

Abstract
The type I insulin-like growth factor receptor (IGF1R) regulates multiple aspects of malignancy and is the target of several drugs currently in clinical trials. While IGF1R’s role in proliferation and survival is well-studied, the regulation of metastasis by IGF1R is not as clearly delineated. Previous work showed that disruption of IGF1R signaling via overexpression of a dominant negative IGF1R inhibited metastasis. To establish a clinically applicable approach to inhibition of metastasis by targeting IGF1R, the effect of an inhibitory antibody against IGF1R, EM164 and its humanized version, AVE1642 on metastasis of cancer cells was examined. EM164 and AVE1642 did not affect primary tumor growth of MDA-435A/LCC6 cells but inhibited metastasis of these cells. Consistent with this inhibition in the formation of metastatic nodules, disruption of IGF1R also resulted in a decreased number of circulating tumor cells in blood of tumor-bearing mice. Disruption of IGF1R with a dominant negative construct or antibody inhibited invasion across Matrigel in vitro. When tumor cells were directly injected into the circulation via the lateral tail vein of mice, IGF1R disruption also resulted in significant reduction of pulmonary nodules, suggesting that regulation of invasion is not the only function of IGF1R signaling. Further, disruption of IGF1R rendered cells more susceptible to anoikis. Thus, IGF1R regulated metastasis independently of tumor growth. The multiple phenotypes regulated by IGF1R must be considered during development of this therapeutic strategy as inhibition of metastasis independent of inhibition of tumor growth is not easily assessed in phase II clinical trials.
Keywords: cancer metastasis, type I IGF receptor, antibodies against IGF1R, invasion, circulating tumor cells, survival
Introduction
The type I insulin-like growth factor receptor (IGF1R) plays important roles in tumor biology and is a new target for cancer therapy (Sachdev and Yee, 2007). Abundant data from cell culture, animal, and human epidemiological studies show that the ligands, insulin-like growth factors (IGFs) -I and -II and IGF1R regulate growth, survival, metabolism, and metastasis of cancer cells. Several antibodies and small molecule inhibitors of IGF1R have been developed (Burtrum et al., 2003; Cohen et al., 2005; Garcia-Echeverria et al., 2004; Haluska et al., 2006; Maloney et al., 2003; Wu et al., 2006). Some of these antibodies are in phase I and II clinical trials in solid tumors including CP-751,871 (Haluska et al., 2007; Karp et al., 2007), IMC-A12 (Higano et al., 2007; Rowinsky et al., 2007) and AVE1642 (Tolcher et al., 2008). Even though these trials are underway, the effects of IGF1R signaling on the malignant phenotype are not completely delineated. A linkage of IGF1R mediated phenotypes to specific pathways regulated by this receptor will enable us to better design trials.
The role of IGF1R in regulating tumor growth is well understood. Activation of IGF1R results in recruitment of adaptor proteins belonging to the insulin receptor substrate (IRS) family and IGF-stimulated phenotypes may be dependent on activation of specific adaptor protein species. Consistent with this notion, activation of IRS-1 has been linked to proliferation and apoptosis while activation of IRS-2 is believed to be linked to motility and metastasis. Liver-specific IGF-I-deficient (LID) mice created by the Cre/loxP recombination system have a 75% reduction in circulating levels of IGF-I compared to control mice (Wu et al., 2003; Yakar et al., 1999) and have decreased growth of both the primary colon tumors and hepatic metastases (Wu et al., 2002). Several studies have also reported that inhibition of IGF1R inhibits not only xenograft growth but also metastasis of various tumor types (Dunn et al., 1998; Lopez and Hanahan, 2002; Reinmuth et al., 2002; Sachdev et al., 2004).
Dunn et al. have shown that a soluble truncated IGF1R inhibits metastasis of MDA-435 cancer cells (Dunn et al., 1998). We have extended this finding using a C-terminally truncated dominant negative IGF1R construct overexpressed in LCC6 cells, a metastatic variant of MDA-435 (Leonessa et al., 1996). We found that cells expressing only the wild-type IGF1R (referred to as LCC6-WT cells) or overexpressing the C-terminally truncated receptor (LCC6-DN cells) form tumors in mice (Sachdev et al., 2004). LCC6-WT cells form metastases in the lungs. In contrast, LCC6-DN cells form no lung metastases even though they grow as primary tumors (Sachdev et al., 2004). Although LCC6 cells have long been considered to be estrogen receptor negative breast cancer cells, it has been reported in recent years that they are likely of melanocytic origin (Rae et al., 2007). Despite their origin, it is clear that LCC6 cells form spontaneous lung metastases after injection into the mammary fat pad of mice and are a useful model of metastasis. Using a similar dominant negative IGF1R approach, it has also been reported that colon cancer cells expressing dominant negative IGF1R fail to form liver metastases following splenic injection or direct injection into the liver of mice (Reinmuth et al., 2002). Furthermore, in a transgenic mouse model of pancreatic islet cell tumorigenesis, RIP1-Tag2 mice expressing high levels of IGF1R exhibit increased invasive carcinomas and lymph node metastases (Lopez and Hanahan, 2002). In contrast, in prostate cancer cells, it has been reported that IGF1R levels are decreased during progression and metastasis of prostate cancer cells (Plymate et al., 1997; Plymate et al., 2004). Based on these results, the data support an important role for IGF1R in cancer metastasis.
While dominant negative constructs are suitable for studying the role of IGF1R in these model systems, they are difficult to translate into an anti-IGF1R therapy. We and others have shown that inhibition of IGF1R with the mouse monoclonal antibody, EM164, retards the xenograft growth of multiple types of cancer cells (Maloney et al., 2003; Sachdev et al., 2006); however, these reports did not examine the effect of IGF1R on metastatic dispersal from the primary tumor site. The goal of this study was to determine the effect of inhibiting IGF1R using a therapeutic approach employing the antibody EM164 and its humanized version, AVE1642. Our data show that IGF1R signaling is not required for growth in the mammary fat pad but is necessary for metastasis in a model of high-risk cancer. Thus, blockade of IGF1R inhibits the metastatic phenotype independently of tumor growth.
Results
EM164 inhibits signaling via IGF1R in LCC6 cells but does not affect proliferation in vitro
We have previously shown that expression of a C-terminally truncated IGF1R construct in LCC6 cells behaves in a dominant negative manner and inhibits signaling via IGFIR in cancer cells (Sachdev et al., 2004). To determine if a blocking monoclonal antibody against IGF1R had similar effects to the dominant-negative construct, LCC6-WT cells were pretreated with EM164 (120 nM) or the humanized version, AVE1642 (120 nM), for 15 minutes prior to stimulation with 5 nM IGF-I or 10 nM IGF-II. Phosphorylation of IRS was used as a readout for signaling via IGF1R. IGF-I and -II phosphorylated IRS and activated the PI3′K pathway in LCC6-WT cells as assayed by an anti-phosphotyrosine immunoblot for activation of the adaptor proteins IRS-1 and IRS-2 and phosphorylation of Akt (Figure 1a upper panels, lanes 2 and 3). In these cells, we have previously shown that IGF-I treatment resulted in both IRS-1 and IRS-2 phosphorylation. Treatment with EM164 inhibited IGF-I and -II stimulated activation of IRS proteins and phosphorylation of Akt (lanes 5 and 6). Levels of total Akt were unchanged (third panel). Similar results were seen with AVE1642, the humanized version of EM164 (Figure 1a, lanes 8 and 9). In LCC6-WT cells, extracellular signal regulated kinase 1/2 (ERK1/2) of the MAPK module are constitutively active. Phosphorylation of ERK1/2 was unaffected by treatment with any of the reagents (Figure 1a, second from bottom panel).
Figure 1. Inhibition of IGF1R disrupts activation of downstream signaling pathways by IGF-I, IGF-II, and insulin.

