

Abstract
During the syntheses and studies of natural iron chelators (mycobactins), we serendipitously discovered that a simple, small molecule, oxazoline-containing intermediate 3 displayed surprising anti-tuberculosis activity (MIC of 7.7 µM, average). Herein we report elaboration of SAR around this hit as well as the syntheses and evaluation of a hundred oxazoline- and oxazole-containing compounds derived from an efficient three step process: 1) formation of β-hydroxy amides with serine or threonine; 2) cyclization to afford oxazolines; and 3) dehydration to give the corresponding oxazoles. A number of compounds prepared by this method were shown to possess impressive activity against M. tuberculosis, extremely low toxicity and therefore high therapeutic indexes, as well as activity against even the more recalcitrant non-replicating form of M. tuberculosis. The uniqueness of their structures and their simplicity should allow them to be further optimized to meet ADME (absorption, distribution, metabolism, excretion) requirements. The syntheses of eight of the most potent in vitro compounds were scaled up and the compounds were tested in an in vivo mouse infection model to evaluate their efficacy before engaging upon more elaborate compound design and optimization.
Keywords: Aryl oxazoline, Aryl oxazole, Benzyl ester, Tuberculosis agents, Structure-activity relationships, Mycobactins
Introduction
The battle against tuberculosis (TB, caused by Mycobacterium tuberculosis) has raged for millennia. [1,2] This disease has claimed the lives of over one billion people and currently infects one third of the world’s population. With 1.7 million deaths in 2006, TB as a single causative agent is the leading killer among infectious diseases [3]. The spread of TB was temporarily interrupted with the advent of several chemotherapeutic agents [2]. However, since the 1980s TB has been on the rise. Presently, over 9 million new cases are reported annually [3]. The rise in TB/HIV co-infection incidence and the spread of strains of TB that are resistant to at least two frontline anti-TB drugs (multidrug-resistant TB or MDR-TB) [1–7] has compounded the challenge for treating new cases. Today only sustained aggressive chemotherapy treatment with a combination of drugs can stop the disease [8]. This increase in infection has become so alarming that in 1993 the World Health Organization (WHO) [9] declared TB a global emergency. The development of multidrug resistant (MDR) and, more recently, extensively drug resistant (XDR) forms of TB have further stimulated research efforts globally [10]. More than ever, there is an urgent need to develop new anti-TB drugs to combat the spread of TB, particularly in its hard-to-kill multidrug-resistant, persistent and latent forms.
One particular strategy to combat TB involves exploiting its need for iron as illustrated by our design, syntheses and studies of siderophore analogs and siderophore-drug conjugates [11–15]. For example, we have demonstrated that synthetic mycobactin S (1), with an (S)-methyl group configuration in the butyrate linker region, inhibits the growth of Mycobacteria tuberculosis H37Rv while mycobactin T, with an (R)-methyl group configuration, promotes growth (Figure 1) and substitution of the butyrate linker of the natural mycobactins with a α–Boc-protected α,β-diaminopropionate linker (2) resulted in potent inhibition of TB growth.
Figure 1.

Mycobactins T and S and the synthetic anti-TB analogs (diaminoproprionate siderophore, ND-005859, and ND-005862).
While effective, this approach involves the syntheses of the structurally complex mycobactin-family [16–18] of siderophores in order to exploit TB’s essential iron assimilation processes. Throughout the syntheses of these siderophore natural products, protected and non-protected intermediates were routinely tested for anti-TB activity. This led to the discovery of ND-005859, 3, a simple, non-iron binding dibenzyl-protected oxazoline, which is a precursor in the syntheses of Mycobactin S and T, as well as the diaminoproprionate siderophore analog (2, Figure 1). When tested in vitro, the structurally simple compound 3 displayed notable anti-TB activity (MIC of 7.7 µM [19]), and furthermore was non-toxic (>128 µM) for VERO cells (green monkey kidney cell line). Indeed the potency of this serendipitous “hit” (compound 3) compared favorably to the natural M. smegmatis siderophore Mycobactin S [20] (1) or the amide analog 2 which boast MIC’s of 3.1 µM and 0.2 µM, respectively [21]. However, after benzyl deprotection of compound 3, the resulting iron binding analog 4 (ND-005862) lost all activity. This suggested that iron assimilation might not be involved in the anti-TB mode of action of this core structure, though it is possible that the benzyl ester of 3 could serve as a TB membrane permeable “prodrug” of the corresponding carboxylic acid which, in turn, might competitively inhibit siderophore biosynthesis [22–25]. While the mechanism of action is currently under investigation, the initial goal of this research was to explore structure-activity-relationships of this structurally simple lead compound in order to improve activity while also providing additional compounds for future mode of action studies.
Given the simplicity of the lead compound, its effectiveness against TB in vitro, and a possibly new mechanism of action, we sought to explore further the potential of compounds of this class as anti-TB agents. Here we report results from the syntheses, characterization, and screening of over 100 compounds against TB. A series of eight compounds were further examined for their toxicity and efficacy in an in vivo mouse model of TB. These simple compounds show promise for future development as a new class of anti-TB agents.
Results and discussion
The goal of this study was to first produce a series of derivatives of compound 3 (Figure 2, Series 1) to explore structure-activity-relationships (SAR) around the head group of the oxazoline core followed by conversion of all active oxazoline analogs to the corresponding oxazoles and comparison of their activity.
Figure 2.

Derivatization strategy of 3, an anti-TB “hit” compound.
