

Abstract
Sarcolipin (SLN) has emerged as an important regulator of the atrial sarcoplasmic reticulum (SR) Ca2+ transport. The inhibitory effect of SLN on cardiac SR Ca2+ ATPase (SERCA) pump can be relieved by β-adrenergic stimulation, which indicates that SLN is a reversible inhibitor. However, the mechanism of this reversible regulation of SERCA pump by SLN is yet to be determined. In the current study using adult rat ventricular myocytes we provide evidence that the threonine 5 (T5) residue at the N-terminus of SLN which is conserved among various species, critically regulates the SLN function. Point mutation of T5→alanine exerts an inhibitory effect on myocyte contractility and calcium transients similar to that of wild-type SLN, whereas mutation of T5→glutamic acid which mimics the phosphorylation abolished the inhibitory function of SLN. Our results showed that T5 can be phosphorylated in vitro by calcium–calmodulin dependent protein kinase II (CaMKII). Blocking the CaMKII activity in WT-SLN overexpressing myocytes using autocamtide inhibitory peptide completely abolished the β-adrenergic response. Taken together, our data suggest that T5 is the key amino acid which modulates SLN function via phosphorylation/dephosphorylation mechanisms through CaMKII pathway.
Keywords: SLN, Sarcolipin, SERCA, Ca2+ ATPase, Phosphorylation, Calcium calmodulin dependent protein kinase II, PLB, Phospholamban
1. Introduction
Sarcolipin (SLN), is a small molecule containing 31 amino acids, having significant homology to phospholamban [1,2] and is found to be expressed in cardiac and skeletal muscles [2–6]. Data shows that sarcolipin co-localizes and physically interacts with cardiac sarcoplasmic reticulum (SR) Ca2+ ATPase 2a (SERCA2a) [3,7,8]. SLN expression is regulated in a developmental and tissue specific manner [3–5,9]; also its expression is altered in a variety of disease conditions that affect cardiac and skeletal muscle [4,5,9–15], in particular SLN level was found to be increased in the atria of dogs in which heart failure was induced by tachypacing [5]. Altered expression of SLN without any changes in SERCA or PLB protein levels in these animal models suggests that the expression levels of SLN could play a critical role in the SR Ca2+ uptake and altered Ca2+ homeostasis observed in the diseased myocardium [5].
The physiological importance of SLN was recently elucidated using genetically engineered mouse models with alterations in SLN protein levels [16–19]. Cardiac specific overexpression of SLN was associated with significant decrease in SR Ca2+ uptake and depressed contractile parameters [16–18]. On the other hand, ablation of SLN was associated with significantly enhanced Ca2+ affinity and velocity of SERCA pump, resulting in an increase in atrial contraction [19]. Interestingly, SLN null atria showed blunted responses to isoproterenol stimulation [19], which suggest that SLN could mediate the β-adrenergic responses in atria. This was further supported by the observation that the inhibitory effect of SLN overexpression was relieved upon β-agonist stimulation [16,18]. In addition, SLN overexpression in the PLB null mice suggested that SLN can inhibit SERCA pump independent of PLB [17]. This inhibition can be reversed upon β-adrenergic stimulation. However, the molecular mechanisms which modulate SLN function are largely unknown.
Based on the analogy of reversible inhibition of SLN and PLB on SERCA2a, we hypothesize that SLN function can be modulated by phosphorylation/dephosphorylation mechanisms. SLN has a conserved threonine residue (T5) at the N-terminus [2,20] that could be phosphorylated by serine/threonine kinases. Co-transfection of SERCA and T5 to alanine (A) mutant SLN in HEK cells resulted in an inhibition of Ca2+ uptake function of SERCA pump, whereas mutation of T5 to glutamic acid (E) resulted in loss of inhibitory effect [17,21]. In addition, Gramolini et al. [17] suggested that T5 can be phosphorylated by a putative serine/threonine protein kinase 16 by co-expression studies in HEK cells. All these studies were carried out in a heterologous cell system and the role of T5 in modulating SLN function in cardiac myocytes is yet to be confirmed.
Therefore, a major goal of this study was to demonstrate the role of T5 in modulating SLN function in cardiac myocytes. Using site-directed mutagenesis and adenoviral gene transfer, we studied the role of T5 in adult rat ventricular myocytes which express very low levels of SLN and eliminates the effect of endogenous SLN [3–5,19].
2. Materials and methods
2.1. Mutagenesis and generation of recombinant adenovirus
The amino acid mutations of Thr 5→Ala (ACT→GCA) and Thr 5→Glu (ACT→GAG) were introduced into rat SLN cDNA by polymerase chain reaction (PCR). The adenoviral construct containing either N-terminal FLAG tagged wild-type SLN (NF-SLN) [3] or N-terminal FLAG tagged SLN mutants (NF-SLNT5A or NF-SLNT5E) were generated as described previously [3]. Briefly, mutant or wild-type SLN was cloned in frame to FLAG tag and subcloned into the shuttle vector, pAdTrack-CMV (Stratagene). This vector also drives the expression of green florescent protein (GFP) under separate CMV promoter and hence the virus-infected cells can be tracked by green florescence. The shuttle vector containing the cDNA of interest was transformed into BJ5183-AD1 to produce recombinant adenoviral plasmids. The recombinant plasmids were transfected into AD-293 HEK cells and amplified by 3–4 passages. Cells were collected after full cytopathic effect became evident, 2–3 days after plaque appearance. Virus was released by 3 cycles of freezing and thawing, and virus was collected by centrifuging at 2500 rpm for 10 min and collecting the supernatant that contained the viral stock. The final viral titers were determined by plaque assay with the final yield at 109 to 1010 pfu/ml [22].
