

Abstract
A recombinant adeno-associated virus (rAAV) vector capable of infecting cells and expressing rat glial cell line-derived neurotrophic factor (rGDNF), a putative central nervous system dopaminergic survival factor, under the control of a potent cytomegalovirus (CMV) immediate/early promoter (AAV-MD-rGDNF) was constructed. Two experiments were performed to evaluate the time course of expression of rAAV-mediated GDNF protein expression and to test the vector in an animal model of Parkinson’s disease. To evaluate the ability of rAAV-rGDNF to protect nigral dopaminergic neurons in the progressive Sauer and Oertel 6-hydroxydopamine (6-OHDA) lesion model, rats received perinigral injections of either rAAV-rGDNF virus or rAAV-lacZ control virus 3 weeks prior to a striatal 6-OHDA lesion and were sacrificed 4 weeks after 6-OHDA. Cell counts of back-labeled fluorogold-positive neurons in the substantia nigra revealed that rAAV-MD-rGDNF protected a significant number of cells when compared with cell counts of rAAV-CMV-lacZ-injected rats (94% vs. 51%, respectively). In close agreement, 85% of tyrosine hydroxylase-positive cells remained in the nigral rAAV-MD-rGDNF group vs. only 49% in the lacZ group. A separate group of rats were given identical perinigral virus injections and were sacrificed at 3 and 10 weeks after surgery. Nigral GDNF protein expression remained relatively stable over the 10 weeks investigated. These data indicate that the use of rAAV, a noncytopathic viral vector, can promote delivery of functional levels of GDNF in a degenerative model of Parkinson’s disease.
Glial cell line-derived neurotrophic factor (GDNF) has been identified as a potent neurotrophic molecule for nigral dopaminergic neurons both in vitro (1, 2) and in vivo (3–11). A trophic molecule capable of halting on-going degeneration of the dopaminergic neurons of the substantia nigra (SN) would be valuable for the treatment of Parkinson’s disease (PD), because no treatment is currently available that impedes the progress of the disease. Indeed, GDNF has shown efficacy in several different animal models of PD (3–10) including primate models (11).
To take advantage of GDNF’s dopaminergic neurotrophic activity for the development of a treatment for PD, several protein delivery issues must be resolved. First, the optimal anatomical target for GDNF must be defined. Thus far, the in vivo data regarding the trophic effects of GDNF in animal models of PD suggest that GDNF is most efficacious when delivered directly to the SN (3, 4, 6–9). Direct GDNF delivery to the SN for PD also would avoid any potential side-effects that might arise from the biological activity of GDNF in brain areas containing GDNF receptors that are not directly associated with disease processes (12–14). Moreover, it is uncertain whether the GDNF protein, which cannot cross the blood–brain barrier (15), could reach the cells in the SN if delivered intracerebroventricularly (but see ref. 16).
The second issue of importance for a GDNF-based treatment for PD is to determine the therapeutic levels of GDNF and to define the therapeutic window. Currently, there is little directly relevant data available to assess this important issue. However, the data available from intranigral injection dose–response studies (4), behavioral effects generated via intrastriatal infusions (5), studies in primates (11), and GDNF tissue levels measured from nigral GDNF expression mediated by a recombinant adenoviral injection (17, 18) suggest that the therapeutic window for GDNF may be quite wide, similar to that seen in studies of the effects of nerve growth factor on central cholinergic cells, as judged by the variety of nerve growth factor doses and treatment regimens that exert positive influences (19, 20).
Finally, acceptable temporal patterns of GDNF delivery capable of halting on-going PD-related dopamine (DA) degeneration must be identified. PD is a progressive disease where DA neurons continue to die over a period of decades. Recent data observed in the Sauer and Oertel (21) progressive 6-hydroxydopamine (6-OHDA)-induced degeneration model in rats suggest that intermittent delivery (22) or short-term post hoc nigral GDNF delivery (9) is sufficient to spare DA neurons after an acute 6-OHDA insult for a long period of time. However, because PD is a long-term progressive disorder, the as yet unknown pathogenesis must be continuous or at least repetitive. Thus, the natural history of PD coupled with the animal model data suggest that GDNF would most likely have to be delivered repeatedly, but not necessarily, continuously to successfully treat PD. Because the SN is located in the midbrain, chronic implantation of a cannula-based device to deliver proteins in a PD patient may be technically problematic. Therefore, alternate strategies to deliver GDNF in PD may be required to safely treat PD in this fashion.