Initially, focus was placed on modifications of the head group region (“series 1” analogs, Figure 2) in order to determine possible SAR trends, increase overall potency and identify an appropriate scaffold for eventual further elaboration of SAR by “tail” region (series 2) modifications. The ultimate goal is to combine the most potent head groups, as described in this document, with the most potent tail groups of the series 2 compounds (work to be reported in a future publication) and produce potentially the most potent and metabolically stable, “drug like” series 3 compounds. Thus, a relatively small set of series 1 compounds was prepared while also considering drug-like parameters [molecular weights under 500, a predicted LogP between 2.5 to 4.5, <5 hydrogen bond donors and <10 hydrogen bond acceptors]. It should be noted that this was not a combinatorial approach. Rather, it was intended to be a methodical parallel approach to create a precise, well characterized set of compounds with properties intended to fall within the calculated range of the molecular physiochemical profile of the currently used anti-TB drugs [26]. Syntheses of all compounds were easily scaled up for in vivo testing due to a short straight forward synthetic plan that involved an EDC mediated coupling of the desired benzoic acid 5 with L-serine benzyl ester and then dehydrative cyclization of the β-hydroxy amide 7 with bis(2-methoxyethyl)amino-sulfur trifluoride (DAST) as reported by Wipf et. al. [27] to form the oxazoline core 8 as products 12 – 56 (Scheme 1). Additionally, analog 57 was easily prepared by reduction of nitro analog 28 with indium, ammonium chloride and ethanol at 100°C. Using this effective procedure, over 45 oxazolines (8) were prepared in overall good yields (as described in the experimental section).
Scheme 1a.

Synthesis of oxazoline analogs.
aReagents: (a) L-Serine-OBn, EDC, DMAP, ACN, room temp, 14 h; (b) Oxalyl chloride, DCM, DMF (catalytic), 4 h, room temp; (c) L-Serine-OBn, DIPEA, DCM, 14 h, 50°C; (d) DAST, K2CO3, −78°C, 1 h.
Additionally, when an oxazoline compound displayed some activity against TB, it was immediately converted to the corresponding oxazole by a mild BrCCl3/DBU oxidation [28] procedure (Scheme 2). Overall, the oxazole analogs, 58 – 100, were, on average, more potent than the corresponding oxazolines despite having a simple, “unoptimized”, benzyl ester tail moiety. Additionally, when scale up was required for in vivo testing the initial amide coupling was performed with an easily prepared or purchased acid chloride 6 resulting in fast, efficient production of multi-gram quantities of 15, 29 and others.
Scheme 2a.

Conversion of oxazoline analogs to oxazoles.
aReagents: (a) DBU, BrCCl3, DCM, 0°C to RT, 1 h to 14 h.
To evaluate the importance of substitution on the oxazoline and oxazole cores a small set of threonine derived analogs were prepared utilizing the EDC coupling of the desired benzoic acid 5 with threonine benzyl ester. Then cyclization of general structure 9 with DAST gave the methyl substituted oxazolines 10 as analogs 101 – 105. Lastly, the corresponding oxazoles (11) were produced by the BrCCl3/DBU oxidation to give final compounds 106 – 111 (Scheme 3).
Scheme 3a.

Preparation of threonine derived oxazoline and oxazole analogs.
aReagents: (a) L-Threonine-OBn, EDC, DMAP, ACN, room temp, 14 h; (b) Oxalyl chloride, DCM, DMF (catalytic), 4 h, room temp; (c) L-Threonine-OBn, DIPEA, DCM, 14 h, 50°C (d) DAST, K2CO3, −78°C, 1 h. (e) DBU, BrCCl3, DCM, 0°C to RT, 1 h.
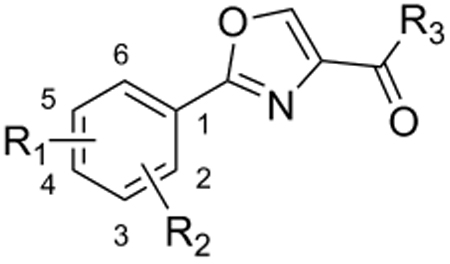
To determine structure-activity-relationships (SAR) around our hit compound 3, the previously mentioned rational design approach was carried out with variation of the head group to produce 47 oxazoline compounds which were tested for inhibition of M. tuberculosis H37Rv (Table 1) in two different culture media, GAS and GAST. GAST contains Tween 80 and is a more iron deficient medium than the GAS (which has added iron but lacks the Tween 80). Surprisingly, these different media had statistically relevant affects upon the MIC of our compounds as well as on the positive control rifampin (MIC of 0.11 µM and <0.01 µM in the GAS to GAST assays, respectively). Interpretation of the anti-TB data along with the VERO cytotoxicity assay data assisted in our selection of compounds to advance for testing in vivo.
Table 1.
SAR of oxazoline (series 1) compounds.