2.2. Myocyte isolation and culture
Cardiac myocytes were isolated from adult Sprague Dawley rats as described earlier with modifications [23,24]. Briefly, the heart was removed from an adult male rat, mounted on a modified Langendorff perfusion apparatus and perfused with Krebs–Henseleit buffer (KHB) (118 mmol/L NaCl, 4.8 mmol/L KCl, 25 mmol/L HEPES, 1.25 mmol/L K2HPO4, 1.25 mmol/L MgSO4, and 11 mmol/L glucose) containing 1 mM Ca2+ for 5 min. The perfusion was then switched to Ca2+-free KHB with 20 μM EGTA. After 10 min, collagenase (Worthington) and bovine serum albumin (BSA-Sigma) were added each at 1 mg/mL to the Ca2+-free KHB, and the perfusion continued. After 20 min, the Ca2+ concentration in the perfusing solution was raised to 1 mM, in 4 stepwise increments. The ventricle was removed and minced gently in a sterile beaker containing enzyme solution (1 mM Ca2+-KHB+ 1 mg/mL collagenase+1 mg/mL BSA) and incubated in a 37 °C water bath for 5 min with gentle swirling. Digests were collected by centrifugation and the cells were resuspended in 1 mM Ca2+-KHB+ 2% BSA. The cells were allowed to gravity settle, the supernatant was removed, and the cells were resuspended in DMEM+5% FBS+1% penicillin/streptomycin (P/S) and plated on laminin coated coverslips for 2 h prior to replacing the media with serum-free DMEM containing recombinant adenovirus. Myocytes were infected with either wild-type (Ad.NF-SLN) or mutant (Ad.NF-SLNT5A or Ad.NF-SLNT5E) adenovirus, at a multiplicity of infection (MOI) of 100 for 48 h. Uninfected cardiac myocytes maintained in culture for 48 h, referred as control(s) were also used for all subsequent experiments.
2.3. Western blot analysis
Cultured ventricular myocytes were lyzed in 6× sample buffer [(30% glycerol, 10% SDS, 600 mM dithiothreitol, 350 mM Tris–Cl ph 6.8, trace of Bromophenol blue) supplemented with NaF (6 mM) and protease inhibitors] and were immediately frozen in liquid N2 [25]. Protein samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and protein loading was normalized using Coomassie staining and to GAPDH levels. For Western blot analysis, myocyte protein samples were electrophoretically separated on SDS-PAGE (10% for SERCA2a and calsequestrin (CSQ), 14% for FLAG and PLB) and transferred to nitrocellulose membrane. Membranes were immunoprobed with following primary antibodies: anti-rabbit SERCA2a, anti-rabbit PLB, anti-rabbit CSQ (ABR), anti-mouse FLAG (SIGMA), anti-rabbit S16 or T17 PLB antibody (Badrilla, UK). Signals were detected by Super Signal WestDura substrate (Pierce) and quantitated by densitometry.
2.4. Immunostaining of isolated cardiac myocytes
Isolated rat ventricular myocytes were infected with either wild-type Ad.NF-SLN or mutant Ad.NF-SLNT5A or Ad.NF-SLNT5E adenovirus (at 100 MOI) for 48 h and processed for immunostaining and confocal imaging [24,3]. Cells were incubated in primary antibody (rabbit polyclonal SERCA2a, mouse monoclonal FLAG antibody, [Sigma]) in phosphate buffered saline (PBS) containing 2% normal goat serum and 1% Triton-X-100 for 1.5 h. The glass coverslips were washed three times with PBS containing 1% Triton-X-100 (PBS-T). Cells were incubated with Texas Red conjugated goat anti-rabbit and FITC-conjugated goat anti-mouse (Molecular Probes) secondary antibodies for 1 h, followed by three washes with PBS-T. After immunostaining, cells were visualized by excitation at 488 nm for FITC and 543 nm for Texas Red using the Zeiss Laser Scanning Microsope (LSM510).
2.5. Myocyte shortening measurements
Cardiac myocyte shortening measurements were done as described earlier [26,27] using the IonOptix system. The myocytes placed in tissue chambers (Cell microcontrols), were kept on the stage of the inverted microscope (Nikon TE-2000S). Myocytes were field- stimulated at 1 Hz and superfused with Tyrode solution (in mM/L: – 140 NaCl, 4 KCl, 1 MgCl2, 1 CaCl2, 10 glucose, and 5 HEPES (pH 7.4). Myocytes used for all functional experiments were rod-shaped without spontaneous contraction and could react to pacing and to different reagents throughout the experiment. After being equilibrated in Tyrode’s solution, 5–6 cells were studied randomly in each preparation for each group. Cells from 3–4 different preparations were studied for each group. The soft-edge software (IonOptix) was used to capture changes in cell length during shortening and re-lengthening. Data were expressed as percent of resting cell length (%RCL). To determine the β-adrenergic receptor mediated contraction, the isoproterenol dose–response curve was generated. Similarly, for CaMKII inhibition, cardiac myocytes with or without the adenovirus were incubated with 20 μM of a cell permeable CaMKII inhibitor (autocamtide inhibitor peptide, AIP, Calbiochem) for 30 min and contractility was measured in the absence or presence of 1 μM isoproterenol.
2.6. Calcium transient measurements
Different myocytes were used for cell shortening and calcium transient measurements to avoid interference of Fluo-4 AM loading on myocyte contractility [34]. For calcium transients myocytes were loaded with Fluo-4 AM (5 μM, Molecular Probes, Eugene, OR) for 10 min at 22 °C. Myocytes were placed in a chamber on the stage of a Nikon-TE2000S inverted microscope and imaged through a Fluor 40× oil objective. Myocytes were field-stimulated at 1 Hz and superfused with Tyrode’s solution containing 1 mmol/L CaCl2. Intracellular calcium [Ca2+]i was measured by Fluo-4 epifluorescence with excitation at 480±20 nm and emission at 535±25 nm. The illumination field was restricted to a small spot to get emission from a single cell. 5–6 cells were studied randomly in each preparation for each group. Cells from 3–4 different preparations were studied for each group. Calcium transients were analyzed using the IonOptix software. Data were expressed as F/Fo, where F is the fluorescence intensity and Fo is the intensity at rest. To avoid experimental bias, all measurements and analysis were done by more than one blinded observers.