In vivo gene therapy, or the ability to locally express or overexpress a de novo gene by injecting a recombinant virus directly into the brain, has recently achieved some success (17, 18). Thus, several groups using different vector systems have reported the ability to restore DA to a previously denervated striatum in the rat (23–25) and two groups have reported nigral neuron sparing after injection of a recombinant replication defective adenovirus (Ad) encoding the cDNA for rat GDNF by using the Sauer and Oertel progressive degeneration PD model in rats (17, 18). The present study uses a viral vector derived from the nonpathogenic human parvovirus, adeno-associated virus (AAV) (26), to deliver rat GDNF (rGDNF) directly to the nigra by using the Sauer and Oertel progressive degeneration model in rats. The recombinant AAV (rAAV) vector was chosen because of its low liability for reversion to replication competency and the lack of apparent neurotoxicity that is likely because of the complete absence of wild-type viral genes in the vector (24, 26). These safety features of rAAV render this vector more clinically practical compared with first generation Ad vectors, because first-generation Ad-mediated transgene expression is transient (27–29), but long-term rAAV-mediated transgene expression has been demonstrated in several animal models of human disease (30, 31). The data demonstrate that the levels of rGDNF produced after intranigral injection of rAAV-rGDNF are sufficient to spare nearly 100% of the neurons from a 6-OHDA lesion, which normally induces 50% nigral cell loss, and that rGDNF gene expression persists for at least 10 weeks.
METHODS
Recombinant AAV Vector Production.
The CMV-lacZ rAAV vector (pdx-31 lacZ, where CMV is cytomegalovirus) was described previously (32). pMD-rGDNF was constructed with a cassette (33) containing a CMV immediate early promoter/enhancer, a human β-globin second intervening sequence, a rGDNF cDNA (a gift of J. E. Springer, University of Kentucky) containing the entire coding sequences from nucleotides 1 to 633 (34, 35), and the β-globin poly(A) inserted between the AAV inverted terminal repeats of psub201 (36).
rAAV vectors were prepared according to ref. 30 with modifications. Briefly, subconfluent 293 cells were cotransfected with the vector plasmid and the AAV helper plasmid pACG 2–1 (37) by using the calcium phosphate method. Cells were then infected with Ad Ad5dl312 (an E1A− mutant) at a multiplicity of infection (MOI) of 2 and the infection was allowed to proceed for 60–72 hr. Cells were harvested and three freeze/thaw cycles were carried out to lyse the cells. The nucleic acid in the lysate was digested with Benzonase (Nycomed, Oslo; 250 units/ml) at 37°C for 10 min and then centrifuged at 1500 × g to pellet the cellular debris. The cell lysate then was fractionated by ammonium sulfate precipitation, and the rAAV virions were isolated on two sequential continuous CsCl gradients. The gradient fractions containing rAAV were dialyzed against sterile PBS, heated for 45 min at 56°C, and stored at −80°C. The purity of the rAAV virus with regard to cellular contaminants, infectious Ad, and contaminating wild-type AAV were evaluated as reported (30). The final particle titer of the rAAV-CMV-lacZ was 1.4 × 1012 viral particles per ml and the rAAV-MD-rGDNF was 1.0 × 1012 viral particles per ml as estimated via dot blot.
Cell Culture and in Vitro Transductions.
Twelve-well culture plates were seeded with 1.2 × 105 HeLa cells per well in DMEM supplemented with 10% fetal bovine serum and antibiotics. The next day increasing MOIs of rAAV-MD-rGDNF virus in the presence or absence of helper Ad (dl309, MOI = 3 to 10 infectious units per cell) was added in Iscove’s modified Dulbecco’s medium supplemented with 10% fetal bovine serum and antibiotics. The MOI of rAAV ranged from 5 to 5 × 104 particles per cell. Virus was left on for 24 hr before medium was replaced with DMEM growth medium. After 20 hr, medium was collected and analyzed for immunoreactive GDNF by ELISA (see below).
Subjects.
Twenty-nine Fischer 344 male rats weighing approximately 220 g were obtained from Harlan–Sprague–Dawley, housed with access to ad libitum food and water on a 12-hr light/12-hr dark cycle, maintained, and treated in accordance with published National Institutes of Health guidelines. All stereotaxic surgical procedures were performed with the rats under isoflurane gas anesthesia and aseptic procedures.
Intrastriatal Injection of Fluorogold (FG).
The retrograde tracer FG (0.2 μl of 4% FG in 0.9% NaCl), was injected bilaterally into the striatum (AP +1.0, LAT ±3.0, DV −5.0) via a 1-μl Hamilton syringe at a rate of 0.05 μl/min for 4 min in 14 of the rats (21). At the cessation of the injection, the needle was allowed to remain in the brain for 10 min before being retracted. The rate of injection for all stereotaxic surgeries described in this study was precisely controlled by an infusion pump (Razel Scientific Instruments, Stamford, CT) that pushed a piston that in turn depressed the plunger on the syringe.
Intranigral Injection of AAV Vectors.
After the rats are placed in the stereotaxic frame (Kopf Instruments, Tujunga, CA), 2 μl of rAAV (either rAAV-MD-rGDNF or rAAV-CMV-Lac-Z) suspended in PBS was injected into the vicinity of the SN [AP −5.4, LAT −2.2, DV −7.5 with the incisor bar set at −3.3 mm below the intra-aural line (9, 38, 39)] through a 5-μl Hamilton syringe fitted with a 30-gauge beveled hypodermic needle for 2 min at a rate of 0.5 μl/min. One minute after the cessation of the injection, the needle was retracted 1 mm and then left in place for 4 min before being slowly withdrawn from the brain.
6-OHDA Lesions.