(a) LCC6-WT cells were untreated or treated with 120 nM EM164 or AVE1642 for 15 minutes followed by stimulation with 5 nM IGF-I or 10 nM IGF-II for 10 minutes. Cell lysates were separated by SDS-PAGE and immunoblotted for phosphorylation of IRS proteins with an anti-phosphotyrosine antibody (pTyr), phosphorylation of Akt (pAkt), total Akt, phosphorylation of p44/p42 MAPK (pMAPK), and total MAPK. EM164 and AVE1642 inhibit signaling pathways activated by IGF1R.
(b) LCC6-WT cells were untreated or treated with 5 nM IGF-I, 120 nM EM164, 120 nM AVE1642, or 120 nM control IgG for 15 minutes or 24 hours. Cell lysates were separated by SDS-PAGE and immunoblotted for IGF1Rβ and total MAPK. Levels of total MAPK were used as loading control. EM164 and AVE1642 down-regulate IGF1R levels after 24h of treatment.
(c) LCC6-WT cells were untreated (SFM) or treated with 20 nM insulin for 5 minutes (lane 2). LCC6-DN cells were treated with 5 nM IGF-I (lane 5) or 10, 20 or 50 nM insulin for the times indicated (lanes 6–12). Cell lysates were separated by SDS-PAGE and immunoblotted for phosphorylation of IRS proteins with a phosphotyrosine specific antibody. DN-IGF1R blocks phosphorylation of IRS proteins by both ligands.
All experiments were repeated three times with similar results.
We and others have previously demonstrated that a major mechanism of action of antibodies against IGF1R is receptor downregulation (Cohen et al., 2005; Maloney et al., 2003; Sachdev et al., 2003). Consistent with this observation, 24-hour treatment with EM164 or AVE1642 downregulated IGF1R levels in LCC6-WT cells (Figure 1b, lanes 8 and 9) compared to no treatment or 24-hour treatment with IGF-I or a control Ab (lanes 6, 7 and 10). Thus, EM164 and AVE1642 inhibited biochemical pathways activated by IGF1R and downregulated receptor.
In LCC6-WT cells, both IGF-I and insulin treatment activated IGF1R as measured by total phosphotyrosine immunoblotting (Figure 1a and 1c, lane 2). LCC6-DN cells overexpressing truncated IGF1R also inhibited the ability of insulin to phosphorylate IRS (Figure 1c, lanes 6–12) indicating that the effect of dominant negative IGF1R may also be due to inhibition of hybrid receptors and/or insulin receptor signaling. Similarly, we have shown that antibodies against IGF1R also may inhibit insulin signaling following prolonged treatment (Sachdev et al., 2006).
We have previously shown that inhibition of IGF1R in LCC6 cells does not affect in vitro proliferation because these cells do not depend on IGF-I for proliferation (Sachdev et al., 2004). In this study, LCC6-WT cell proliferation was also not affected by IGF-II or insulin (Figure 2). Similar to LCC6-DN cells, blockade of IGF1R by EM164 did not affect basal growth in serum in vitro (Figure 2).
Figure 2. Inhibition of IGF1R with EM164 does not affect in vitro proliferation of LCC6-WT cells.

1 × 105 LCC6-WT cells were plated in 24-well plates. Following serum starvation, cells were untreated (SFM) or treated with 5 nM IGF-I, 10 nM IGF-II, 10 nM insulin, or 10% FBS in the absence or presence of 120 nM EM164. Cell numbers were estimated by MTT on day 5 and are shown as the mean absorbance at 570 nm ± SEM of quadruplicate samples. EM164 does not inhibit proliferation of LCC6-WT cells. The experiment was repeated four times with similar results and a representative experiment is shown.
EM164 does not inhibit xenograft growth of LCC6 cells
We next determined the effect of EM164 on xenograft growth of LCC6-WT cells. 5×106 LCC6-WT cells were injected into the second mammary fat pad of female athymic mice as described previously (Sachdev et al., 2003). Mice were randomized to receive 0.9% NaCl, EM164, or scFv-Fc, another antibody against IGF1R (Li et al., 2000; Sachdev et al., 2003) starting on day three after inoculation of cells. Neither EM164 nor scFv-Fc had an effect on the xenograft growth of LCC6-WT cells (Figure 3a). IGF1R levels were downregulated in LCC6-WT tumors harvested from mice treated with EM164 compared to tumors treated with 0.9% NaCl (Figure 3b). Furthermore, EM164 inhibited IGF1R activation and phosphorylation of Akt in the tumor in response to bolus administration of IGF-I (data not shown). Thus, EM164 inhibited the biochemical pathways activated by IGF-I in vivo and downregulated IGF1R levels, but failed to inhibit xenograft growth of LCC6-WT cells.
Figure 3. Antibodies against IGF1R do not inhibit xenograft growth of LCC6-WT cells.

(a) 5 × 106 cells in 60 μl of phenol-red free IMEM were injected into the mammary fat pad of 4–5 week old female athymic mice. On day 3 following injection of cells, mice were treated with either 0.9% NaCl, 500 μg of scFv-Fc, or 800 μg of EM164 every three days. Tumor growth was measured every three days and tumor volume calculated using the formula – length × breadth2/2. Tumor growth is represented as tumor volume in mm3 versus days. Neither scFv-Fc nor EM164 inhibit xenograft growth of LCC6-WT tumors. Experiment was repeated three times with similar results and a representative one is shown.
(b) At the end of the experiment in (a), tumors were harvested, snap frozen in liquid nitrogen, and homogenized. Tumor extracts were separated by SDS-PAGE and immunoblotted for IGF1R levels. Levels of total MAPK were used as loading control. EM164 downregulates IGF1R levels in the tumors.
EM164 inhibits pulmonary metastases
Because the antibodies against IGF1R failed to inhibit tumor growth (Figure 3a), we next examined the effect of EM164 on pulmonary metastases of LCC6-WT cells. LCC6-WT cells were injected into the mammary fat pad of mice and mice were treated with EM164 or an isotype-matched control antibody beginning on day three. Tumors were resected when the volumes were ~300 mm3 and the mice were followed for another 37–38 days. At the end of this time period, lungs were harvested, fixed in formalin, and analyzed for macroscopic metastases. Representative photographs of lungs from mice bearing LCC6-WT tumors that were treated with EM164 or the control antibody are shown in Figure 4. Lungs from mice treated with the control antibody had abundant well-circumscribed pulmonary nodules, whereas lungs from mice treated with EM164 had no visible macroscopic pulmonary nodules and only two had microsopic metastatic deposits (Figure 4, table).
Figure 4. EM164 inhibits metastases of LCC6-WT cells.

LCC6-WT cells were injected into the mammary fat pad of 4–5 week old female athymic mice. On day 3 following injection of cells, mice were treated with either an isotype-matched control antibody (n=10) or 800 μg of EM164 (n=10) every three days. Tumors were resected surgically when they reached a volume of ~300 mm3, which usually occurred between days 17–19. Mice were then treated for another 36–37 days. At day 55–57 following injection of cells, mice were sacrificed, lungs harvested and fixed in 10% neutral buffered formalin. Photographs are representative of lungs from a mouse with LCC6-WT tumors treated with an isotype matched control antibody (left) or EM164 (right). The table summarizes the number of mice with macroscopic and microscopic pulmonary nodules. Lungs from mice bearing LCC6-WT tumors treated with a control antibody have abundant well-circumscribed pulmonary nodules while lungs from mice with LCC6-WT tumors that were treated with EM164 have no visible macroscopic pulmonary nodules.