2.7. In vitro CaMKII phosphorylation
Wild-type and T5A mutant human and mouse SLN were cloned into the bacterial expression vector pET23d (Novagen) and the bacterially expressed protein was purified according to the manufacturer’s instructions. In vitro phosphorylation was carried out with 10 μg protein each using a CaMKII phosphorylation kit (New England Biolabs) according to manufacturer’s instructions. Briefly, prior to SLN protein phosphorylation, CaMKII was activated by autophosphorylation by incubating for 10 min at 30 °C in a CaMKII buffer (in mM/L 50 Tris–HCl, 10 MgCl2, 2 dithiothreitol and 0.1 Na2 EDTA) supplemented with 100 μMATP, 1.2 μM calmodulin and 2 mM CaCl2. Equal amounts of WT and T5A mutant SLN proteins were labeled with 100 μCi/μmol γ-P32 ATP and phosphorylated using activated CaMKII. 10 and 20 μL of labeled proteins were resolved on 14% SDS-PAGE and autoradiographed.
2.8. Statistics
Data are presented as mean ± SEM. Statistical significance (P<0.05) was determined between groups using an ANOVA (followed by Neuman–Keuls test) for multiple groups or a Student’s paired t-test for two groups.
3. Results
3.1. Mutant SLN targets to the SR and co-localizes with SERCA2a
The infection efficiency of the WT or mutant SLN adenoviruses was assessed by the green fluorescent protein expression. After 48 h of infection, 90% of cells were fluorescent and indicated the transgene expression. To determine if the mutant SLN targets into the SR membrane and co-localizes with SERCA2a, we performed immunostaining of myocytes infected with adenovirus carrying N-terminally flagged WT or mutant SLN using FLAG and SERCA2a antibodies. Confocal imaging showed a distinct horizontal and vertical pattern for FLAG antibody (Fig. 1; green) in all three groups of myocytes infected with WT, T5A or T5E SLN adenovirus. Further, this pattern is indistinguishable from the SERCA2a antibody staining (Fig. 1; red) as seen from the overlay of these two images (Fig. 1; orange). These results indicate that the overexpressed WT or mutant SLN is co-localized with SERCA2a and the point mutation at T5 did not affect the targeting of SLN onto SR membrane.
Fig. 1.
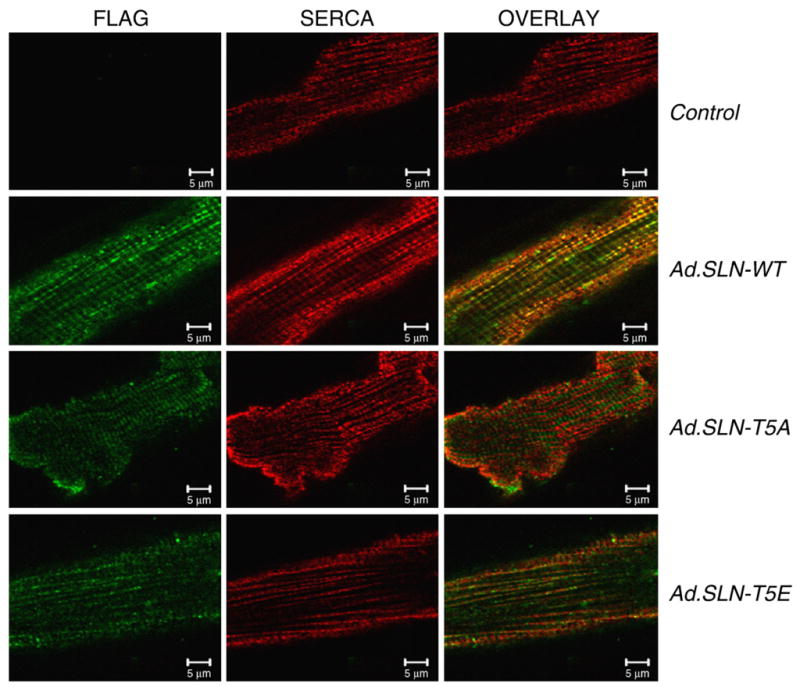
Confocal microscopic images of rat ventricular myocytes, showing co-localization of WT and mutant SLN with SERCA2a. Adult rat ventricular myocytes were infected with Ad. WT-SLN, Ad.SLN-T5A or Ad.SLN-T5E or left uninfected and stained with FLAG antibody (green) and SERCA2a (red) antibody. Orange-overlay of images shows co-localization.
3.2. Overexpression of mutant SLN does not alter PLB levels or its functional status in cardiac myocytes
To determine the expression of the transgene, total protein extract from WT and mutant SLN infected myocytes were analyzed by Western blot using FLAG antibody. As seen in Fig. 2, the wild-type and mutant SLN proteins were expressed at similar levels indicating the uniform rate of infection.
Fig. 2.