Three weeks after virus injections, unilateral intrastriatal infusions of 6-OHDA lesions were performed with the rats under isoflurane anesthesia by stereotaxic injection of 6-OHDA HBr [20 μg/μl; calculated as free base, dissolved in ascorbate (2 mg/ml)/0.9% NaCl]. Four microliters of 6-OHDA were injected into the striatum at the same coordinates used for the FG injections (AP +1.0, LAT −3.0, DV −5.0) over 8 min at a rate of 0.5 μl/min as described (21). The needle was allowed to remain in the brain for 5 min before being retracted at the end of the 6-OHDA infusion.
Histology.
Each rat was perfused through the aorta with PBS followed by ice-cold 4% paraformaldehyde while under deep pentobarbital anesthesia. Forty-micrometer nigral sections were immunocytochemically stained for tyrosine hydroxylase (TH) with a mAb against TH (Chemicon) or for lacZ expression with a polyclonal antibody against β-galactosidase (Cappel). Detection used the VIP chromogen procedure (Vector) as described (40). For visualization of FG fluorescence, the 40 μM sections were mounted and coverslipped without further processing (21).
Cell Counting.
TH-stained striatal sections were examined to evaluate the relative homogeneity of the extent of the 6-OHDA lesions before cell counting (data not shown). One rAAV-CMV-lacZ animal was removed from the study because of lack of a lesion in the vector-injected hemisphere (see Fig. 4). Fluorogold-positive (FG+) cells were counted under epi-illuminecsence throughout the entire extent of the SN for each animal in both hemispheres with 200 μm between each section. Percentage survival of FG+ cells was calculated as the number of cells counted in the vector injected per lesioned hemisphere divided by the cells counted in the contralateral side of the same sections × 100, as described (9). For TH+ cell counting, three or four sections from the central portion of the SN as defined by the presence of the medial terminal nucleus of the accessory optic tract were chosen as described (9). The percent survival of TH+ cells was calculated identically to the FG data. All sections were counted by two blinded observers who had an interrater reliability of >0.9, and the data are expressed as the mean of the two counts.
Figure 4.
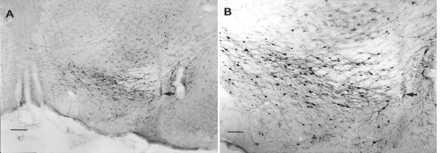
Photomicrographs of nigral β-galactosidase expression 7 weeks after rAAV-CMV-lacZ injection. The nigral sections shown were taken from an rAAV-CMV-lacZ-injected rat that showed no evidence of a 6-OHDA lesion, and the rat was removed from the study. rAAV-CMV-lacZ-injected animals that were included in the study were found to have little or no β-galactosidase staining, probably because of the destruction of transduced cells by the subsequent striatal 6-OHDA lesion. (A) Low-power micrograph of the SN. The arrow indicates the injection site. (Bar = 200 μm.) (B) Higher-power micrograph of the same field. The arrow indicates the injection site. As has been observed (24, 41) most of the cells expressing β-galactosidase appear to be neurons. (Bar = 100 μm.)
GDNF Protein Levels Measured by ELISA.
Fifteen rats received injections of either rAAV-CMV-lacZ (n = 6) or rAAV-MD-rGDNF (n = 9) in the vicinity of the SN as described above. Three or 10 weeks after vector injection, the animals were deeply anesthetized with sodium pentobarbital and decapitated, their brains rapidly dissected and placed on dry ice, and a 2-mm-diameter punch of the area surrounding the SN pars compacta on each side was taken from a 3-mm thick coronal slice that contained the rAAV vector injection site. Tissue levels of rGDNF were determined from tissue homogenates by ELISA using a commercial kit (Promega, G3240). The punch was sonicated (Branson sonifier model 250, output setting 2, 30% duty cycle for 10 sec) in a homogenization buffer (1.2 mM Hepes, pH 7.2/10% glycerol/1% Triton X-100/1 mM phenymethylsulfonyl fluoride/0.6 μM leupeptin) at a tissue concentration of 50 mg/ml (wet wt/vol). Samples were assayed at a final tissue concentration of 1–2 mg/ml. Results were expressed as the total ng of rGDNF per punch to be comparable to levels reported in a previous study using Ad (17).
RESULTS
rAAV-MD-rGDNF was added to HeLa cell cultures to confirm that measurable levels of rGDNF could be produced by this vector. Fig. 1 shows that rGDNF was detected in the medium by using ELISA when 1 × 108 rAAV particles (MOI = 50 particles per cell) or greater were used to transduce the cultures.
Figure 1.
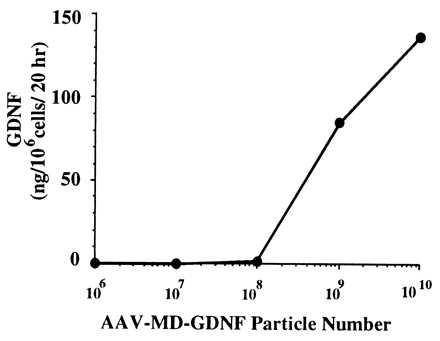
Production of GDNF in vitro from rAAV-MD-GDNF-transduced HeLa cells. Released GDNF levels were measured after addition of increasing amounts of rAAV-MD-GDNF vector to cultures containing 3 × 105 HeLa cells. The GDNF was measured by ELISA from samples taken after 20 hr from the culture medium.