Disruption of IGF1R signaling inhibits circulating tumor cells
To form metastases at a distant site, cancer cells must acquire the ability to invade the basement membrane, intravasate into blood or lymphatic vessels, survive in the harsh environment in the circulation, extravasate into a distant site, and finally colonize a distant organ. Since both dominant negative IGF1R and EM164 inhibited pulmonary metastases, we next determined the effect of inhibition of IGF1R on circulating tumor cells (CTC) as a measure of tumor cell intravasation. To measure CTC, blood was collected before sacrificing mice, mixed with heparin, and the buffy coat separated using Ficoll-Paque centrifugation as described (Boyum, 1964). The recovered nucleated cells were mixed with agar at a concentration of 0.5% and overlaid on 0.8% bottom agar. Only tumor cells form colonies under these conditions as blood mononuclear cells do not. Photographs of colonies in soft agar were taken at day 4. All of the mice bearing LCC6-WT tumors had abundant colony growth reflective of CTCs, but in striking contrast, none of the mice with LCC6-DN tumors had any colonies (Figure 5a). Similarly, mice bearing LCC6-WT tumors treated with an isotype matched control antibody had abundant CTC in their blood, whereas mice bearing LCC6-WT tumors treated with EM164 (800 μg) every three days had a striking inhibition of detectable CTC (Figure 5b). Numerous macroscopic and microscopic metastases were evident in H&E stained sections from lungs of all mice bearing LCC6-WT tumors treated with the control Ab; however, micrometastases were infrequently seen in the lungs of mice treated with EM164 (Figure 5c).
Figure 5. Disruption of IGF1R inhibits circulating tumor cells (CTC) in mice bearing LCC6 tumors.
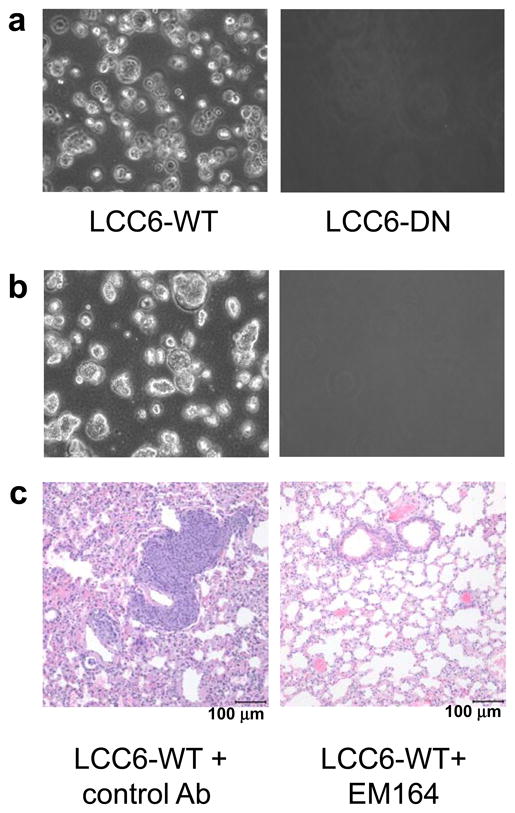
(a) Colonies in soft agar formed by CTC from mice bearing LCC6-WT (left) or LCC6-DN (right) tumors. Blood from mice bearing either LCC6-WT (n=5) or LCC6-DN (n=5) tumors was collected and mixed with 100 μl of heparin. Buffy coat was isolated by density centrifugation through Ficoll-Paque. Nucleated cells in the buffy coat were mixed with SeaPlaque agar at a final concentration of 0.5% agar and overlaid on a layer of 0.8% bottom agar in a 6-well dish. Colonies in soft agar were photographed 4–5 days later. CTC were detected in all mice with LCC6-WT tumors and in none of the mice with LCC6-DN tumors. Experiment was repeated two times with similar results.
(b) Colonies in agar from mice bearing LCC6-WT tumors treated with control Ab (left) or EM164 (right). Colonies were cultured as above. EM164 inhibits CTC in mice.
(c) Lungs from the mice in (b) were harvested, fixed in 10% neutral buffered formalin, sectioned, and stained with H&E. Sections of lungs from mice treated with control Ab (left) or EM164 (right). Results are representative of images of H&E stained lung sections collected from 10 mice.
These results are in agreement with the data described in Figure 4, in which pulmonary nodules were seen in mice with LCC6-WT tumors treated with a control antibody but not in EM164-treated mice. Although it is possible that the sensitivity of this assay prevented us from detecting very small numbers of CTC, it is clear that inhibition of IGF1R resulted in a striking decrease in the number of CTC. These results suggest that IGF1R is required for the metastatic process. Because IGF1R inhibition affected the number of CTC, our data suggest that invasion across the basement membrane, intravasation into the vasculature, or survival in the circulation requires functional IGF1R signaling.
Disruption of IGF1R inhibits invasion across Matrigel in vitro
To determine the mechanism by which IGF1R disruption inhibits metastasis and CTC, we next examined IGF1R regulated invasion in vitro. LCC6-WT and LCC6-DN cells were placed on PVDF membranes with 0.4 micron pores and coated with Matrigel. Cells invading through the membrane after four hours were counted. IGF-I stimulated invasion of LCC6-WT cells compared to unstimulated LCC6-WT cells while IGF-I did not affect invasion of LCC6-DN cells (Figure 6a,). Basal invasion was also decreased in LCC6-DN cells as compared to LCC6-WT cells (Figure 6a). EM164 also inhibited IGF-I stimulated invasion of LCC6-WT cells while an isotype-matched control antibody had no such effect (Figure 6b). Thus, the absence of CTC after IGF1R disruption could be due to the inhibition of invasion. However, since we measured CTC at the end of our experiments, it is plausible that the lack of detectable CTC may also be due to decreased survival in the circulation after IGF1R inhibition.
Figure 6. Disruption of IGF1R inhibits invasion of LCC6 cells in vitro.

(a) LCC6-WT and LCC6-DN cells were placed on Matrigel coated inserts. Cells were untreated or stimulated with IGF-I for 4h. Cells invading to the other side of the membrane were counted. Data is represented as the number of invading cells counted in 3 random fields using a microscope with a grid. IGF-I stimulates invasion of LCC6-WT cells, whereas IGF1R disruption in LCC6-DN cells inhibits invasion in vitro.
(b) LCC6-WT cells were untreated (none), pretreated with an isotype-matched control antibody (control), or pretreated with120 nM EM164 (EM164) for 30 minutes. Cells were incubated with IGF-I or 5% FBS or without (SFM). IGF-I stimulates invasion of LCC6-WT cells, and inhibition of IGF1R with EM164 inhibits invasion in vitro.
Experiments in (a) and (b) were repeated two times with similar results. Representative data are shown here. Statistical significance was analyzed using one-way ANOVA with Bonferrroni’s post test. Significance is denoted by * (for p<0.01) or ** (for p< 0.001).
Disruption of IGF1R inhibits colonization in the lungs
To determine if IGF1R regulation of invasion was the only factor in the decreased metastases, we injected cells directly into the circulation via the tail vein to bypass the requirement for the cells to invade in vivo. To confirm that cells injected into the tail vein reached the lungs via the circulation in all mice, LCC6-WT and LCC6-DN cells were engineered to express firefly luciferase (LCC6-WT/pFBLuc and LCC6-DN/pFBLuc, respectively) and injected into the tail vein of mice. One hour after injection of cells, luciferase expression was seen in the lungs of all mice (Figure 7, left panels), indicating that both LCC6-WT/pFBLuc and LCC6-DN/pFBLuc cells reached the lungs. These mice were then monitored for metastases on days 12, 21, 30 and 46. Mice with LCC6-WT/pFBLuc cells (3/3) showed metastases in the lungs at day 30 (Figure 7a). Treatment with AVE1642 inhibited lung colonization of LCC6-WT/pFBLuc mice (Figure 7b). Similarly, mice injected with LCC6-DN/pFBLuc cells had inhibition of lung colonization (Figure 7c). These data show that disruption of IGF1R inhibits metastastic dispersal by decreasing either cell survival in the circulation or colonization of the lungs. To confirm the above results from BLI and perform semi-quantitative analyses of the effect of disruption of IGF1R on lung colonization, mice were injected with LCC6-WT or LCC6-DN cells in the tail vein and monitored for approximately 60 days. Mice were then sacrificed, lungs were fixed, and photographed. The number of nodules in six lung sections stained with H&E/lung were counted and averaged. Mice injected with LCC6-WT cells had abundant nodules visible macroscopically in the whole lung, as compared to far fewer nodules in lungs from mice injected with LCC6-DN cells (Figure 8a). Analyses of metastatic lesions in H&E stained lung sections revealed fewer and smaller pulmonary nodules in mice injected with LCC6-DN cells (Figure 8b). Similarly, mice injected with LCC6-WT cells and treated with AVE1642 every three days had fewer macroscopic and microscopic lung nodules (Figure 9a and b) compared to untreated mice. Thus, therapeutic inhibition of IGF1R with AVE1642 also inhibited colonization of lungs after direct injection into the circulation. This result could be due to decreased cell survival in the circulation or impaired colonization of the lungs.