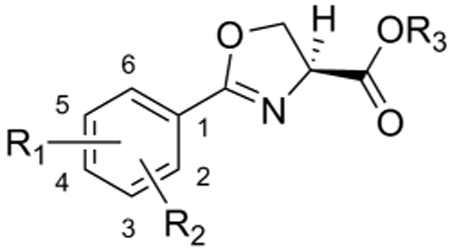 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | MolWt | ClogPa | GAS (µM) |
GAST (µM) |
VERO Cells: IC50 (µM) |
NRP-TB: LORA (µM) |
| 3 | H | 2-OBn | Bn | 387.44 | 5.77 | 12.6* | 7.69* | >128 | 67.3 |
| 4 | H | 2-OH | H | 207.19 | 4.06 | >128 | >128 | ||
| 12 | H | 2-OBn | Me | 311.34 | 4.06 | 118 | >128 | ||
| 13 | H | H | H | 191.19 | 1.93 | >128 | >128 | ||
| 14 | H | 2-OH | Me | 221.21 | 1.71 | >128 | >128 | ||
| 15 | H | H | Bn | 281.31 | 4.08 | 7.20* | 4.42* | >128 | 63 |
| 16 | H | 2-Cl | Bn | 315.75 | 4.79 | 49.9 | 15.2 | ||
| 17 | H | 3-Cl | Bn | 315.76 | 4.79 | 11.6 | 1.80 | >128 | |
| 18 | 4-Cl | H | Bn | 315.75 | 4.79 | 61.0 | 14.9 | ||
| 19 | H | 3-Cl | Me | 239.66 | 3.09 | >128 | 68.7 | ||
| 20 | H | 3-Cl | H | 225.63 | 3.09 | >128 | 106 | ||
| 21 | 4-OMe | H | Bn | 311.34 | 4.00 | 26.1 | 2.29 | >128 | |
| 22 | 4-OEt | H | Bn | 325.36 | 4.53 | 60.6 | 30.5 | ||
| 23 | 4-OMe | 3-Cl | Bn | 345.78 | 4.62 | 60.7 | 7.58 | ||
| 24 | 4-OMe | 3-F | Bn | 329.32 | 4.08 | 50.5 | 3.91 | ||
| 25 | H | 3-F | Bn | 299.3 | 4.22 | 60.7 | 6.86 | ||
| 26 | 4-Cl | 3-F | Bn | 333.74 | 4.94 | 52.0 | 6.49 | ||
| 27 | 4-NO2 | H | Me | 250.21 | 2.12 | >128 | >128 | ||
| 28 | 4-NO2 | H | Bn | 326.31 | 3.82 | 30.6 | 7.59 | >128 | |
| 29 | 4-F | 3-NO2 | Bn | 344.29 | 3.67 | 6.24 | 0.95 | >128 | 26 |
| 30 | 4-NO2 | 3-F | Bn | 344.29 | 3.67 | 30.2 | 3.68 | >128 | 59 |
| 31 | 3-Cl | 2-OBn | Bn | 421.87 | 6.39 | 6.30 | 34.0 | ||
| 32 | 4-F | 2-NO2 | Bn | 344.29 | 3.97 | 30.7 | 40.4 | ||
| 33 | 4-NO2 | 2-F | Bn | 344.29 | 3.97 | 13.0 | 8.70 | ||
| 34 | 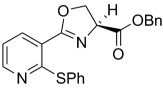 |
390.45 | 5.50 | 17.8 | 13.4 | ||||
| 35 | 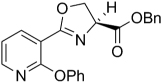 |
374.39 | 4.68 | 58.8 | >128 | ||||
| 36 | 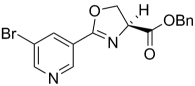 |
361.19 | 3.53 | 27.6 | 26.6 | ||||
| 37 | 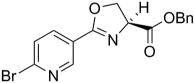 |
361.19 | 3.53 | 22.9 | 21.1 | ||||
| 38 | 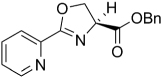 |
282.29 | 2.58 | 110 | 127 | ||||
| 39 | 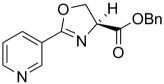 |
282.29 | 2.58 | 53.3 | 92.9 | ||||
| 40 | 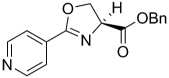 |
282.29 | 2.58 | 55.6 | 81.3 | ||||
| 41 | 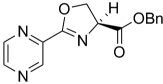 |
283.28 | 1.63 | 61.2 | >128 | ||||
| 42 | 4-OMe | 3-OMe | Bn | 341.36 | 3.74 | 39.0 | 21.6 | ||
| 43 | 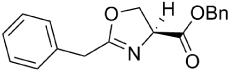 |
295.33 | 3.26 | >128 | >128 | ||||
| 44 | 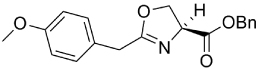 |
325.36 | 4.01 | >128 | >128 | ||||
| 45 | H | 2-OMe | Bn | 311.33 | 4.00 | 37.0 | 29.2 | ||
| 46 | 4-OPr | H | Bn | 339.39 | 5.06 | 2.97 | 22.4 | ||
| 47 | 4-OiPr | H | Bn | 339.39 | 4.84 | 3.2 | 14.9 | ||
| 48 | 4-OBu | H | Bn | 353.41 | 5.59 | 2.72 | 5.11 | ||
| 49 | 4-CH3 | H | Bn | 295.33 | 4.58 | 15.5 | 27.3 | ||
| 50 | 4-NO2 | 2-OBn | Bn | 432.43 | 5.81 | 15.6 | 4.86 | ||
| 51 | 4-OBn | 3-F | Bn | 405.42 | 5.85 | 0.77 | 1.42 | >128 | |
| 52 | H | 3-OMe | Bn | 311.33 | 4.00 | 24.8 | 42.5 | ||
| 53 | H | 3-CN | Bn | 306.32 | 3.51 | 47.2 | 75.1 | ||
| 54 | H | 3-Br | Bn | 360.20 | 4.94 | 24.2 | 52.5 | ||
| 55 | 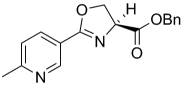 |
296.32 | 3.08 | 73.0 | 122 | ||||
| 56 |  |
358.39 | 4.47 | 12.5 | 29.8 | ||||
| 57 | 4-NH2 | H | Bn | 296.32 | 2.85 | 25.4 | 26.8 | ||
| Rifampin (control) | 822.94 | 0.06* | 0.01* | 125* | |||||
Average of assay repeats.