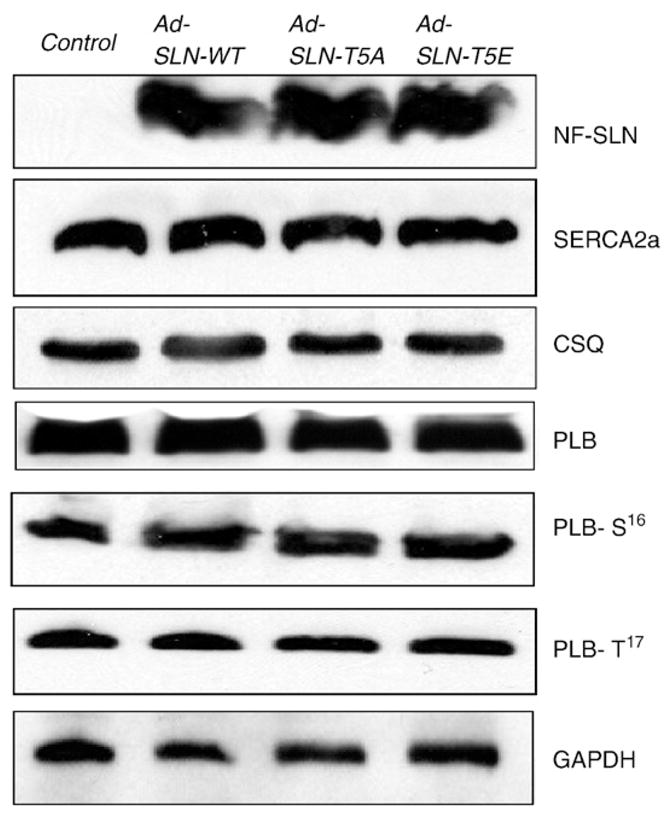
Expression of Ca handling proteins in control and Ad.SLN-WT, Ad.SLN-T5A or Ad. SLN-T5E-infected myocytes. Representative western blots showing the SERCA and PLB monomer protein levels in control and virus-infected myocytes. The expression levels of CSQ and GAPDH were used as loading controls. Total protein isolated from myocytes after 48 h of infection with the respective adenoviruses or untreated control myocytes were resolved on SDS-PAGE and immunoblotted with antibodies specific for FLAG (to detect FLAG tagged SLN), SERCA2a, PLB, CSQ and GAPDH. Phosphorylation status of PLB at Ser 16 (PLB-S16) and Thr 17 (PLB-T17) was determined using site specific antibodies.
To determine if the overexpression of WT or mutant SLN has any effect on the ratio of SERCA2a to PLB, the protein levels were quantitated by Western blot analysis. The SERCA2a and monomeric PLB protein levels were not altered in myocytes infected with either WT or mutant SLN (Fig. 2). Further, the basal phosphorylation of PLB at Ser 16 or Thr 17 was not significantly different from control as well as WT-SLN expressing myocytes. Calsequestrin (CSQ) and GAPDH were used as loading controls.
3.3. T5E mutant SLN has no effect on the myocyte contractility
To determine how the T5 mutations modulate SLN function and myocyte contractility, we measured cell shortening by edge detection method. After 48 h infections with recombinant adenovirus, myocytes were paced at 1 Hz and cell shortening was measured. Fig. 3 shows that the overexpression of T5A mutant SLN significantly decreased the cell shortening compared to uninfected control myocytes. The inhibitory effect of T5A mutant SLN was similar to the effect of WT SLN on myocyte contractility. The T5E mutant SLN has no significant effect on cell shortening (Fig. 3A; %RCL: Control — 6.84±0.52%, WT — 3.81±0.54%, T5A — 3.90±0.34% and T5E — 5.78±0.49%). Consistent with cell shortening, WT and T5A mutant SLN expressing myocytes showed significantly prolonged time to 50% relaxation (RT50), whereas the RT50 values for cells expressing T5E mutant were similar to uninfected myocytes (Fig. 3(B)). (RT50: - Control=0.10± 0.01 s, WT=0.13±0.01 s, T5A=0.14±0.01 s and T5E=0.10± 0.006 s). The time to peak contraction was not significantly different between the groups (data not shown). Further, rates of cell shortening (+dL/dT) and relaxation (−dL/dT) were also significantly lower in WT and T5A expressing myocytes while the expression of T5E mutation did not alter the dL/dT values (Fig. 3C). (+dL/dT: Control = 82.01± 10.68 μm/s, WT=40.60±5.14 μm/s, T5A= 49.87±9.63 and T5E=94.47±13.72 μm/s; − dL/dT: - Control= − 128.5±18.54 μm/s, WT = − 44.21±5.38 μm/s, T5A = − 58.64± 8.74 μm/s, T5E = − 122.4±17.95 μm/s). These results indicate that the SLN T5E mutant which mimics the phosphorylated form lost its inhibitory effect on myocyte contractility.
Fig. 3.
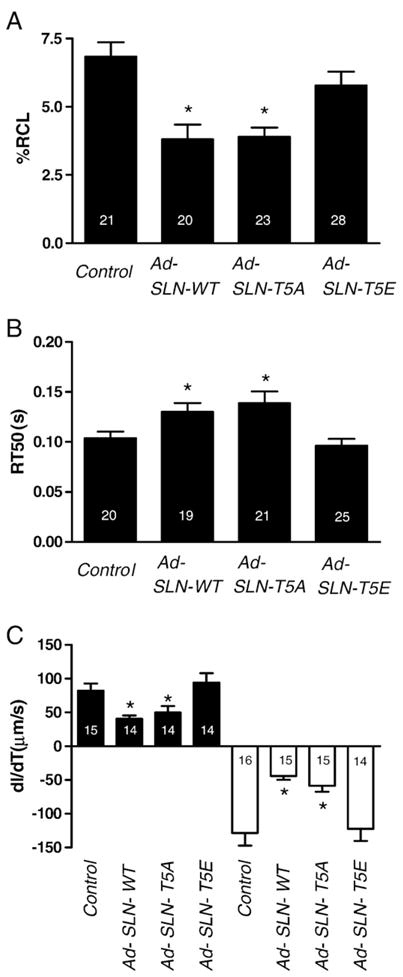
Mutation of T5 alters myocyte contractility— contraction amplitude (A) and time to 50% relaxation (B) in control myocytes and myocytes infected with Ad.SLN-WT, Ad. SLN-T5A or Ad.SLN-T5E adenovirus. (C) Rates of shortening (+dL/dT) are represented on the positive axis (closed bars) and relaxation (−dL/dT) on the negative axis (open bars). Myocyte contractility was analyzed after 48 h by pacing at 1 Hz. *Significantly different from control myocytes (p<0.05). Number of cells (N) indicated within each bar graph.