Figure 7. Inhibition of IGF1R with AVE1642 or the dominant negative approach inhibits colonization of lungs by LCC6 cells as assayed by bioluminescence imaging (BLI).
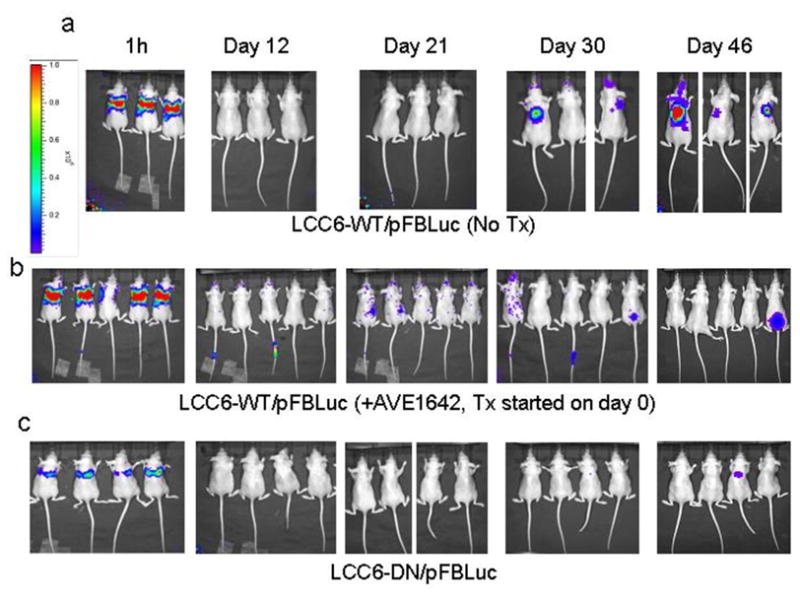
LCC6 cells were engineered to express firefly luciferase toenable monitoring of metastases by BLI. 1 × 105 LCC6-WT/pFBluc or LCC6-DN/pFBluc cells were injected into the tail vein. At the indicated times after injection of cells, bioluminescence images were acquired and analyzed using the IVIS imaging system and Living Image acquisition and analysis software. The intensity of the signal measured as photon flux is indicated as a color scale.
(a–b) Mice injected with LCC6-WT/pFBLuc cells were randomized to no treatment (a) or treatment with 800 μg of AVE1642 every three days (b).
(c) Mice injected with LCC6-DN/pFBLuc cells were not treated.
All mice showed presence of luciferase expressing cells an hour after injection into the tail vein indicating that the cells reached the lungs of all mice. Mice injected with LCC6-WT/pFBLuc cells and left untreated (n=3) show pulmonary metastases. Pulmonary metastases are inhibited in mice injected with LCC6-WT/pFBLuc cells and then treated with AVE1642 (n=5) or injected with LCC6-DN/pFBLuc cells (n=4).
Figure 8. Disruption of IGF1R inhibits ability of LCC6 cells to metastasize to the lungs following direct injection of cells into the circulation.

1 × 106 LCC6-WT or LCC6-DN cells were injected into the tail vein of 4–5 week old female athymic mice. At day 62, mice were sacrificed, lungs harvested and fixed in 10% neutral buffered formalin. H&E stained lung sections were analyzed for metastases.
(a) Representative images of lungs from mice injected with LCC6-WT (left) and LCC6-DN (right) cells.
(b) Metastases were quantified by counting pulmonary nodules in six serial sections and averaged. Nodules are grouped by size. Pulmonary nodules in mice injected with LCC6-DN cells (n=7) in the tail vein are significantly fewer in number and smaller in size compared to nodules in the lungs of mice injected with LCC6-WT cells (n=6). Statistical analysis was performed with one-way ANOVA and significance is indicated by * (for p<0.01) or ** (for p< 0.001).
The experiment was repeated twice with similar results and a representative experiment is shown.
Figure 9. Inhibition of IGF1R with AVE1642 inhibits lung colonization of cells injected into the circulation.

1 × 106 LCC6-WT cells were injected into the tail vein of 4–5 week old female athymic mice. Mice were randomized to no treatment (n=9) or AVE1642 (n=9) every three days beginning on day 2. At day 62, lungs for H&E staining were collected and analyzed as in Figure 7.
(a) Representative images of lungs from mice with LCC6-WT cells (left) or LCC6-WT cells treated with AVE1642 (right).
(b) Metastases were quantified by counting pulmonary nodules in six serial sections and averaged. Pulmonary nodules in lungs of mice injected with LCC6-WT cells in the tail vein and treated with AVE1642 are fewer in number compared to the lungs of mice injected with LCC6-WT cells that were not treated.
The experiment was repeated twice with similar results and a representative experiment is shown.
Inhibition of IGF1R enhances sensitivity of cancer cells to apoptotic stimuli
To determine if functional IGF1R was necessary for survival, we next assessed if inhibition of IGF1R rendered LCC6 cells more susceptible to apoptosis. LCC6-WT and LCC6-DN cells were treated with increasing concentrations of doxorubicin and the number of cells was measured 48 h later. LCC6-DN cells were more sensitive to doxorubicin at lower concentrations compared to LCC6-WT cells (Figure 10a). Furthermore, LCC6-DN (Figure 10b) or LCC6-WT cells treated with EM164 (Figure 10c) resulted in increased apoptosis compared to untreated LCC6-WT cells as measured by cleavage of poly(ADP ribose) polymerase (PARP) 48 h after serum withdrawal. This result suggests that inhibition of IGF1R enhanced sensitivity to apoptotic stimuli in vitro. However, cells must survive in an anchorage independent manner in the circulation; therefore, we next studied the effect of IGF1R inhibition on anoikis.
Figure 10. Disruption of IGF1R enhances susceptibility to apoptotic stimuli in vitro.

(a) LCC6-WT and LCC6-DN cells were plated in 24 well plates in growth medium. Cells were serum starved the next day and treated with increasing doses of doxorubicin from 0.1 to 1000 ng/ml. Cell numbers were determined by uptake of MTT on day 4. Data is represented as the percent of day 0 absorbance at 570 nm versus concentration of doxorubicin. Dominant negative IGF1R renders LCC6 cells more sensitive to doxorubicin.
(b) LCC6-WT and LCC6-DN cells were plated in 60 mm dishes in growth medium containing 10% serum. Cells were subjected to serum withdrawal the next day. Cells were harvested and lysed at the time points indicated. Cellular proteins were subjected to SDS-PAGE and immunoblotted for PARP (upper panel) with an antibody that recognizes both the full-length and cleaved PARP and with total MAPK (lower panel) as a loading control. Dominant negative IGF1R renders LCC6 cells more sensitive to apoptosis following serum withdrawal.
(c) The experiment was performed as in (b) except LCC6-WT cells were treated with 120 nM EM164 for 48 h. EM164 treatment results in increased cleavage of PARP in LCC6-WT cells. The experiments were repeated four times with similar results and representative data are shown.