The bioefficacy of rAAV vector delivered rGDNF was investigated via intranigral injection of rAAV-MD-rGDNF that resulted in significant protection from subsequent intrastriatal 6-OHDA administration for both back-labeled FG+ cells (Figs. 2A and 3 E–H) and TH+ dopaminergic cells (Figs. 2B and 3 A–D) as compared with rAAV-CMV-lacZ-injected rats 4 weeks after the lesion. Moreover, GDNF delivery produced a similar magnitude of the effect for the two different histological indices of nigral cell survival [FG vs. TH staining; ANOVA: F(1, 22) = 0.4, P > 0.5]. As can be seen in Figs. 3 and 4, there was little disruption of the tissue observable 7 weeks after the injection of rAAV. Inspection of TH-stained striatal sections revealed partial DA denervations of the striatum, and perinigral injection of rAAV-MD-rGDNF had no effect on TH+ striatal terminals as compared with rAAV-CMV-lacZ-treated animals (data not shown), as reported (9, 22).
Figure 2.
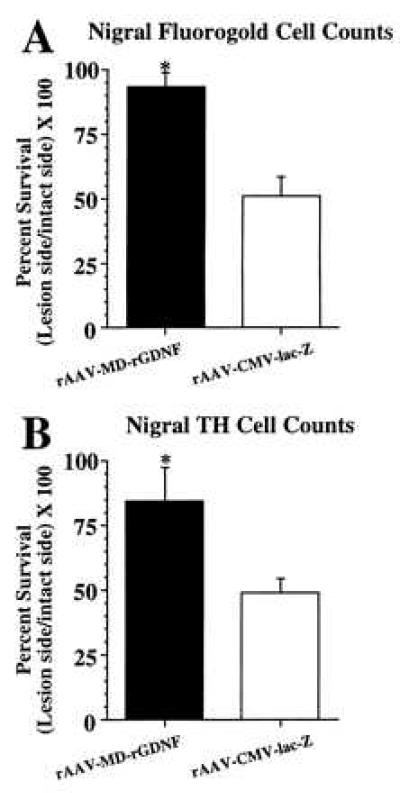
Nigral cell survival 4 weeks after a striatal 6-OHDA lesion. (A) Cell survival as estimated from cell counts of nigral FG+ cells. Nigral injection of rAAV-MD-rGDNF (solid bar, n = 6) resulted in a significantly greater number of FG+ cells surviving (indicated by *) after the striatal 6-OHDA lesion as compared with the rAAV-CMV-lacZ [open bar, n = 7, F(1, 11) = 20.1, P < 0.001]. (B) Cell survival estimated from cell counts of nigral TH+ cells. Nigral injection of rAAV-MD-rGDNF (solid bar, n = 6) resulted in a significantly greater number of TH+ cells surviving (indicated by *) after the striatal 6-OHDA lesion as compared with the rAAV-CMV-lacZ [open bar, n = 7, F(1, 11) = 6.9, P = 0.02]. Error bars represent +1 SEM.
Figure 3.
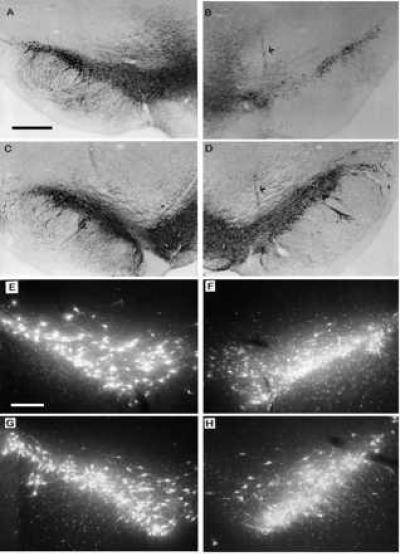
Photomicrographs of intact and lesioned/vector-injected SN 4 weeks after a striatal 6-OHDA lesion. (A and C) Photomicrographs of TH immunocytochemical staining of intact SN from a representative rAAV-CMV-lacZ-injected animal and a representative rAAV-MD-rGDNF-injected rat, respectively. (B and D) Photomicrographs of TH staining of the SN from lesioned and vector-injected hemisphere of representative rAAV-CMV-lacZ-injected animal and a representative rAAV-MD-rGDNF-injected rat, respectively. The arrows indicate a vector injection site. The cell loss due to the 6-OHDA lesion seen in B was significantly diminished (see Results) by pretreatment with rAAV-MD-rGDNF as demonstrated in D. The bar in A = 200 μm and applies to A–D. (E and G) Photomicrographs of FG back-labeled SN from the intact hemisphere from a representative rAAV-CMV-lacZ-injected animal and a representative rAAV-MD-rGDNF-injected rat, respectively. (F and H) Photomicrographs of FG back-labeled SN from the lesioned and vector-injected hemisphere from a representative rAAV-CMV-lacZ-injected animal and a representative rAAV-MD-rGDNF-injected rat, respectively. In addition to the 6-OHDA-induced cell loss demonstrated in F, there are numerous shrunken cells, punctae, and fibers with varicosities that are not apparent in the rAAV-MD-rGDNF-injected nigra shown in H. This pattern of 6-OHDA-induced damaged in FG back-labeled SN has been reported previously (17, 21, 22). The bar in E = 100 μm and applies to E–H.