To determine the effect of isoproterenol on mutant and WT-SLN overexpressing myocytes, cells were treated with an increasing concentration of isoproterenol and the contractility was measured. As depicted in Fig. 4, the control uninfected myocytes and SLN T5E expressing myocytes respond to increasing amounts of ISO by increasing %RCL in a dose dependent manner. The inhibitory effect of WT SLN was relieved only upon high ISO concentration (10−6 M); whereas, the myocytes expressing T5A mutant SLN continue to exert a significant inhibition on myocyte contractility even at high ISO concentration. (%RCL for Control is [− ISO=7.33±0.54%, 10−9 M ISO = 9.89 ± 1.02%, 10−6 M ISO = 14.1 ± 1.97%]; WT SLN [− ISO = 4.88 ± 0.83%, 10−9 M ISO = 5.93 ± 0.82%, 10−6 M ISO=9.9±1.35%]; T5A [− ISO=4.58±1.2%, 10−9 M ISO=6.34± 1.49%, 10−6 M ISO=8.08±1.52%; T5E [− ISO=7.23±1.24%, 10−9 M ISO=11.6±1.64%, 10−6 M ISO=13.2±1.82%].
Fig. 4.
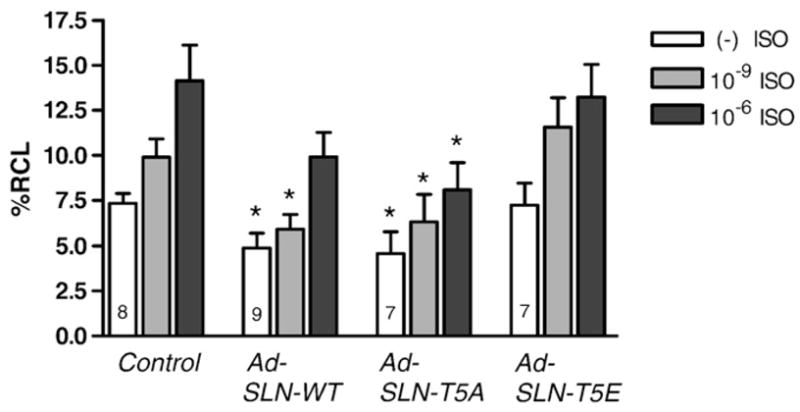
Isoproterenol dose–response in WT and mutant SLN. Contractility of myocytes infected with either Ad.SLN-WT, Ad. SLN-T5A or Ad. SLN-T5E was measured in the presence of increasing amounts of isoproterenol [— ISO (open bars), 10−9 M ISO (grey bars) or 10−6 M ISO (dark grey bars)] in comparison to uninfected control. Data expressed as mean±S.E.M. *Indicates significantly different from corresponding treatment of control myocytes (p<0.05).
These results suggest that the inhibitory effect of SLN can be relieved only upon stimulation with high ISO concentration and the β-adrenergic response observed in SLN infected myocytes are independent of PLB.
3.4. Mutation of T5 alters the rate and amplitude of Ca2+ transients
To evaluate the effect of T5A or T5E mutation on [Ca2+]i transients, we measured the amplitude, time to 50% relaxation (RT50) and time to peak value (TTp) of calcium transients. Results in Fig. 5 indicate that the [Ca2+]i transients decreased significantly in myocytes expressing WT-SLN and T5A mutant SLN compared to the control uninfected myocytes. On the other hand, T5E mutant SLN had no significant effect on [Ca2+]i transients (ΔF/Fo Control=0.10±0.014, WT=0.05±0.01, T5A=0.05±0.01, T5E=0.09±0.01) (Fig. 5A). The time to 50% decay of Ca2+ (Fig. 5B) was significantly prolonged and to a similar extent in myocytes expressing WT or T5A mutant SLN, whereas SLN T5E had no effect on Ca2+ decay. (Control=0.08±0.004 s, WT=0.102±0.01 s, T5A=0.12±0.01 s and T5E=0.08±0.004 s). Time to 50% peak amplitude (Fig. 5C) was not significantly different between the groups. (Control=0.04±0.004 s, WT=0.04±0.002 s, T5A=0.05± 0.003 s, and T5E=0.04±0.001).
Fig. 5.
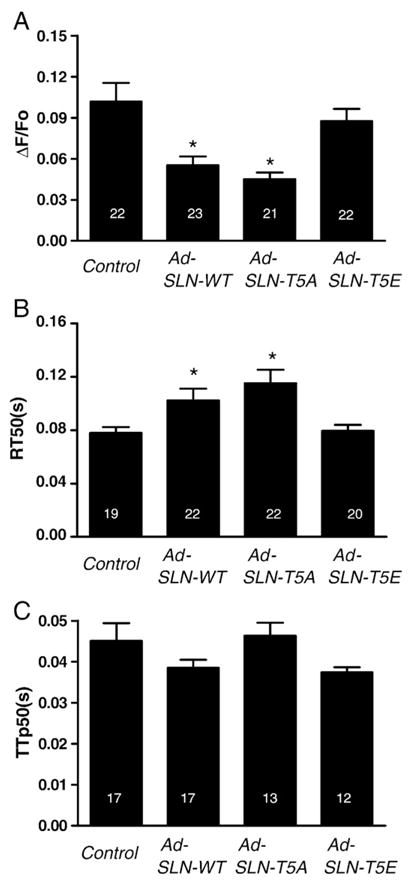
T5 mutation in SLN alters cardiac myocyte calcium transient — summary data (expressed as mean±S.E.M.) of (A) — Ca2+ transient amplitude (B) — time to 50% peak amplitude and (C) of Ca2+ transient relaxation (RT50). *Significantly different from control (p<0.05).
3.5. CaMKII is a potential modulator of SLN function
To identify T5 as a phosphorylation site for CaMKII, in vitro phosphorylation of purified mouse and human WT and T5A mutant SLN were carried out using γ32P-ATP and exogenous CaMKII. The phosphorylated proteins were analyzed by SDS-PAGE and autoradiography (Fig. 6A). Results show that both mouse and human WT SLN were phosphorylated by CaMKII and this phosphorylation was abolished by the T5 to alanine point mutation.