Inhibition of IGF1R enhances anoikis of LCC6 cells
To examine the effect of IGF1R inhibition on anoikis, cells were cultured on a layer of 0.9% agar over 48 hours and viability was determined using trypan blue exclusion. A greater loss of viability was seen in LCC6-DN cells compared to LCCT-WT cells at 24 hours (Figure 11a). Thus, LCC6-DN cells exhibited enhanced susceptibility to anoikis. IGF-I enhanced survival of LCC6-WT cells under anchorage-independent conditions in vitro and AVE1642 dramatically reversed IGF-I-enhanced survival of LCC6-WT cells (Figure 11b). Thus, these results show that functional IGF1R signaling can increase survival of LCC6 cells in the circulation. The data described here however, do not rule out the possibility that IGF1R may also be required for growth at the metastatic site. Ongoing studies are examining the requirement for functional IGF1R for growth at metastatic sites.
Figure 11. Disruption of IGF1R enhances anoikis in vitro.

(a) 200,000 LCC6-WT or LCC6-DN cells were plated on top of 0.9% SeaPlaque agar solidified in the bottom of 6 well plates in 0.5% FBS. At the time points indicated, cells were trypsinized, collected and surviving cells counted using trypan blue exclusion. The number of surviving cells is represented versus time. LCC6-DN cells have a decreased number of surviving cells compared to LCC6-WT cells. The experiment was repeated three times with similar results.
(b) LCC6-WT cells treated without or with AVE1642 for 30 minutes were mixed without or with 5 nM IGF-I and plated on top of 0.9% agar in 6 well plates. Twenty-four hours later, cells were trypsinized, collected, and surviving cells were counted with trypan blue exclusion. The data are represented as cell number. IGF-I enhances survival of LCC6-WT cells and inhibition of IGF1R with AVE1642 decreases cell survival. The experiment was repeated twice with similar results.
Statistical significance was analyzed using one-way ANOVA with Bonferrroni’s post test. Significance is denoted by ** (for p<0.001) or *** (for p< 0.0001).
Discussion
Metastasis is a highly inefficient process that requires the co-ordinated regulation of a series of complex processes with only a small percent of tumor cells acquiring the characteristics necessary to escape from the primary tumor and successfully colonize distant sites. We have used LCC6 cells as a model of high-risk metastatic disease. We have previously shown that a dominant negative IGF1R inhibited pulmonary metastases of these cells without affecting growth of the primary tumor in the mammary fat pad of athymic mice (Sachdev et al., 2004). Herein, we demonstrate that a therapeutic inhibition of IGF1R with an antibody against IGF1R also inhibited pulmonary metastases without affecting primary tumor growth (Figures 2 and 3). EM164 down-regulated IGF1R levels in the tumors (Figure 3b) and also inhibited IGF-I stimulated phosphorylation of IRS and downstream Akt, suggesting that EM164 inhibited the biochemical pathways activated by IGF1R but did not affect primary tumor growth. Based on these data, we have shown that IGF1R can affect metastasis without affecting tumor growth in vivo.
This regulation of metastasis independent of tumor growth is not isolated to LCC6 cells only. Our in vitro data with other breast cancer cell lines showed similar findings. In MDA-MB231BO cells (Yoneda et al., 2001), a metastatic variant of the MDA-MB231 breast cancer cells, we have shown that IGF-I does not stimulate proliferation but enhances motility in vitro (Jackson et al., 2001). We have also shown that neutralization of IGF-I with IGF binding protein-1 (IGFBP-1) inhibits IGF-I stimulated motility but not proliferation of MDA-MB-231BO cells (Zhang and Yee, 2002). Similarly in T47D-YA cells (a variant of T47D breast cancer cells), ectopic expression of the adaptor protein IRS-2 results in IGF-I stimulated motility but not proliferation of the T47D-YA/IRS-2 cells (Byron et al., 2006). Furthermore in T47D-YA/IRS-2 cells, inhibition of IGF1R with the antibody αIR3 inhibits motility in response to IGF-I (Byron et al., 2006). In fact, this has also been observed by other groups in other types of cancers such as neuroblastomas (van Golen et al., 2006). Thus, these results indicate that regulation of metastasis independently of tumor growth may be seen in a wide variety of cancers. This has important clinical implications for the development of therapies against IGF1R for clinical use as discussed later.
Various in vitro and in vivo models have suggested that activation of IGF1R regulates various steps in the metastatic cascade. In this study, we found that disruption of IGF1R inhibited invasion across Matrigel in vitro, suggesting that IGF1R regulates invasion of these cells. When IGF1R was inhibited, we observed no difference in the activity of metalloproteinases or expression of the chemokine receptor CXCR4 (data not shown) which has been shown to be important for organotropism of breast cancer metastasis to organs such as bone and lungs (Muller et al., 2001). Furthermore, we show here that disruption of IGF1R either with a dominant negative construct or EM164 significantly inhibited the presence of CTC in the blood of mice. Pulmonary metastases were also inhibited when IGF1R was disrupted following direct injection of cells into the circulation. Thus, our data suggest that functional IGF1R is required at multiple steps in the metastatic process including cell invasion and survival in the circulation.
How does IGF1R regulate metastasis independently of tumor growth? One possibility is that functional IGF1R is essential for survival in the circulation as discussed earlier. This is supported by evidence showing that indeed IGF1R is essential for anchorage-independent survival once cells are free of adhesion to the extracellular matrix (ECM) in vitro (Baserga, 2005; Valentinis et al., 1999). Further, activation of IGF1R has been shown to mediate resistance to anoikis or enhance survival in the absence of attachment to ECM in cancer cells (Ravid et al., 2005). Thus, surviving in the circulation or at a secondary site is one of the critical steps in the metastatic cascade as tumor cells are subjected to some of the harshest environment in the host in the circulation (Townson et al., 2003). Additionally, our data do not rule out the possibility that functional IGF1R is a critical growth regulator at a secondary site or is required to co-opt other signals promoting tumor cell survival in the lungs. Indeed, a key regulator of metastasis is the regulation of growth of tumor cells at the secondary site (Chambers et al., 2002).
Work from several groups has demonstrated that there is a mutually sustaining reciprocity between tumor epithelial cells and their surrounding stroma (Gupta and Massague, 2006). Many of the genes that comprise metastasis signatures have included genes expressed by the stroma (Allinen et al., 2004; Minn et al., 2005b). As tumor cells invade across the basement membrane, they can initiate development of the reactive stroma, further propelling metastasis (Radisky and Radisky, 2007). Thus, it has become increasingly clear that the microenvironment influences the invasion and metastasis of tumor cells and that the local microenvironment impacts tumor cells as they escape. Our data suggest that disruption of IGF1R does not affect tumor growth of LCC6 cells in the mammary fat pad of mice. Perhaps this is due to the fact that in the mammary fat pad of the mouse, the microenvironment provides the necessary signals for growth, and IGF1R is not needed in this pathway.
In contrast, LCC6 cells need functional IGF1R for survival in the circulation and colonization of metastatic sites. Thus, it is probable that the microenvironment at the metastatic site in the lungs is insufficient to provide all the cues needed for successful establishment of metastatic growth in the lungs. Although we have only studied metastatic growth in the lungs, it is possible that IGF1R also regulates growth in the bone microenvironment. However, it has been shown that only six genes are shared in common between the bone metastasis signature (Kang et al., 2003; Minn et al., 2005b) and lung metastasis signature (LMS) of breast cancer cells (Gupta et al., 2005; Minn et al., 2005a). Therefore, it is possible that our results are specific for metastasis to the lungs. Ongoing studies are examining differential expression of genes in LCC6 cells without and with disruption of IGF1R that may further provide a better insight into how IGF1R regulates metastasis independent of tumor growth.