CLogP was calculated using the Cambridge Soft ChemDraw Ultra, v 8.0 software. Activity against non-replicating M. tuberculosis was determined using the Low Oxygen Recovery Assay (LORA).
The SAR of the oxazoline compounds showed an overwhelming increase in potency when the phenyl ring had a 3-chloro substitution (17) compared to the 2-chloro (16) and 4-chloro (18) analogs (1.80 µM, 15.2 µM and 14.9 µM in the GAST medium, respectively). However, analogs with other electron withdrawing groups like the 3-fluoro (25), 3-bromo (54), and 3-cyano (53) were all less active (6.86, 52.5, and 75.1 µM in the GAST medium, respectively) than those with the 3-chloro substitution (17, 1.80 µM in the GAST medium). Incorporation of the electron donating, 3-methoxy group (52, 42.5 µM GAST medium) significantly decreased activity relative to the 3-chloro analog (17, 1.80 µM in GAST medium). Similarly, analog 57 with the 4-amino group (26.8 µM in the GAST medium) was considerably less active than 4-nitro analog 28 (7.59 µM in the GAST medium). It was also evident that the benzyl ester was important for activity since the corresponding methyl ester analogs 12, 19, and 27 were considerably less active. Similarly, homologation between the oxazoline core and the phenyl head group was not tolerated as shown by diminished activity of analogs 43 and 44. The most potent oxazoline compounds contained two substituents on the phenyl ring. For instance, 4-fluoro, 3-nitro analog 29 was very active with an MIC of 0.95 µM in the GAST medium! Compound 30, with a reversal of the substituents relative to 29, was 3-fold less active (3.7 µM in the GAST medium) and further displacement, as in analogs 32 and 33, resulted in additional reduction of activity (40.4 and 8.7 µM in the GAST medium, respectively). It should also be noted that compounds 29 and 30 showed moderate activity (26 µM and 59 µM, respectively) in the LORA assay. This assay was specifically designed to simulate non-replicating M. tuberculosis during persistence and/or latency. Analog 51 with the 4-benzyl and 3-fluoro substituents was the most active in this series with MIC’s of 0.77 µM and 1.4 µM in the GAS and GAST assays. Additionally, to validate the importance of the heterocyclic core, all oxazoline precursors of general structure 7 (non-cyclized L-serine amides) were screened and the overwhelming majority of compounds were found to be inactive (>128 µM).
Next, since many of the oxazoline analogs showed relatively potent anti-TB activity, the corresponding oxazole derivatives were produced to test what effects a planer, aromatic heterocyclic core would have on anti-TB activity (Table 2). Remarkably, the majority of oxazoles synthesized had improved in vitro activity, with seven compounds 59, 65, 66, 68, 70, 78 and 89 having sub micromolar (<1 µM) MIC’s! Interestingly, there was little difference in activity of the ortho-, meta-, and para-methoxy substituted analogs (93, 94, and 60) and the corresponding chloro derivatives (68, 59, and 87), though the ortho- and meta-chloro compounds were the most active. Of these, most compounds tested were active in the LORA non-replicating assay and were non-toxic in the VERO cytotoxicity assay (>121 µM). Replacement of the phenyl ring with a pyridyl substituent gave oxazoles with significant activity (76, 77, 79 as the 2-pyridyl, 3-pyridyl, 4-pyridyl, respectively) with MIC’s between 1 – 4 µM in the GAS assay, whereas the analogous oxazoline compounds (38, 39, and 40) had weak activities at best (MIC’s >50 µM in the GAS medium). In contrast, oxazoline 51 (MIC’s ~1 µM in both assays) was three-fold more active than the corresponding oxazole, 72 (MIC ~3 µM in both assays). As with the xazoline core, removal of the benzyl ester or transesterification to the methyl, ethyl or t-butyl esters resulted in nearly complete loss of activity (data not shown). This loss of activity after transesterification as was surprising considering that the ethyl ester was key to the potency in a series of related 3,5-disubstituted isoxazolines reported by Lee et. al. [29] and the quinoline-isoxazole series described by Kozikowski et. al. [30].
Table 2.