Fig. 6.
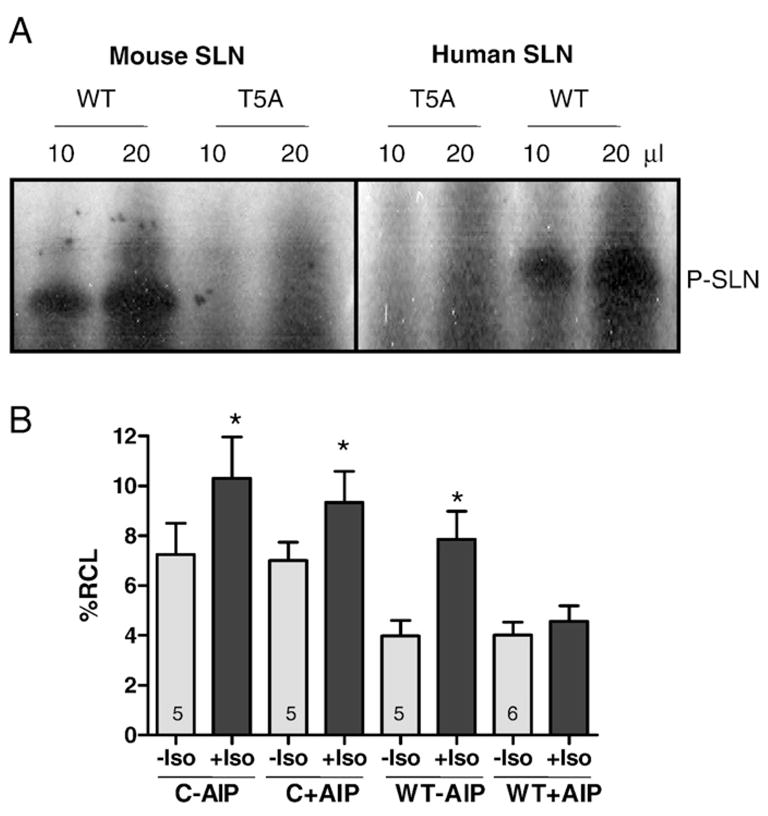
SLN is a target for CaMKII phosphorylation. (A) In vitro phosphorylation of mouse and human SLN by CaMKII — representative autoradiogram showing phosphorylated SLN corresponding to mouse and human SLN. (Details are in Section 2 Materials and methods). (B) Effect of CaMKII inhibitor autocamtide inhibitor peptide (AIP) on myocyte contractility — Contractility of control and Ad.SLN-WT infected myocytes was measured in response to isoproterenol in the presence and absence of 20 μM AIP. Data expressed as mean±S.E.M. * Indicates that contractility is significantly different from corresponding untreated myocytes. (p<0.05).
To further confirm that CaMKII modulates the SLN function during β-adrenergic stimulation, we studied the effect of CaMKII inhibition on the isoproterenol (ISO) stimulated contractility of ventricular myocytes expressing WT SLN. As depicted in Fig. 6B, overexpression of WT SLN inhibits the myocyte contractility and this inhibitory effect was relieved upon ISO treatment. Pretreatment with CaMKII inhibitor autocamtide inhibitory peptide (AIP) blocks the ISO effect on cells expressing WT SLN but not on control cells (Fig. 6B); %RCL: 7.24± 1.26% (− ISO – AIP) 10.3±1.67% (+ISO − AIP); 7.01±0.73% (− ISO+ AIP) 9.3±1.25 (+ISO+AIP) in control and 3.98±0.62% (− ISO – AIP) 7.87±1.12% (+ISO − AIP); 4.02±0.52% (− ISO+AIP) 4.57±0.62% (− ISO+AIP) in WT SLN). Isoproterenol treatment significantly (p<0.05) increased the contractility in all the groups except in myocytes expressing WT SLN pre-treated with AIP. These results suggest that CaMKII is involved in the regulation of SLN function during β-adrenergic stimulation.
4. Discussion
In this study, for the first time, we demonstrated the role of threonine 5 phosphorylation in modulating SLN function on cardiac myocyte contractility and calcium transients. By co-expressing SERCA2 and SLN in HEK 293 cells, MacLennan et al. have shown that T5 mutation on SLN can affect the calcium uptake function [8]. In the present study, using site-directed mutagenesis and adenoviral over-expression in ventricular myocytes, we identified T5 of SLN as a phosphorylation site for CaMKII and mediator of the β-adrenergic responses.
The use of ventricular myocytes to define the SLN function and its physiological relevance on myocyte contractility is always questionable as they express very low levels of SLN [5]. However, studies using transgenic and knockout mouse models have shown that the function of SLN is the same in atria and in the ventricles [16–19]. Thus the data obtained using ventricular myocytes should have the same functional relevance as of atrial myocytes. Further, the use of ventricular myocytes allowed us to determine the effect of T5 mutation on SLN without the interference of endogenous SLN.
The SR membrane targeting and co-localization of mutant SLN with SERCA2a indicate that N-terminal FLAG or the point mutation has no effect on the structural integrity of the mutant proteins. Overexpression of WT or mutant SLN did not alter the expression and functional status of SERCA and PLB indicating that the functional changes observed were due to SLN overexpression. The inhibitory effect of T5A on myocyte contractility and calcium transients indicate that T5A mutation did not affect the SLN function. However, this inhibitory effect was not relieved upon ISO stimulation. On the other hand, T5E mutant SLN, which mimics the phosphorylated form, lost its inhibitory function on myocyte contractility and calcium transients. Our results are consistent with an earlier report [21] which showed that the point mutation of SLN T5 to glutamic acid abolished the inhibitory effect of SLN on Ca2+ uptake function in a heterologous (HEK 293) cell system. Amino acid comparison studies have shown that T5 is the only conserved phosphorylation site in the kinase accessible cytoplasmic domain of SLN among various species [2,20 and 6] which further supports the role of T5 on SLN function. Taken together, our data suggest that T5 is the critical amino acid which modulates SLN function through phosphorylation/dephosphorylation mechanisms.