Recently, the notion of self-seeding has been postulated to explain metastasis where circulating tumor cells return to the primary site to enhance growth (Norton and Massague, 2006). As a result, increased growth of the primary tumor is accompanied by decreased metastases at distant sites. Our data argue against this model; disruption of IGF1R did not affect “primary” tumor growth but effectively reduced metastases. Thus, it seems unlikely that self-seeding was responsible for the growth of these cells in the mammary fat pad.
Our results have important implications for the clinical development of IGF1R targeted strategies. First, this metastatic phenotype regulated by IGF1R will be difficult to measure in clinical trials. Second, biochemical inhibition of the target does not automatically lead to inhibition of tumor growth. Inhibition of IGF1R kinase activity is an important biodynamic marker, but may not be linked to a clinical outcome measured in a phase 2 drug study. Thus, suitable biomarkers need to be developed to identify tumors responsive to anti-IGF1R therapy. We have previously shown that IRS-1 activation is associated with proliferation of cancer cells (Jackson et al., 1998), while IRS-2 is associated with motility of breast cancer cells (Jackson et al., 2001). These observations have also been confirmed in vivo by studies showing that IRS-2 null animals, but not IRS-1 null animals, have significantly decreased incidence of metastasis compared to wild-type mice expressing polyoma virus middle T antigen (PyV-MT) in the mammary gland (Gibson et al., 2007; Nagle et al., 2004). This suggests that one biomarker could be expression of phosphorylated IRS-1 compared to IRS-2. However, it has recently also been shown that both IRS-1 and IRS-2 overexpression can lead to metastasis (Dearth et al., 2006). Perhaps the identification of an “IGF1R driven metastasis signature” would be useful in identifying and monitoring patients who could benefit from IGF1R targeted therapy for inhibition of metastatic disease and we are currently investigating this.
In the present study, we show that IGF1R regulates metastasis independently of tumor growth. As such, our study has several important implications for clinical trials with agents targeting IGF1R. Clinical trials of this therapeutic strategy may need to adjust their endpoints for assessing benefit by taking into account that inhibition of IGF1R signaling may inhibit metastasis but not primary tumor growth.
Materials and Methods
Materials
All reagents and chemicals were purchased from Sigma (St. Louis, MO) and cell culture reagents were purchased from Invitrogen/Life Technologies (Rockville, MD) unless otherwise noted. IGF-I and -II were purchased from Novozymes GroPep (Thebarton, Australia). Human insulin was from Eli Lilly (Indianapolis, IN). The antibody against the β-subunit of IGF1R (IGF1Rβ) was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-phosphotyrosine antibody conjugated to horseradish-peroxidase was from BD Transduction Laboratories (Lexington, KY). Antibodies against p44/p42 ERK 1/2 (phosphospecific and total) and Akt (phosphospecific and total) were purchased from Cell Signaling (Beverly, MA). The antibody against PARP was from Roche (Indianapolis, IN). Anti-rabbit secondary antibodies conjugated to HRP were from GE Biosciences (Piscataway, NJ). Heparin was obtained from Hospira, Inc. (Lake Forest, IL) and Ficoll-Paque was purchased from Gallard-Schlessinger (Carle Place, NY). SeaPlaque agar was obtained from Lonza Biologicals (Rockland, ME). scFv-Fc was purified as described previously (Li et al., 2000). EM164 was obtained from Immunogen Inc. and the humanized version AVE1642 was obtained from sanofi-aventis.
Cell stimulation
Cells were grown in 6 cm dishes in regular growth medium. Cells at 70% confluence were washed two times with phosphate-buffered saline (PBS) and serum deprived for 24 hours in serum free media (SFM) as described previously (Jackson et al., 1998). For treatment, medium was replaced with SFM containing either 5 nM IGF-I, 10 nM IGF-II for 10 minutes or 20 nM insulin for 5 minutes. To determine if EM164 inhibited IGF-I or IGF-II mediated activation of IGF1R or other downstream pathways, cells were first pretreated with antibodies for 15 minutes and then with the various ligands for additional 10 minutes. To determine the effect of the antibodies on IGF1R levels, cells were untreated or treated with the different antibodies for 15 minutes or 24 hours.
Cell lysis
Cells were washed three times with ice-cold PBS on ice and lysed with 300μl/6 cmdish lysis buffer TNESV (50 mM Tris-Cl pH 7.4, 1% NP-40, 2 mM EDTA pH 8.0, 100 mM NaCl, 10 mM Na orthovanadate, 1 mM PMSF, 20 μg/ml leupeptin, and 20μg/ml aprotinin).
Immunoblotting
40 μg of cellular proteins were subjected to reducing SDS-PAGE on 8% polyacrylamide gels following the Laemmli system (Laemmli, 1970). After SDS-PAGE, proteins were transferred to nitrocellulose membrane and immunoblotting performed. For detecting phosphorylated proteins, membranes were incubated with 1:2000 dilution of PY20-HRP anti-phosphotyrosine antibody in TBST for 1 hour at room temperature. Chemiluminescence was done using SuperSignal West Pico substrate (Pierce, Rockford, IL). IGF1Rβ, phospho-Akt (Ser473), total Akt, phospho-p44/p42 ERK 1/2 (Thr 202/Tyr 204) and total p44/p42 ERK 1/2 antibodies were used as per manufacturer’s instructions. The polyclonal antibody against PARP was used at 1:2000 dilution.
Proliferation assays
Cells were plated in 24-well plates with 10,000 cells/well in regular growth medium. Cells were switched to SFM for 24 hours and then treated as indicated in the Figure legends. All treatments were done in quadruplicate. Growth was measured 4–5 days after treatment by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay as described previously (Twentyman and Luscombe, 1987).
Invasion assay
Invasion was determined using the Boyden chamber assay as described previously (Jackson et al, 2000) with the exception that the membrane was coated with Matrigel to allow the ability of cells to invade through Matrigel to be assayed. The number of cells invading to the bottom of the membrane were counted in three random fields using a grid in the eyepiece of the microscope.
Apoptosis assay
The susceptibility of cells to apoptosis in vitro was measured by cleavage of PARP in response to serum withdrawal using an antibody that recognizes both the full-length and cleaved PARP.
Anoikis assay
Anoikis was measured by growth on 0.9% agar. 100,000 cells in medium with 0.5% FBS were plated on top of agar solidified in the bottom of 6-well plates. At the times indicated in the figure, cell numbers were counted using trypan blue exclusion. The data are represented as mean ± SEM of triplicate samples.
Xenograft growth
All animal protocols were approved by the University of Minnesota Institutional Animal Care and Use Committee. 5×106 LCC6-WT or LCC6-DN cells were injected into the mammary fat pad of 4–5 week old female athymic mice.
Three days after injection of cells, mice with LCC6-WT cells were treated with an isotype-matched control antibody, EM164, or scFv-Fc every three days. Tumor growth was measured bidirectionally. Tumor volumes were calculated using the formula length × breadth2/2
Metastasis models
Metastasis was investigated using two different models. The first model used was described by us previously (Sachdev et al, 2004). Briefly, xenograft tumors were grown in the second mammary fat pad of athymic female mice, the tumors were surgically resected when the volumes were ~300 mm3 (between days 17–19 after injection of cells). Mice were then sacrificed between days 55–57, and the lungs examined macroscopically.
The second model of metastasis used was the tail vein injection model. To visualize cells following tail vein injection, LCC6-WT and LCC6-DN cells were engineered to express luciferase using retroviral infection. 1 × 105 LCC6-WT/pFBLuc or LCC6-DN/pFBLuc cells were injected into the tail vein and metastases were monitored every 7–10 days by noninvasive bioluminescence imaging (BLI).
To measure the ability of cells to colonize the lungs following direct injection into the circulation, 1 × 106 cells were injected into the tail vein of 5–6 week old female athymic mice. At approximately day 60–62 after cells were injected, the mice were sacrificed and lungs analyzed macroscopically and microscopically for metastases.