SAR of oxazole compounds.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Comp d |
R1 | R2 | R3 | MolWt | ClogPa | GAS (µM) |
GAST (µM) | VERO Cells: IC50 (µM) |
NRP-TB: LORA (µM) |
| 58 | 4-NO2 | H | OBn | 324.29 | 3.49 | 1.79 | 42.3 | ||
| 59 | H | 3-Cl | OBn | 313.74 | 4.19 | 1.25 | 0.99 | >128 | |
| 60 | 4-OMe | H | OBn | 309.32 | 3.49 | 1.72 | 1.80 | >128 | |
| 61 |  |
383.36 | 3.13 | 35.8 | 49.5 | >128 | |||
| 62 |  |
372.8 | 4.09 | 118 | 48.1 | ||||
| 63 | H | 2-OBn | OBn | 385.41 | 4.69 | 62.6 | 10.3 | >128 | |
| 64 | 3-Cl | 2-OBn | OBn | 419.86 | 5.26 | 5.40 | >128 | ||
| 65 | H | H | OBn | 279.29 | 3.45 | 0.47 | 0.49 | 121 | 3.80 |
| 66 | 4-F | 3-NO2 | OBn | 342.28 | 3.34 | 1.10 | 0.48 | >128 | 31.9 |
| 67 | 4-NO2 | 3-F | OBn | 342.28 | 3.34 | 35.4 | 2.66 | >128 | |
| 68 | H | 2-Cl | OBn | 313.74 | 3.94 | 0.73 | 1.69 | >128 | 11.4 |
| 69 | 4-F | 2-NO2 | OBn | 342.28 | 3.64 | 2.40 | 9.90 | >128 | |
| 70 | 4-NO2 | 2-F | OBn | 342.28 | 3.64 | 0.70 | 1.65 | >128 | 11.6 |
| 71 | 4-NO2 | 2-OBn | OBn | 430.41 | 4.68 | >128 | >128 | ||
| 72 | 4-OBn | 3-F | OBn | 403.4 | 5.27 | 3.03 | 3.86 | ||
| 73 | 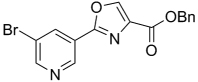 |
359.17 | 2.96 | 2.37 | 2.24 | >128 | >128 | ||
| 74 | 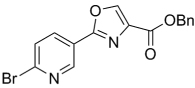 |
359.17 | 2.96 | 2.54 | 1.84 | >128 | 25.1 | ||
| 75 | 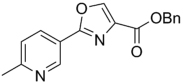 |
294.30 | 2.55 | 3.01 | 10.2 | ||||
| 76 | 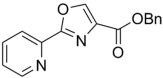 |
280.28 | 2.25 | 2.00 | 2.47 | ||||
| 77 | 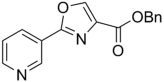 |
280.28 | 2.04 | 3.82 | 6.20 | ||||
| 78 | 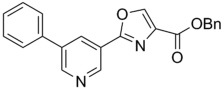 |
356.37 | 3.93 | 1.49 | 0.98 | >128 | |||
| 79 | 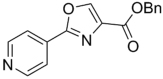 |
280.28 | 2.04 | 1.42 | 6.10 | ||||
| 80 | 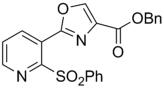 |
420.44 | 3.74 | 66.2 | >128 | ||||
| 81 | 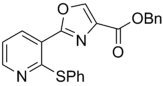 |
388.44 | 4.98 | 8.00 | >128 | ||||
| 82 | 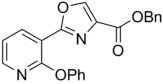 |
389.44 | 3.59 | 6.86 | 91.4 | ||||
| 83 | 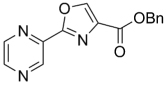 |
281.27 | 1.29 | 7.32 | 22.5 | ||||
| 84 | H | 2-PO(Ph)2 | OBn | 479.86 | 4.22 | 61.8 | 61.5 | ||
| 85 | 4-OMe | 3-OMe | OBn | 339.34 | 3.20 | 16.0 | 17.5 | ||
| 86 | 4-OEt | H | OBn | 323.34 | 4.02 | 15.5 | 1.97 | >128 | 29.7 |
| 87 | 4-Cl | H | OBn | 313.74 | 3.99 | 18.9 | 7.77 | >128 | 56.2 |
| 88 | 4-NH2 | H | OBn | 294.3 | 2.63 | 1.98 | 3.67 | >128 | 68.6 |
| 89 | H | 3-F | OBn | 297.28 | 3.59 | 0.91 | 1.42 | >128 | |
| 90 | 4-OMe | 3-Cl | OBn | 343.76 | 3.87 | 2.35 | 5.33 | >128 | |
| 91 | 4-Cl | 3-F | OBn | 331.73 | 4.15 | 2.87 | 22.4 | ||
| 92 | 4-OMe | 3-F | OBn | 327.31 | 3.47 | 5.55 | 6.99 | >128 | |
| 93 | H | 2-OMe | OBn | 309.32 | 3.49 | 3.88 | 7.54 | ||
| 94 | H | 3-OMe | OBn | 309.32 | 3.49 | 6.04 | 3.76 | ||
| 95 | H | 3-CN | OBn | 304.3 | 2.92 | 25.2 | 60.5 | ||
| 96 | H | 3-Br | OBn | 358.19 | 4.34 | 1.93 | 6.08 | ||
| 97 | 4-CH3 | H | OBn | 293.32 | 3.97 | 12.84 | 7.68 | ||
| 98 | 4-OPr | H | OBn | 337.37 | 4.54 | 3.48 | 1.94 | ||
| 99 | 4-OiPr | H | OBn | 337.37 | 4.32 | 1.95 | 3.21 | ||
| 100 | 4-OBu | H | OBn | 351.40 | 5.07 | 1.76 | 3.83 | ||
| Rifampin (control) | 822.94 | 0.06* | 0.01* | 125* | |||||
Average of assay repeats.
CLogP was calculated using the Cambridge Soft ChemDraw Ultra, v 8.0 software. Activity against non-replicating M. tuberculosis was determined using the Low Oxygen Recovery Assay (LORA).
Next, in an attempt to determine whether simple methyl substitution on the oxazoline (Table 3) and oxazole (Table 4) cores would affect anti-TB activity, a small focused set of threonine-derived compounds was prepared and tested. Preliminary results indicated that methyl substituted oxazoline compounds were significantly less active compared to the analogous oxazoline parents. For instance, compounds 102 and 103 were completely inactive (>128 µM) relative to compounds 3 and 15 (7.69 µM and 4.42 µM in the GAST assay, respectively). The threonine-derived oxazole compounds also had greatly diminished activity, as seen, for example, by comparison of compound 108 (>128 µM) to compound 3 (7.69 µM in the GAST assay). In fact, the more potent head group analogs, 109, 110, and 111 all had diminished anti-TB activity as methyl substituted oxazole analogs compared to the non-methyl bearing oxazole analogs (65, 60, 66). Furthermore, compound 109 had greatly increased toxicity in the VERO assay. In short, even simple methyl substitution on the oxazoline and oxazole cores resulted in diminished potency and often increased toxicity toward the VERO cells.
Table 3.
SAR of threonine-derived oxazoline.