To determine the persistence of rGDNF transgene expression, by using a separate group of animals, GDNF protein levels were determined at 3 weeks and 10 weeks after rAAV injection from nigral punches taken from either rAAV-MD-rGDNF or rAAV-CMV-lacZ-injected rats. Injection of rAAV-CMV-lacZ resulted in only background levels of GDNF expression as defined by the GDNF levels measured in the contralateral intact SN regardless of the interval between vector injection and sacrifice (Table 1). In contrast, punches taken from rAAV-MD-rGDNF-injected animals contained significantly elevated GDNF levels even at 10 weeks after the original vector injection (Table 1). Although, the GDNF protein levels present at 10 weeks after injection in the rAAV-MD-rGDNF injected animals were 40% less than those measured at 3 weeks, this decline was not statistically significant (Table 1).
Table 1.
GDNF protein levels measured by ELISA at 3 and 10 weeks after intranigral rAAV injection
| Intranigral rAAV vector | n | Time after rAAV, weeks | GDNF ipsilateral to vector injection, pg | GDNF contralateral to vector injection, pg |
|---|---|---|---|---|
| lacZ | 2 | 3 | 180.0 ± 78.0 | 185.0 ± 17.0 |
| GDNF | 6 | 3 | 1,234.5 ± 180.0*,** | 125.0 ± 34.4 |
| lacZ | 4 | 10 | 124.8 ± 63.8 | 167.8 ± 80.5 |
| GDNF | 3 | 10 | 747.0 ± 364.9*,** | 38.0 ± 22.0 |
Data are the mean ± SEM.
Significant difference between the rAAV-CMV-lacZ injected group and the rAAV-MD-rGDNF-injected group [F(1, 22) = 10.0, P < 0.005].
Significant difference between GDNF protein measured on the vector-injected hemisphere relative to the intact contralateral hemisphere [F(1, 22) = 14.2, P = 0.001]. There was no significant reduction of GDNF levels between 3 and 10 weeks [main effect of survival time F(1, 22) = 1.9, P = 0.18].
TH-stained nigral sections also were examined by using low levels of visible light simultaneously with epiluminescence, which allowed visualization of both the nigral FG back-labeling and TH staining. Inspection of the TH-stained sections in this manner revealed that some nigral fields, in several rAAV-MD-rGDNF-injected animals but not in rAAV-CMV-lacZ-injected rats, contained a full complement of FG+ cells but relatively few TH+ cells. The presence of nigral FG+ cells that are back-labeled from striatum and survive the 6-OHDA lesion but do not stain for TH may explain the slight difference in survival rate estimated from nigra FG+ cell counting (93.7%) as compared with the survival rate estimated from nigral TH+ cell counts (84.5%, Fig. 2).
Fig. 4 contains an example of a section at the rAAV-CMV-lacZ injection site stained for β-galactosidase. This pattern of infection confirms that the injections were slightly dorsal to the SN pars compacta, which was the intended target.
DISCUSSION
The present study used a well-characterized animal model of progressive degeneration of nigrostriatal dopaminergic neurons, first described by Sauer and Oertel (21), to demonstrate that rAAV-mediated nigral GDNF delivery is efficacious in an animal model of PD. In this striatal 6-OHDA lesion model, nigrostriatal dopaminergic cells die progressively between 1 and 4 weeks after the lesion (21). Proof that the dopaminergic cells have actually degenerated rather than having merely lost their TH+ phenotype is demonstrated by the concomitant loss of nigral neurons that are labeled with the retrograde fluorescent tracer FG (21). Relatively long-term delivery of GDNF protein (10 μg in 1 μl every other day for 4 weeks after 6-OHDA) in the vicinity of the SN has been demonstrated to protect nigrostriatal DA cells from degeneration in the Sauer and Oertel model (9, 22). Furthermore, significant protection was observed when animals were examined 4 months after the cessation of GDNF by using a similar intermittent treatment regimen (22). Therefore, the Sauer and Oertel 6-OHDA-induced progressive degeneration model provides a method to study the nigrostriatal DA system during on-going degeneration, which has conclusively been demonstrated to respond to GDNF (3, 22). Similar to the data obtained by using GDNF protein injections (3, 22), in the present study, GDNF delivered near the SN via rAAV injections was able to protect nigral DA cells from 6-OHDA-induced degeneration when administered 3 weeks before 6-OHDA. In contrast to the sparing of nigral neurons from 6-OHDA-induced degeneration, neither treatment with perinigral rAAV nor GDNF protein injections were able to protect striatal TH+ fibers (3, 9, 22). Moreover, the lack of recovery of striatal TH+ fibers is still evident, after demonstrated protection of nigral neurons, 4 months after nigral GDNF injections and there was no observed recovery of motor function (22). Thus, these data suggest that striatal dopaminergic recovery is probably necessary for functional improvement. In support of this contention, striatal Ad-GDNF injections produced improvements in striatal 6-OHDA lesion-induced amphetamine-induced rotational asymmetry (18). It will be important to determine whether continued GDNF delivery during a prolonged post-6-OHDA lesion interval would be enough trophic stimulus or whether striatal rAAV-rGDNF injections are necessary to support striatal TH+ fiber recovery.