BLI and analyses of metastases
For BLI, mice were anesthetized and injected intraperitoneally with 150 mg/kg of D-luciferin in PBS. Bioluminescence images were acquired with the IVIS Imaging System (Caliper Life Sciences, Mountain View, CA) 10 minutes after injection of luciferin. Images were acquired for 60 seconds. Metastases were visualized using Living Image software (Caliper Life Sciences) by measuring the photon flux in photons/s/cm2/steradian. For histological analyses of metastases, lungs were harvested, fixed in 10% neutral buffered formalin, paraffin embedded, sectioned, and stained with H&E. Metastatic lesions were quantified by averaging the number of pulmonary nodules in six serial sections per mouse in all mice in the experiments.
Detection of circulating tumor cells
At the end of the experiment, mice were anesthetized and blood collected by cardiac puncture. Blood elements and tumor cells were isolated by density centrifugation using Ficoll-Paque. 1 ml of blood was diluted with 2 ml DMEM + 5% FBS, overlaid gently on a 3 ml cushion of Ficoll-Paque, and centrifuged at 1200 rpm for 30 minutes with the brakes off. The buffy coat with cells was collected and washed once with DMEM + 5% FBS. The cell pellet was resuspended in 0.3 ml media, mixed with 0.5 ml of 0.8% agarose, and plated on a bottom layer of 0.8% agar solidified in the bottom of a well of a 6-well plate. Colonies were photographed after 4–5 days.
Acknowledgments
This work was supported by NIH R01 CA074285 (D. Yee), NIH P30 CA077598PHS Cancer Center Support Grant P30 CA77398 from the National Cancer Institute and the Prospect Creek Foundation.
We acknowledge Immunogen Inc. for providing us with EM164 and the isotype-matched control antibody (anti-CD20), sanofi-aventis for AVE1642 (the humanized version of EM164), and the National Cell Culture Center (NCCC), a division of Biovest International, Fridley, MN for producing supernatant containing scFv-Fc from NSO 3b1Fc 1F12 cells in spinner cultures. We would like to thank Nicole Kirchoff and the staff of the Comparative Pathology Shared Resource of the Masonic Cancer Center, University of Minnesota. We thank Sarah Thuriot and Alissa Pelzer for assistance with the care of the mice. We would also like to thank Michael Franklin for editorial assistance.
Footnotes
Conflict of interest: Drs. Sachdev, Zhang, Matise and Yee declare no conflict of interest. Dr. Gaillard-Kelly is an employee of sanofi-aventis.
References
- Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6:17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Baserga R. The insulin-like growth factor-I receptor as a target for cancer therapy. Expert Opin Ther Targets. 2005;9:753–68. doi: 10.1517/14728222.9.4.753. [DOI] [PubMed] [Google Scholar]
- Boyum A. Separation of White Blood Cells. Nature. 1964;204:793–4. doi: 10.1038/204793a0. [DOI] [PubMed] [Google Scholar]
- Burtrum D, Zhu Z, Lu D, Anderson DM, Prewett M, Pereira DS, et al. A fully human monoclonal antibody to the insulin-like growth factor I receptor blocks ligand-dependent signaling and inhibits human tumor growth in vivo. Cancer Res. 2003;63:8912–21. [PubMed] [Google Scholar]
- Byron SA, Horwitz KB, Richer JK, Lange CA, Zhang X, Yee D. Insulin receptor substrates mediate distinct biological responses to insulin-like growth factor receptor activation in breast cancer cells. Br J Cancer. 2006;95:1220–8. doi: 10.1038/sj.bjc.6603354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–72. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- Cohen BD, Baker DA, Soderstrom C, Tkalcevic G, Rossi AM, Miller PE, et al. Combination therapy enhances the inhibition of tumor growth with the fully human antitype 1 insulin-like growth factor receptor monoclonal antibody CP-751,871. Clin Cancer Res. 2005;11:2063–73. doi: 10.1158/1078-0432.CCR-04-1070. [DOI] [PubMed] [Google Scholar]
- Dearth RK, Cui X, Kim HJ, Kuiatse I, Lawrence NA, Zhang X, et al. Mammary tumorigenesis and metastasis caused by overexpression of insulin receptor substrate 1 (IRS-1) or IRS-2. Mol Cell Biol. 2006;26:9302–14. doi: 10.1128/MCB.00260-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn SE, Ehrlich M, Sharp NJ, Reiss K, Solomon G, Hawkins R, et al. A dominant negative mutant of the insulin-like growth factor-I receptor inhibits the adhesion, invasion, and metastasis of breast cancer. Cancer Res. 1998;58:3353–61. [PubMed] [Google Scholar]
- Garcia-Echeverria C, Pearson MA, Marti A, Meyer T, Mestan J, Zimmermann J, et al. In vivo antitumor activity of NVP-AEW541-A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell. 2004;5:231–9. doi: 10.1016/s1535-6108(04)00051-0. [DOI] [PubMed] [Google Scholar]
- Gibson SL, Ma Z, Shaw LM. Divergent roles for IRS-1 and IRS-2 in breast cancer metastasis. Cell Cycle. 2007;6:631–7. doi: 10.4161/cc.6.6.3987. [DOI] [PubMed] [Google Scholar]
- Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–95. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Gupta GP, Minn AJ, Kang Y, Siegel PM, Serganova I, Cordon-Cardo C, et al. Identifying site-specific metastasis genes and functions. Cold Spring Harb Symp Quant Biol. 2005;70:149–58. doi: 10.1101/sqb.2005.70.018. [DOI] [PubMed] [Google Scholar]
- Haluska P, Carboni JM, Loegering DA, Lee FY, Wittman M, Saulnier MG, et al. In vitro and in vivo antitumor effects of the dual insulin-like growth factor-I/insulin receptor inhibitor, BMS-554417. Cancer Res. 2006;66:362–71. doi: 10.1158/0008-5472.CAN-05-1107. [DOI] [PubMed] [Google Scholar]
- Haluska P, Shaw HM, Batzel GN, Yin D, Molina JR, Molife LR, et al. Phase I dose escalation study of the anti insulin-like growth factor-I receptor monoclonal antibody CP-751,871 in patients with refractory solid tumors. Clin Cancer Res. 2007;13:5834–40. doi: 10.1158/1078-0432.CCR-07-1118. [DOI] [PubMed] [Google Scholar]
- Higano CS, Yu EY, Whiting SH, Gordon MS, LoRusso P, Fox F, et al. Phase I study of weekly IMC-A12, a human insulin like growth factor-I receptor antibody, in patients with advanced solid tumors. ASCO. 2007 Abst #648. [Google Scholar]
- Jackson JG, White MF, Yee D. Insulin receptor substrate-1 is the predominant signaling molecule activated by insulin-like growth factor-I, insulin, and interleukin-4 in estrogen receptor-positive human breast cancer cells. J Biol Chem. 1998;273:9994–10003. doi: 10.1074/jbc.273.16.9994. [DOI] [PubMed] [Google Scholar]
- Jackson JG, Zhang X, Yoneda T, Yee D. Regulation of breast cancer cell motility by insulin receptor substrate- 2 (IRS-2) in metastatic variants of human breast cancer cell lines. Oncogene. 2001;20:7318–25. doi: 10.1038/sj.onc.1204920. [DOI] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–49. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- Karp DD, Paz-Ares LG, Blakely LJ, Kreisman H, Cohen RB, Langer CJ, et al. ASCO. 2007. Efficacy of the anti-insulin like growth factor I receptor (IGF-IR) antibody CP 751,871 in combination with paclitaxel and carboplatin as first-line treatment for NSCLC. Abst #7506. [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Leonessa F, Green D, Licht T, Wright A, Wingatelegette K, Lippman J, et al. MDA435/LCC6 and MDA435/LCC6(MDR1): ascites models of human breast cancer. British Journal of Cancer. 1996;73:154–161. doi: 10.1038/bjc.1996.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SL, Liang SJ, Guo N, Wu AM, Fujita-Yamaguchi Y. Single-chain antibodies against human insulin-like growth factor I receptor: expression, purification, and effect on tumor growth. Cancer Immunol Immunother. 2000;49:243–52. doi: 10.1007/s002620000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez T, Hanahan D. Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell. 2002;1:339–53. doi: 10.1016/s1535-6108(02)00055-7. [DOI] [PubMed] [Google Scholar]