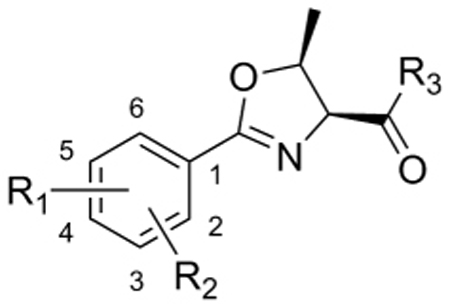 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | MolWt | ClogPa | GAS (µM) |
GAST (µM) |
VERO Cells: IC50 (µM) |
NRP-TB: LORA (µM) |
| 101 | H | H | OH | 205.21 | 2.44 | >128 | >128 | ||
| 102 | H | H | OMe | 219.24 | 2.89 | >128 | >128 | ||
| 103 | H | 2-OBn | OBn | 401.45 | 6.29 | >128 | >128 | ||
| 104 | 4-OMe | H | OBn | 325.35 | 4.51 | 54.1 | 37.8 | ||
| 105 | 4-F | 2-NO2 | OBn | 358.32 | 4.18 | 12.5 | 4.11 | 38.1 | |
| Rifampin (control) | 822.94 | 0.06* | 0.01* | 125* | |||||
Average of assay repeats.
CLogP was calculated using the Cambridge Soft ChemDraw Ultra, v 8.0 software. Activity against non-replicating M. tuberculosis was determined using the Low Oxygen Recovery Assay (LORA).
Table 4.
SAR of threonine-derived oxazole.
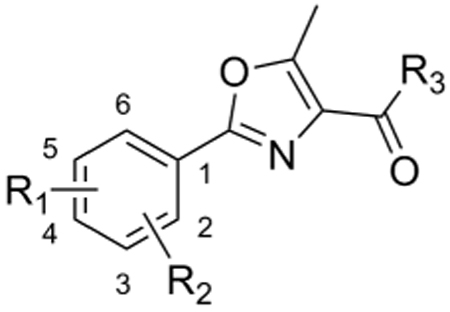 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | MolWt | ClogPa | GAS (µM) |
GAST (µM) |
VERO Cells: IC50 (µM) |
NRP-TB: LORA (µM) |
| 106 | H | H | OH | 203.2 | 2.46 | >128 | >128 | ||
| 107 | H | 2-OH | OH | 219.2 | 1.49 | >128 | >128 | ||
| 108 | H | 2-OBn | OBn | 399.45 | 4.94 | 28.3 | >128 | 110 | |
| 109 | H | H | OBn | 293.32 | 3.74 | 3.92 | 15.2 | 35.0 | >128 |
| 110 | 4-OMe | H | OBn | 323.34 | 3.76 | 47.4 | 15.8 | ||
| 111 | 4-F | 2-NO2 | OBn | 356.3 | 3.34 | 15.0 | 4.62 | >128 | >128 |
| Rifampin (control) | 822.94 | 0.06* | 0.01* | 125* | |||||
Average of assay repeats.
CLogP was calculated using the Cambridge Soft ChemDraw Ultra, v 8.0 software. Activity against non-replicating M. tuberculosis was determined using the Low Oxygen Recovery Assay (LORA).
Encouraged by the anti-TB data, several compounds were subsequently tested against a diverse panel of nontuberculosis mycobacteria and other bacteria (Gram positive and negative) as well as select strains of yeast. This panel included Bacillus subtitlis ATCC 6633, Staphylococcus aureus SG 511, Mycobacterium luteus ATCC 10240, E. coli SG 458, Proteus vulgaris OX 19, Mycobacterium smegmatis SG 987, Mycobacterium aurum SB 66, Mycobacterium aurum DSM 43536, Mycobacterium vaccae 10670, Mycobacterium fortuitum B, Sporobolomyces salmonicolor SBUG 549, Candida albicans C.A., Glomerella, and Mycobacterium avium strains MAC 100, 101, 108, 109, 116. In all cases, compounds 1, 15, 29, 60, 63, 65, 66, and 68 were inactive (MIC’s equal to negative control) despite showing some zones of inhibition in the agar diffusion assay against Mycobacterium vaccae 10670 and Mycobacterium fortuitum B. This remarkably selective inhibition of TB suggests a unique mode of action for the slower growing M. tuberculosis. A microarray experiment [31] with compound 3 revealed up regulation of the Mtb genes (responsible for mycobacin/exochelin synthesis) coupled with the down regulation of the bacterioferritin (bfr) genes (data not shown). These results suggested a condition of iron stress, possibly due to inhibition of the mycobacterial siderophore biosynthesis pathway and in turn interference of iron trafficking [22–25]. Further mode of action studies are ongoing and will be reported in due course.
Animal Testing
In vivo studies to assess toxicity were performed using compounds 3, 15, 29, 60, 63, 65, 66, and 68. Mice were given 100 mg/kg po once a day in 100 ul 0.5% carboxymethylcellulose (CMC) for five days, followed by 2 days of rest and then 200 mg/kg for five days. All compounds were well tolerated with respect to body weight, gross appearance and behavior with the exception of compound 66 where the average weight loss was 12% at the end of the two week period (from 21 g to 18.5 g). Subsequently, all compounds were tested for efficacy in BALB/c mice infected with M. tuberculosis. At days 5, 60 and 85 following a lose dose aerosol infection with M. tuberculosis Erdman, mean cfu (SD) in mouse lungs of untreated controls were 2.3 (2.5) × 102, 1.8 (1.0) × 106 and 4.7 (2.3) × 106, respectively. Beginning on day 60 post-infection, groups of 8 mice received a single oral dose at 200 mg/kg (except compound 66 which was dosed at 50 mg/kg due to a 10% loss in body weight during tolerance testing) in 200 µL of 0.5% CMC 5 days per week for three weeks. Untreated controls received the CMC vehicle only and the positive drug control consisted of rifampin (RMP) at 10 mg/kg dosed in an identical manner and vehicle as the test compounds. After two days of no treatment (in order to reduce the probability of carry over of lung tissue-associated compound on the plating medium), on day 85 post-infection mice were sacrificed and lung tissue homogenates plated to determine bacterial load. Whereas the rifampin control effected a reduction in bacterial load of greater than 2 logs versus the untreated control, none of the test compounds showed efficacy with this dosing regimen and mode of administration due potentially to the metabolic liability of the benzyl ester moiety. The stability of the benzyl ester was then evaluated in vitro in simulated gastric fluid and rat liver microsomes. Not surprisingly, the benzyl esters were rapidly cleaved to their corresponding inactive carboxylic acids in simulated gastric fluid assay (Table 5).