The nigral DA neuron protection data presented herein are strikingly similar to recent data obtained by using the Sauer and Oertel model after nigral or striatal injection of an E1–E3 deleted recombinant Ad encoding human GDNF (17, 18). Although TH+ cell survival was not quantified in one report (17) and FG back-labeling was not performed in the other (18), the back-labeled FG+ cell survival observed herein is in agreement with that reported for the Ad-mediated nigral GDNF delivery (17), and the TH+ cell survival seen herein is somewhat greater than reported for striatal adenoviral GDNF delivery [85% vs. 60% (18)]. The greater nigral cell survival found after perinigral Ad (17) or rAAV vector-mediated GDNF delivery may be because of a lower efficiency of nigral GDNF delivery when Ad-GDNF is placed in striatum (18).
Greater levels of GDNF protein were reported at both 1 and 4 weeks after injection (13,000 pg and 4,700 pg, respectively) in the nigral Ad study (17) than those measured at 3 and 10 weeks in the present study (1,235 pg and 747 pg, respectively; see Table 1) as a result of AAV transduction. It is difficult to directly compare the measured GDNF protein values between the present study and the Ad vector study (17) because much smaller punches taken in the present study may have missed some of the extent of the transduced area. As pointed out in the nigral Ad study, both reported GDNF values are well above the GDNF EC50 for embryonic mesencephalic DA cells in culture if even a modest fraction of the measured GDNF levels are assumed to be secreted (17, 34). Although there is no direct data available to estimate which fraction of the total tissue GDNF is extracellular at any given time, in vitro data presented herein from HeLa cell transductions (Fig. 1) confirm that GDNF can escape from cells that have no special secretory mechanisms.
The GDNF levels dropped a significant 65% between 1 and 4 weeks in the nigral Ad vector study (17) as compared with a nonsignificant 35% reduction observed between 3 and 10 weeks in the present study. First-generation E1–E3-deleted Ad vectors have been used in human trials and have been shown to support only transient transgene expression because of a rapid host immune response against either viral or transgene derived antigens expressed by the vector (27). In contrast to the lack of long-term transgene expression achieved with first-generation Ad vectors (27), rAAV-mediated transgene expression has been shown to persist for up to 1 year in the rat striatum although at levels that are reduced compared with early time points (R.J.M. and S.E.L., unpublished observations and ref. 31). Because wild-type AAV is a nonpathogenic virus and rAAV-CMV-lacZ has been demonstrated to express transgene for at least 4 months in a nonhuman primate (41), vectors based on rAAV to be used for gene therapy of neurodegenerative disorders may be presently more clinically relevant than first-generation E1− E3− Ad vectors.
The data regarding a GDNF-based gene therapy presented herein, as well as those reported previously (17), suggest that therapeutic nigral protein delivery via in vivo injection of an appropriately safe vector may be a viable strategy for the treatment of PD. The present data indicate that expression of relatively low levels of GDNF are sufficient to provide significant protection against 6-OHDA-induced nigrostriatal degeneration. Thus, with the absence of significant GDNF-induced toxicity after repeated bolus injections of the protein (3, 22), Ad vector delivery (17, 18) or rAAV transduction (see Results) further suggests that there is a wide therapeutic window for the trophic effects of GDNF on nigrostriatal DA neurons. Finally, long-term transgene expression has been conclusively demonstrated for rAAV (31) and lentivectors (42) in the brain, which indicates that vector transduction in the nigra may support continuous local GDNF delivery over a clinically relevant duration. Thus, vector-mediated delivery of GDNF may fulfill most of the requirements necessary to qualify as a therapeutic strategy for PD: (i) biological efficacy, (ii) long-term expression, and (iii) lack of toxicity coupled with a wide therapeutic window. Thus, the next steps necessary to continue toward development of an in vivo gene therapy for the treatment of PD are long-term studies that demonstrate both transgene expression and safety in nonhuman primates and the addition of the ability to regulate transgene expression (43–45) as a safety feature or for dosage control.
Acknowledgments
This work was performed at Somatix Therapy Corporation, which has merged with Cell Genesys, Inc. We gratefully acknowledge the technical assistance of Heidi Oline, Ming Chen, Dorothy Clevenger, Terry Jaret, Fang-Fang Wu, Judith John, Dea Nagy, Matthew Morton, Joyce Conway, Ya-Li Lee, Xiadong Xu, Brian Kaspar, and Barbara Sloan. Special thanks go to Larry Cohen for his support of our research program.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: rAAV, recombinant adeno-associated virus; GDNF, glial cell line-derived neurotrophic factor; rGDNF, rat GDNF; PD, Parkinson’s disease; DA, dopamine; SN, substantia nigra; 6-OHDA, 6-hydroxydopamine; CMV, cytomegalovirus; TH, tyrosine hydroxylase; MOI, multiplicity of infection; FG, fluorogold; Ad, adenovirus.