- Maloney EK, McLaughlin JL, Dagdigian NE, Garrett LM, Connors KM, Zhou XM, et al. An anti-insulin-like growth factor I receptor antibody that is a potent inhibitor of cancer cell proliferation. Cancer Res. 2003;63:5073–83. [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005a;436:518–24. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest. 2005b;115:44–55. doi: 10.1172/JCI22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- Nagle JA, Ma Z, Byrne MA, White MF, Shaw LM. Involvement of insulin receptor substrate 2 in mammary tumor metastasis. Mol Cell Biol. 2004;24:9726–35. doi: 10.1128/MCB.24.22.9726-9735.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton L, Massague J. Is cancer a disease of self-seeding? Nat Med. 2006;12:875–8. doi: 10.1038/nm0806-875. [DOI] [PubMed] [Google Scholar]
- Plymate SR, Bae VL, Maddison L, Quinn LS, Ware JL. Reexpression of the type 1 insulin-like growth factor receptor inhibits the malignant phenotype of simian virus 40 T antigen immortalized human prostate epithelial cells. Endocrinology. 1997;138:1728–1735. doi: 10.1210/endo.138.4.5071. [DOI] [PubMed] [Google Scholar]
- Plymate SR, Tennant MK, Culp SH, Woodke L, Marcelli M, Colman I, et al. Androgen receptor (AR) expression in AR-negative prostate cancer cells results in differential effects of DHT and IGF-I on proliferation and AR activity between localized and metastatic tumors. Prostate. 2004;61:276–90. doi: 10.1002/pros.20099. [DOI] [PubMed] [Google Scholar]
- Radisky ES, Radisky DC. Stromal induction of breast cancer: inflammation and invasion. Rev Endocr Metab Disord. 2007;8:279–87. doi: 10.1007/s11154-007-9037-1. [DOI] [PubMed] [Google Scholar]
- Rae JM, Creighton CJ, Meck JM, Haddad BR, Johnson MD. MDA-MB-435 cells are derived from M14 melanoma cells--a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res Treat. 2007;104:13–9. doi: 10.1007/s10549-006-9392-8. [DOI] [PubMed] [Google Scholar]
- Ravid D, Maor S, Werner H, Liscovitch M. Caveolin-1 inhibits cell detachment-induced p53 activation and anoikis by upregulation of insulin-like growth factor-I receptors and signaling. Oncogene. 2005;24:1338–47. doi: 10.1038/sj.onc.1208337. [DOI] [PubMed] [Google Scholar]
- Reinmuth N, Fan F, Liu W, Parikh AA, Stoeltzing O, Jung YD, et al. Impact of insulin-like growth factor receptor-I function on angiogenesis, growth, and metastasis of colon cancer. Lab Invest. 2002;82:1377–89. doi: 10.1097/01.lab.0000032411.41603.c2. [DOI] [PubMed] [Google Scholar]
- Rowinsky EK, Youssoufian H, Tonra JR, Solomon P, Burtrum D, Ludwig DL. IMC-A12, a human IgG1 monoclonal antibody to the insulin-like growth factor I receptor. Clin Cancer Res. 2007;13:5549s–5555s. doi: 10.1158/1078-0432.CCR-07-1109. [DOI] [PubMed] [Google Scholar]
- Sachdev D, Hartell JS, Lee AV, Zhang X, Yee D. A dominant negative type I insulin-like growth factor receptor inhibits metastasis of human cancer cells. J Biol Chem. 2004;279:5017–24. doi: 10.1074/jbc.M305403200. [DOI] [PubMed] [Google Scholar]
- Sachdev D, Li SL, Hartell JS, Fujita-Yamaguchi Y, Miller JS, Yee D. A chimeric humanized single-chain antibody against the type I insulin-like growth factor (IGF) receptor renders breast cancer cells refractory to the mitogenic effects of IGF-I. Cancer Res. 2003;63:627–35. [PubMed] [Google Scholar]
- Sachdev D, Singh R, Fujita-Yamaguchi Y, Yee D. Down-regulation of insulin receptor by antibodies against the type I insulin-like growth factor receptor: implications for anti-insulin-like growth factor therapy in breast cancer. Cancer Res. 2006;66:2391–402. doi: 10.1158/0008-5472.CAN-05-3126. [DOI] [PubMed] [Google Scholar]
- Sachdev D, Yee D. Disrupting insulin-like growth factor signaling as a potential cancer therapy. Mol Cancer Ther. 2007;6:1–12. doi: 10.1158/1535-7163.MCT-06-0080. [DOI] [PubMed] [Google Scholar]
- Tolcher AW, Patnaik A, Till E, Takimoto CH, Papadopoulos KP, Massard C, et al. A phase I study of AVE1642, a humanized monoclonal antibody IGF-1R (insulin like growth factor1 receptor) antagonist, in patients(pts) with advanced solid tumor(ST) J Clin Oncol. 2008;26 Abstr 3582. [Google Scholar]
- Townson JL, Naumov GN, Chambers AF. The role of apoptosis in tumor progression and metastasis. Curr Mol Med. 2003;3:631–42. doi: 10.2174/1566524033479483. [DOI] [PubMed] [Google Scholar]
- Twentyman PR, Luscombe M. A study of some variables in a tetrazolium dye (MTT) based assay for cell growth and chemosensitivity. Br J Cancer. 1987;56:279–85. doi: 10.1038/bjc.1987.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentinis B, Morrione A, Peruzzi F, Prisco M, Reiss K, Baserga R. Anti-apoptotic signaling of the IGF-I receptor in fibroblasts following loss of matrix adhesion. Oncogene. 1999;18:1827–36. doi: 10.1038/sj.onc.1202471. [DOI] [PubMed] [Google Scholar]
- van Golen CM, Schwab TS, Kim B, Soules ME, Su Oh S, Fung K, et al. Insulin-like growth factor-I receptor expression regulates neuroblastoma metastasis to bone. Cancer Res. 2006;66:6570–8. doi: 10.1158/0008-5472.CAN-05-1448. [DOI] [PubMed] [Google Scholar]
- Wu JD, Haugk K, Coleman I, Woodke L, Vessella R, Nelson P, et al. Combined in vivo effect of A12, a type 1 insulin-like growth factor receptor antibody, and docetaxel against prostate cancer tumors. Clin Cancer Res. 2006;12:6153–60. doi: 10.1158/1078-0432.CCR-06-0443. [DOI] [PubMed] [Google Scholar]
- Wu Y, Cui K, Miyoshi K, Hennighausen L, Green JE, Setser J, et al. Reduced circulating insulin-like growth factor I levels delay the onset of chemically and genetically induced mammary tumors. Cancer Res. 2003;63:4384–8. [PubMed] [Google Scholar]
- Wu Y, Yakar S, Zhao L, Hennighausen L, LeRoith D. Circulating insulin-like growth factor-I levels regulate colon cancer growth and metastasis. Cancer Res. 2002;62:1030–5. [PubMed] [Google Scholar]
- Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, et al. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A. 1999;96:7324–9. doi: 10.1073/pnas.96.13.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda T, Williams PJ, Hiraga T, Niewolna M, Nishimura R. A bone-seeking clone exhibits different biological properties from the MDA-MB-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J Bone Miner Res. 2001;16:1486–95. doi: 10.1359/jbmr.2001.16.8.1486. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yee D. Insulin-like growth factor binding protein-1 (IGFBP-1) inhibits breast cancer cell motility. Cancer Res. 2002;62:4369–75. [PubMed] [Google Scholar]