Table 5.
Compound stability to simulated gastric juices and microsomes.
| Gastric Stability |
Microsomes | |
|---|---|---|
| Compound | % Remaining (2 h.) |
% Remaining (15 min.) |
| 3 | 1.4 | 8.4 |
| 15 | 0.4 | 5.7 |
| 29 | 2.2 | 13 |
| 60 | 0.4 | 14 |
| 63 | 4.2 | 13 |
| 65 | 0.5 | 15 |
| 66 | 2.1 | 8.1 |
| 68 | 4.2 | 8.8 |
Furthermore, these compounds were metabolically unstable (Table 5). The in vitro metabolism of 15 was further studied and the major metabolite was the corresponding carboxylic acid, providing an additional reason why these fairly potent in vitro compounds failed to show in vivo efficacy. As such, plans are now underway to improve the pharmacokinetic properties of the lead compounds through the use of more metabolically stable replacements of the simple benzyl esters to hopefully impart enhanced in vivo stability and possibly efficacy. The experimental section profiles the syntheses and characterization of these eight compounds evaluated in vivo, as well as by microsomal and simulated gastric fluid assays.
Conclusion
On a planet in which over 1/3 of the population is infected with tuberculosis, it is more than ever prudent to evaluate new anti-tuberculosis agents in vitro and in vivo. Herein we described simple serine derived oxazoline and oxazole templates with highly selective anti-TB activity. SAR studies demonstrate that, in general, oxazoles are more potent than their oxazoline precursors and that alkyl substitution on either the oxazoline or oxazole diminishes antiTB activity. Activity can also be influenced by the type and position of substituents on the pendant phenyl group, with electron withdrawing chloro and combination of chlroro and nitro groups being somewhat advantageous though clear correlations require more study. The corresponding acids and simple alkyl esters are substantially less active than the corresponding benzyl esters. While the metabolic lability of the benzyl esters precludes their use as orally active agents, many of these agents like compound 65 (with an MIC of <0.5 µM against replicating Mycobacterium tuberculosis H37Rv) can be made on multiple gram scale in just three steps, thus further studies to extend both SAR and ADME properties are merited.
Experimental Section
All anhydrous solvents, reagent grade solvents for chromatography and starting materials were purchased from either Aldrich Chemical Co. (Milwaulkee, WI) or Fisher Scientific (Suwanee, GA). Water was distilled and purified through a Milli-Q water system (Millipore Corp., Bedford, MA). L-Glutathione reduced form (GSH) and nicotinamide adenine dinucleotide phosphate reduced form (NADPH) were obtained from Sigma-Aldrich (St. Louis, MO). Male Sprague-Dawley rat liver microsomes were purchased from BD Gentest (Woburn, MA). General methods of purification of compounds involved the use of silica cartridges purchased from AnaLogix, Inc. (Burlington, WI; www.ana-logix.com) and/or recrystallization. The reactions were monitored by thin-layer chromatography (TLC) on precoated Merck 60 F254 silica gel plates and visualized using UV light (254 nm) reported in Rf values. All compounds were analyzed for purity and characterized by 1H and 13C NMR using Varian 300MHz NMR and/or Varian 500MHz NMR spectrometers. Chemical shifts are reported in ppm (δ) relative to the residual solvent peak in the corresponding spectra; chloroform 7.26 ppm and 77.23 ppm, methanol 3.31 ppm and 49.00 ppm, methylene chloride 5.32 ppm and 54.00 ppm and coupling constants (J) are reported in hertz (Hz) (where, s = singlet, bs = broad singlet, d = doublet, dd = double doublet, bd = broad doublet, ddd = double doublet of dublet, t = triplet, tt – triple triplet, q = quartet, m = multiplet) and analyzed using MestReC NMR data processing. High resolution mass spectra (“HRMS”) by fast atom bombardment (“FAB”) were taken on a JEOL AX505HA magnetic sector mass spectrometer and values are reported as m/z Melting points (“mp”) were measured on a Thomas-Hoover capillary melting point apparatus and are uncorrected. FTIR (“IR”) spectra were obtained using a Thermo-Nicolet IR200 spectrometer and absorption frequencies were reported in reciprocal centimeters (cm−1). The high pressure liquid chromatography (“HPLC”) analyses were carried out on a Waters 2695 instrument consisting of a photodiode array detector 996, using the Waters symmetry C18 5 µm reverse phase column (Waters, Milford, MA, www.waters.com). Mobile phases: (a) 50% HPLC grade acetonitrile in Millipore purified water at a flow rate of 1.0 mL min−1 and UV diction at 254 nm. (b) 60% HPLC grade acetonitrile in Millipore purified water at a flow rate of 1.0 mL min−1 and UV diction at 254 nm. A typical run time was 20 minutes. The liquid chromatography mass spectrum (“LC/MS”) analyses were carried out on Waters ZQ instrument consisting of chromatography module Alliance HT, photodiode array detector 2996, and mass spectrometer Micromass ZQ, using a 3 × 50 mm Pro C18 YMC reverse phase column. Mobile phases: 10 mM ammonium acetate in HPLC grade water (A) and HPLC grade acetonitrile (B). A gradient was formed from 5% to 80% of B in 10 minutes at 0.7 mL/min. The MS electrospray source operated at capillary voltage 3.5 kV and a desolvation temperature 300°C. Elemental analyses were performed by Midwest Microlabs, LLC (Indianapolis, IN). All reactions were conducted under argon unless otherwise noted. Solvents were removed in vacuo on a rotary evaporator. Abbreviations: DCM = dichloromethane; DMF = dimethylformamide; ACN = acetonitrile EtOAc = ethyl acetate; HOAc = acetic acid; DAST = Bis(2-methoxyethyl)amino-sulfur trifluoride; EDC = N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride.