Our recent findings show that ISO can relieve the inhibitory effect of SLN in a transgenic mouse model overexpressing SLN in the heart [16,18] and blunted response to ISO by SLN knockout atria [19] provide strong evidence that SLN could be the mediator of β-adrenergic responses in atria. Although atria express both PLB and SLN, it has been shown that PLB contributes only in part, to the β-adrenergic mediated atrial contractility [31,32] further supporting our view. The present study also pointed out the importance of T5 phosphorylation in modulating SLN function during β-adrenergic stimulation. However, the signaling pathway which mediates SLN phosphorylation during β-adrenergic stimulation is yet to be studied.
A recent study suggests that a serine–threonine protein kinase, STK16 could phosphorylate SLN at T5 [17]. However, the physiological relevance of STK16 in cardiac function and its role during β-adrenergic stimulation are not known. On the other hand, the role of CaMKII in mediating β-adrenergic responses in heart is better studied. CaMKII has been shown to phosphorylate phospholamban at Thr17 during β-adrenergic receptor stimulation and could be important for cardiac function [28–30]. In the current study, we provide data for the first time to suggest that SLN can be phosphorylated by CaMKII at T5. Point mutation of T5 to alanine abolished the CaMKII phosphorylation in vitro suggesting that T5 is the target for CaMKII. Pretreatment of myocytes overexpressing WT SLN, with the CaMKII inhibitor AIP abolished the ISO mediated increase in myocyte contraction further supporting the view that CaMKII could phosphorylate SLN. AIP had no effect on the ISO mediated contractility of control, uninfected ventricular myocytes. This is an interesting finding which suggests that the β-adrenergic response in ventricular myocytes is not mediated through a CaMKII pathway. It is well known that phosphorylation of PLB at Ser16 through a PKA pathway mediates the β-adrenergic responses in the ventricle [28,30,33]. Since the phosphorylation of Ser16 precedes that of Thr 17, inhibition of CaMKII alone is not sufficient to abolish ISO response in control myocytes [33]. Further, overexpression of T5A, the phosphorylation mutant of SLN showed a significantly blunted response to ISO even at high concentrations and continued to exert a significantly inhibitory effect on contractility compared to control myocytes. This suggests that SLN mediated response to β-adrenergic stimulus is independent of PLB. Taken together, our results suggest that the major signaling pathway which mediates the β-adrenergic response is chamber specific.
In summary, our studies suggest that threonine 5 is the critical amino acid which modulates SLN function through phosphorylation/dephosphorylation and mediates the β-adrenergic response possibly through CaMKII pathway.
Glossary
- SLN
sarcolipin
- SR
sarco(endo)plasmic reticulum
- SERCA
SR Ca2+ ATPase
- CSQ
calsequestrin
- PLB
phospholamban
- T5
Threonine 5
- A
alanine
- E
glutamic acid
- CaMKII
calcium–calmodulin dependent kinase
Footnotes
This work was supported by National Institutes of Health Grant HL-64140 (to M. P.).
References
- 1.Wawrzynow A, et al. Sarcolipin, the “proteolipid” of skeletal muscle sarcoplasmic reticulum, is a unique, amphipathic, 31-residue peptide. Arch Biochem Biophys. 1992;298(2):620–3. doi: 10.1016/0003-9861(92)90457-8. [DOI] [PubMed] [Google Scholar]
- 2.Odermatt A, et al. Characterization of the gene encoding human sarcolipin (SLN), a proteolipid associated with SERCA1: absence of structural mutations in 3ve patients with Brody disease. Genomics. 1997;45(3):541–53. doi: 10.1006/geno.1997.4967. [DOI] [PubMed] [Google Scholar]
- 3.Babu GJ, et al. Overexpression of sarcolipin decreases myocyte contractility and calcium transient. Cardiovasc Res. 2005;65(1):177–86. doi: 10.1016/j.cardiores.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 4.Vangheluwe P, et al. Sarcolipin and phospholamban mRNA and protein expression in cardiac and skeletal muscle of different species. Biochem J. 2005;389(Pt 1):151–9. doi: 10.1042/BJ20050068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babu GJ. Differential expression of sarcolipin protein during muscle development and cardiac pathophysiology. J Mol Cell Cardiol. 2007;43(2):215–22. doi: 10.1016/j.yjmcc.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhupathy P, Babu GJ, Periasamy M. Sarcolipin and phospholamban as regulators of cardiac sarcoplasmic reticulum Ca2+ ATPase. J Mol Cell Cardiol. 2007;42(5):903–11. doi: 10.1016/j.yjmcc.2007.03.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asahi M. Regulation of sarco(endo)plasmic reticulum Ca2+ adenosine triphosphatase by phospholamban and sarcolipin: implication for cardiac hypertrophy and failure. Trends Cardiovasc Med. 2003;13(4):152–7. doi: 10.1016/s1050-1738(03)00037-9. [DOI] [PubMed] [Google Scholar]
- 8.MacLennan DH, Asahi M, Tupling AR. The regulation of SERCA-type pumps by phospholamban and sarcolipin. Ann NY Acad Sci. 2003;986:472–80. doi: 10.1111/j.1749-6632.2003.tb07231.x. [DOI] [PubMed] [Google Scholar]
- 9.Minamisawa S. Atrial chamber-specific expression of sarcolipin is regulated during development and hypertrophic remodeling. J Biol Chem. 2003;278(11):9570–5. doi: 10.1074/jbc.m213132200. [DOI] [PubMed] [Google Scholar]
- 10.Uemura N. Down-regulation of sarcolipin mRNA expression in chronic atrial 3brillation. Eur J Clin Invest. 2004;34(11):723–30. doi: 10.1111/j.1365-2362.2004.01422.x. [DOI] [PubMed] [Google Scholar]
