproduction and neuroprotection. J Steroid Biochem Mol Biol. 2007;106:143–50. doi: 10.1016/j.jsbmb.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampei K, Goto S, Alkayed NJ, Crain BJ, Korach KS, Traystman RJ, Demas GE, Nelson RJ, Hurn PD. Stroke in estrogen receptor-alpha-deficient mice. Stroke. 2000a;31:738–43. doi: 10.1161/01.str.31.3.738. [DOI] [PubMed] [Google Scholar]
- Sampei K, Mandir AS, Asano Y, Wong PC, Traystman RJ, Dawson VL, Dawson TM, Hurn PD. Stroke outcome in double-mutant antioxidant transgenic mice. Stroke. 2000b;31:2685–91. doi: 10.1161/01.str.31.11.2685. [DOI] [PubMed] [Google Scholar]
- Shapira S, Sapir M, Wengier A, Grauer E, Kadar T. Aging has a complex effect on a rat model of ischemic stroke. Brain Res. 2002;925:148–58. doi: 10.1016/s0006-8993(01)03270-x. [DOI] [PubMed] [Google Scholar]
- Simpson ER, Clyne C, Rubin G, Boon WC, Robertson K, Britt K, Speed C, Jones M. Aromatase—a brief overview. Annu Rev Physiol. 2002;64:93–127. doi: 10.1146/annurev.physiol.64.081601.142703. [DOI] [PubMed] [Google Scholar]
- Toung TJ, Chen TY, Littleton-Kearney MT, Hurn PD, Murphy SJ. Effects of combined estrogen and progesterone on brain infarction in reproductively senescent female rats. J Cereb Blood Flow Metab. 2004;24:1160–6. doi: 10.1097/01.WCB.0000135594.13576.D2. [DOI] [PubMed] [Google Scholar]
- Toung TJ, Traystman RJ, Hurn PD. Estrogen-mediated neuroprotection after experimental stroke in male rats. Stroke. 1998;29:1666–70. doi: 10.1161/01.str.29.8.1666. [DOI] [PubMed] [Google Scholar]
- Vermeulen A. Andropause. Maturitas. 2000;34:5–15. doi: 10.1016/s0378-5122(99)00075-4. [DOI] [PubMed] [Google Scholar]
- Yamada-Mouri N, Hirata S, Kato J, Hoshi K. Expression and distribution of cortical type aromatase mRNA variant in the adult rat brain. J Steroid Biochem Mol Biol. 1997;60:325–9. doi: 10.1016/s0960-0760(96)00222-1. [DOI] [PubMed] [Google Scholar]
- Yang SH, Perez E, Cutright J, Liu R, He Z, Day AL, Simpkins JW. Testosterone increases neurotoxicity of glutamate in vitro and ischemia-reperfusion injury in an animal model. J Appl Physiol. 2002;92:195–201. doi: 10.1152/jappl.2002.92.1.195. [DOI] [PubMed] [Google Scholar]
- Yeap BB, Almeida OP, Hyde Z, Chubb SA, Hankey GJ, Jamrozik K, Flicker L. Higher serum free testosterone is associated with better cognitive function in older men, while total testosterone is not. The Health In Men Study. Clin Endocrinol (Oxf) 2008;68:404–12. doi: 10.1111/j.1365-2265.2007.03055.x. [DOI] [PubMed] [Google Scholar]