References
- 1.Hou J G G, Lin L F H, Mytilineou C. J Neurochem. 1996;66:74–82. doi: 10.1046/j.1471-4159.1996.66010074.x. [DOI] [PubMed] [Google Scholar]
- 2.Clarkson E D, Zawada W M, Freed C R. Neuroreport. 1995;7:145–149. [PubMed] [Google Scholar]
- 3.Kearns C M, Gash D M. Brain Res. 1995;672:104–111. doi: 10.1016/0006-8993(94)01366-p. [DOI] [PubMed] [Google Scholar]
- 4.Hoffer B J, Hoffman A, Bowenkamp K, Huettl P, Hudson J, Martin D, Lin L F H, Gerhardt G A. Neurosci Lett. 1994;182:107–111. doi: 10.1016/0304-3940(94)90218-6. [DOI] [PubMed] [Google Scholar]
- 5.Shults C W, Kimber T, Martin D. Neuroreport. 1996;7:627–631. doi: 10.1097/00001756-199601310-00060. [DOI] [PubMed] [Google Scholar]
- 6.Bowenkamp K E, Hoffman A F, Gerhardt G A, Henry M A, Biddle P T, Hoffer B J, Granholm A C E. J Comp Neurol. 1995;355:479–489. doi: 10.1002/cne.903550402. [DOI] [PubMed] [Google Scholar]
- 7.Bowenkamp K E, Lapchak P A, Hoffer B J, Bickford P C. Neurosci Lett. 1996;211:81–84. doi: 10.1016/0304-3940(96)12729-4. [DOI] [PubMed] [Google Scholar]
- 8.Opacka-Juffry J, Ashworth S, Hume S P, Martin D, Brooks D J, Blunt S B. Neuroreport. 1995;7:348–352. [PubMed] [Google Scholar]
- 9.Sauer H, Rosenblad C, Björklund A. Proc Natl Acad Sci USA. 1995;92:8935–8939. doi: 10.1073/pnas.92.19.8935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hudson J, Granholm A C, Gerhardt G A, Henry M A, Hoffman A, Biddle P, Leela N S, Mackerlova L, Lile J D, Collins F, Hoffer B J. Brain Res Bull. 1995;36:425–432. doi: 10.1016/0361-9230(94)00224-o. [DOI] [PubMed] [Google Scholar]
- 11.Gash D M, Zhang Z M, Ovadia A, Cass W A, Yi A, Simmerman L, Russell D, Martin D, Lapchak P A, Collins F, Hoffer B J, Gerhardt G A. Nature (London) 1996;380:252–255. doi: 10.1038/380252a0. [DOI] [PubMed] [Google Scholar]
- 12.Treanor J J S, Goodman L, Desauvage F, Stone D M, Poulsen K T, Beck C D, Gray C, Armanini M P, Pollock R A, Hefti F, Phillips H S, Goddard A, Moore M W, Bujbello A, Davies A M, Asai N, Takahashi M, Vandlen R, Henderson C E, Rosenthal A. Nature (London) 1996;382:80–83. doi: 10.1038/382080a0. [DOI] [PubMed] [Google Scholar]
- 13.Jing S Q, Wen D Z, Yu Y B, Holst P L, Luo Y, Fang M, Tamir R, Antonio L, Hu Z, Cupples R, Louis J C, Hu S, Altrock B W, Fox G M. Cell. 1996;85:1113–1124. doi: 10.1016/s0092-8674(00)81311-2. [DOI] [PubMed] [Google Scholar]
- 14.Choi-Lundberg D L, Bohn M C. Dev Brain Res. 1995;85:80–88. doi: 10.1016/0165-3806(94)00197-8. [DOI] [PubMed] [Google Scholar]
- 15.Jiao S, Miller P J, Lapchak P A. Neurosci Lett. 1996;220:187–190. doi: 10.1016/s0304-3940(96)13265-1. [DOI] [PubMed] [Google Scholar]
- 16.Lapchak P A, Jiao S, Collins F, Miller P J. Brain Res. 1997;747:92–102. doi: 10.1016/s0006-8993(96)01265-6. [DOI] [PubMed] [Google Scholar]
- 17.Choi-Lundberg D L, Lin Q, Chang L -N, Chiang L, Hay C M, Mohajeri H, Davidson B L, Bohn M C. Science. 1997;275:838–841. doi: 10.1126/science.275.5301.838. [DOI] [PubMed] [Google Scholar]
- 18.Bilang-Bleuel A, Revah F, Colin P, Locquet I, Robert J-J, Mallet J, Horellou P. Proc Natl Acad Sci USA. 1997;94:8818–8823. doi: 10.1073/pnas.94.16.8818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hefti F, Hartikka J, Knusel B. Neurobiol Aging. 1989;10:1–20. doi: 10.1016/0197-4580(89)90118-8. [DOI] [PubMed] [Google Scholar]
- 20.Venero J L, Hefti F, Knusel B. Mol Pharmacol. 1996;49:303–310. [PubMed] [Google Scholar]
- 21.Sauer H, Oertel W H. Neurosciences. 1994;59:401–415. doi: 10.1016/0306-4522(94)90605-x. [DOI] [PubMed] [Google Scholar]