- 11.Trivieri MG. Cardiac-specific elevations in thyroid hormone enhance contractility and prevent pressure overload-induced cardiac dysfunction. Proc Natl Acad Sci U S A. 2006;103(15):6043–8. doi: 10.1073/pnas.0601072103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimura M. Mechanical stress-dependent transcriptional regulation of sarcolipin gene in the rodent atrium. Biochem Biophys Res Commun. 2005;334(3):861–6. doi: 10.1016/j.bbrc.2005.06.186. [DOI] [PubMed] [Google Scholar]
- 13.Ottenheijm CA. Sarcoplasmic reticulum calcium uptake and speed of relaxation are depressed in nebulin-free skeletal muscle. FASEB J. 2008;22(8):2912–9. doi: 10.1096/fj.07-104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pashmforoush M. Nkx2–5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell. 2004;117(3):373–86. doi: 10.1016/s0092-8674(04)00405-2. [DOI] [PubMed] [Google Scholar]
- 15.Minamisawa S. Post-transcriptional downregulation of sarcolipin mRNA by triiodothyronine in the atrial myocardium. FEBS Lett. 2006;580(9):2247–52. doi: 10.1016/j.febslet.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 16.Babu GJ. Targeted overexpression of sarcolipin in the mouse heart decreases sarcoplasmic reticulum calcium transport and cardiac contractility. J Biol Chem. 2006;281(7):3972–9. doi: 10.1074/jbc.M508998200. [DOI] [PubMed] [Google Scholar]
- 17.Gramolini AO. Cardiac-specific overexpression of sarcolipin in phospholamban null mice impairs myocyte function that is restored by phosphorylation. Proc Natl Acad Sci U S A. 2006;103(7):2446–51. doi: 10.1073/pnas.0510883103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asahi M, et al. Cardiac-specific overexpression of sarcolipin inhibits sarco(endo) plasmic reticulum Ca2+ ATPase (SERCA2a) activity and impairs cardiac function in mice. Proc Natl Acad Sci U S A. 2004;101(25):9199–204. doi: 10.1073/pnas.0402596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Babu GJ, et al. Ablation of sarcolipin enhances sarcoplasmic reticulum calcium transport and atrial contractility. Proc Natl Acad Sci U S A. 2007;104(45):17867–72. doi: 10.1073/pnas.0707722104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gramolini AO, et al. Sarcolipin retention in the endoplasmic reticulum depends on its C-terminal RSYQY sequence and its interaction with sarco(endo)plasmic Ca(2+)-ATPases. Proc Natl Acad Sci U S A. 2004;101(48):16807–12. doi: 10.1073/pnas.0407815101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Odermatt A, et al. Sarcolipin regulates the activity of SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+-ATPase. J Biol Chem. 1998;273(20):12360–9. doi: 10.1074/jbc.273.20.12360. [DOI] [PubMed] [Google Scholar]
- 22.He TC, et al. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95(5):2509–14. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altschuld R, et al. Calcium tolerance of isolated rat heart cells. J Mol Cell Cardiol. 1980;12:1383–95. doi: 10.1016/0022-2828(80)90123-6. [DOI] [PubMed] [Google Scholar]
- 24.Rust EM, Albayya FP, Metzger JM. Identification of a contractile deficit in adult cardiac myocytes expressing hypertrophic cardiomyopathy-associated mutant troponin T proteins. J Clin Invest. 1999;103(10):1459–67. doi: 10.1172/JCI6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huke S, Bers DM. Temporal dissociation of frequency-dependent acceleration of relaxation and protein phosphorylation by CaMKII. J Mol Cell Cardiol. 2007;42(3):590–9. doi: 10.1016/j.yjmcc.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ren J, et al. Depressed contractile function and adrenergic responsiveness of cardiac myocytes in an experimental model of Parkinson disease, the MPTP-treated mouse. Neurobiol Aging. 2004;25(1):131–8. doi: 10.1016/s0197-4580(03)00035-6. [DOI] [PubMed] [Google Scholar]
- 27.Klein AL, Wold LE, Ren J. The cyclooxygenase-2 product prostaglandin E2 modulates cardiac contractile function in adult rat ventricular cardiomyocytes. Pharmacol Res. 2004;49(2):99–103. doi: 10.1016/j.phrs.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 28.MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4(7):566–77. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 29.Simmerman HK, Jones LR. Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol Rev. 1998;78(4):921–47. doi: 10.1152/physrev.1998.78.4.921. [DOI] [PubMed] [Google Scholar]
- 30.Zhao W, et al. Threonine-17 phosphorylation of phospholamban: a key determinant of frequency-dependent increase of cardiac contractility. J Mol Cell Cardiol. 2004;37(2):607–12. doi: 10.1016/j.yjmcc.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Koss KL, et al. Differential phospholamban gene expression in murine cardiac compartments. Molecular and physiological analyses. Circ Res. 1995;77(2):342–53. doi: 10.1161/01.res.77.2.342. [DOI] [PubMed] [Google Scholar]
- 32.Kadambi VJ. Phospholamban modulates murine atrial contractile parameters and responses to beta-adrenergic agonists. JMCC. 1998 Jul;30(7):1275–84. doi: 10.1006/jmcc.1998.0688. [DOI] [PubMed] [Google Scholar]
- 33.Chu G, et al. A single site (Ser16) phosphorylation in phospholamban is sufficient in mediating its maximal cardiac responses to beta-agonists. J Biol Chem. 2000 Dec 8;275(49):38938–43. doi: 10.1074/jbc.M004079200. [DOI] [PubMed] [Google Scholar]
- 34.Spurgeon HA. Simultaneous measurement of Ca2+, contraction, and potential in cardiac myocytes. Am J Physiol. 1990 Feb;258(2 Pt 2):H574–86. doi: 10.1152/ajpheart.1990.258.2.H574. [DOI] [PubMed] [Google Scholar]