- 22.Winkler C, Sauer H, Lee C S, Björklund A. J Neurosci. 1996;16:7206–7215. doi: 10.1523/JNEUROSCI.16-22-07206.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.During M J, Naegele J R, O’Malley K L, Geller A I. Science. 1994;266:1399–1403. doi: 10.1126/science.266.5189.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaplitt M G, Leone P, Samulski R J, Xiao X, Pfaff D W, O’Malley K L, During M J. Nat Genet. 1994;8:148–153. doi: 10.1038/ng1094-148. [DOI] [PubMed] [Google Scholar]
- 25.Horellou P, Vigne E, Castel M N, Barneoud P, Colin P, Perricaudet M, Delaere P, Mallet J. Neuroreport. 1994;6:49–53. doi: 10.1097/00001756-199412300-00014. [DOI] [PubMed] [Google Scholar]
- 26.Muzyczka N. Curr Top Microbiol Immunol. 1992;158:97–129. doi: 10.1007/978-3-642-75608-5_5. [DOI] [PubMed] [Google Scholar]
- 27.Yang Y, Nunes F A, Berencsi K, Furth E E, Gonczol E, Wilson J M. Proc Natl Acad Sci USA. 1994;91:4407–4411. doi: 10.1073/pnas.91.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Y, Li Q, Ertl H C J, Wilson J M. J Virol. 1995;69:2004–2015. doi: 10.1128/jvi.69.4.2004-2015.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai Y, Schwarz E M, Gu D, Zhang W-W, Sarvetnick N, Verma I M. Proc Natl Acad Sci USA. 1995;92:1401–1405. doi: 10.1073/pnas.92.5.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Snyder R O, Miao C H, Patijn G A, Spratt S K, Danos O, Gown A M, Winther B, Meuse L, Cohen L K, Thompson A R, Kay M A. Nat Genet. 1997;16:270–276. doi: 10.1038/ng0797-270. [DOI] [PubMed] [Google Scholar]
- 31.During M J, Leone P. Clin Neurosci. 1996;3:292–300. [PubMed] [Google Scholar]
- 32.McCown T J, Xiao X, Li J, Breese G R, Samulski R J. Brain Res. 1996;713:99–107. doi: 10.1016/0006-8993(95)01488-8. [DOI] [PubMed] [Google Scholar]
- 33.Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage F H, Verma I M, Trono D. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 34.Lin L F, Doherty D H, Lile J D, Bektesh S, Collins F. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- 35.Springer J E, Mu X, Bergmann L W, Trojanowski J Q. Exp Neurol. 1994;167:170. doi: 10.1006/exnr.1994.1091. [DOI] [PubMed] [Google Scholar]
- 36.Samulski R J, Chang L S, Shenk T. J Virol. 1989;63:3822–3828. doi: 10.1128/jvi.63.9.3822-3828.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J, Samulski R J, Xiao X. J Virol. 1997;71:5236–5243. doi: 10.1128/jvi.71.7.5236-5243.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego, CA: Academic; 1987. [DOI] [PubMed] [Google Scholar]
- 39.Hudson J, Granholm A C, Gerhardt G, Hoffman A, Biddle P, Leela N S, Collins F, Hoffer B. Soc Neurosci Abst. 1993;19:652. [Google Scholar]
- 40.Sternberger L A, Hardy P H, Cuculis J J, Meyer H G. J Histochem Cytochem. 1970;18:315–333. doi: 10.1177/18.5.315. [DOI] [PubMed] [Google Scholar]
- 41.Bankiewicz K S, Leff S E, Nagy D, Jungles S, Rokovich J, Spratt K, Cohen L, Libonati M, Snyder R O, Mandel R J. Exp Neurol. 1997;144:147–156. doi: 10.1006/exnr.1996.6401. [DOI] [PubMed] [Google Scholar]
- 42.Blömer U, Naldini L, Kafri T, Trono D, Verma I M, Gage F H. J Virol. 1997;71:6641–6649. doi: 10.1128/jvi.71.9.6641-6649.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gossen M, Bujard H. Proc Natl Acad Sci USA. 1992;89:5547–5557. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.No D, Yao T-P, Evans R M. Proc Nat Acad Sci USA. 1996;93:3346–3351. doi: 10.1073/pnas.93.8.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rivera V M, Clackson T, Natesan S, Pollock R, Amara J F, Keenan T, Magari S R, Phillips T, Courage N L, Cerasoli F, Jr, Holt D A, Gilman M. Nat Med. 1996;2:1028–1032. doi: 10.1038/nm0996-1028. [DOI] [PubMed] [Google Scholar]