General Procedure for Preparation of Serine Amides (7, open L-Serine form)
Method 1
Benzoic acid (1 g, 8.2 mmol, 1.1 equiv) and L-serine benzyl ester hydrochloride (1.7 g, 7.4 mmol, 1 equiv) in 30 mL of acetonitrile (ACN) were treated with N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (EDC-HCl, 2.9 g, 14.9 mmol, 2 equiv) and N,N-dimethylaminopyridine (DMAP, 2.3 g, 18.6 mmol, 2.5 equiv). The resulting solution was stirred for 12 h at room temperature. The reaction mixture was diluted with ethyl acetate (EtOAc), washed with 0.5 N citric acid (2×), water and 10% aqueous NaHCO3 (2×). The organic phase was collected and dried over sodium sulfate (Na2SO4), filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl aceteate : dichloromethane (DCM) gradient solvent system.
Method 2
Benzoyl chloride (4.15 g, 3.4 mL, 28.6 mmol, 1 equiv) was dissolved in 120 mL of dichloromethane (DCM, anhydrous) and then L-serine benzyl ester hydrochloride (7.2 g, 30.1 mmol, 1.05 equiv) and diisopropylamine (DIPEA, 9.3 g, 12.5 mL, 71.6 mmol, 2.5 equiv) was added very slowly to the chilled reaction mixture (0°C, ice bath temperature). The reaction was warmed to room temperature and then heated to reflux, 50°C (bath temperature) for 14 h. The reaction mixture was cooled, washed with water and then 10% aqueous NaHCO3 solution (2×). The organic phase was collected and dried over sodium sulfate (Na2SO4), filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl acetate : DCM gradient solvent system.
General Procedure for Preparation of Oxazoline Products(compounds 3, 4, 8, 12 – 57)
L-serine amide 7 (204 mg, 0.68 mmol, 1 equiv) was dissolved in DCM (5 mL) and cooled to −78°C (bath temp, dry ice-acetone). Bis(2-methoxyethyl)amino-sulfur trifluoride (DAST, 0.1 mL, 0.78 mmol, 1.15 equiv) was added slowly to the cold mixture under argon and the reaction was stirred for 30 min. Next, K2CO3, (254 mg, 1.8 mmol, 2.7 equiv) was added and then the reaction was allowed to warm to room temperature. Once the reaction appeared to be complete by TLC analysis, the reaction mixture was poured into 10% aqueous NaHCO3 solution and was extracted with DCM (2×). The organic layer washed again with the NaHCO3 solution, brine, dried over Na2SO4 , filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl acetate : DCM gradient solvent system.
(S)-Benzyl 2-(2-(benzyloxy)phenyl)-4,5-dihydrooxazole-4-carboxylate (3)
See our previous studies.17,18
2-(Benzyloxy)benzoyl chloride (13.3 g, 52.3 mmol) was dissolved in 250 mL of DCM and then L-serine benzyl ester hydrochloride (13.1 g, 54.9 mmol) and diisopropylethylamine (22.8 mL, 130.7 mmol) were added while the reaction was chilled in an ice bath (0°C bath temperature). The reaction mixture was warmed to room temperature and then heated to reflux, 50°C (bath temperature) for 14 h. The reaction mixture was cooled, washed with 10% aqueous NaHCO3 solution (2×), water, then washed with 0.5 N citric acid (2×) and then brine. The organic phase was collected and dried over sodium sulfate (Na2SO4), filtered and then concentrated in vacuo to give 21.2 g (99%) of (S)-benzyl 2-(2-(benzyloxy)benzamido)-3-hydroxypropanoate as an oil which became an off white solid upon standing. 1H NMR (300 MHz, CDCl3) δ 8.79 ( 1 H, d, J = 7.1 Hz), 8.20 ( 1 H, dd, J = 7.8, 1.8 Hz), 7.52-7.27 ( 10 H, m), 7.07 ( 2 H, dd, J = 15.9, 8.0 Hz), 5.23-5.14 ( 4 H, m), 4.93-4.85 ( 1 H, m), 3.92 ( 2 H, dd, J = 3.9, 1.6 Hz). (S)-Benzyl 2-(2-(benzyloxy)benzamido)-3-hydroxypropanoate (21.2 g, 54 mmol) was dissolved in DCM (450 mL) and then cooled to −78°C (bath temp, dry ice-acetone). Bis(2-methoxyethyl)amino-sulfur trifluoride (DAST, 8.2 mL, 62.1 mmol) was added slowly to the cold mixture under argon where it was stirred for 30 mins. Then K2CO3 (20.2 g, 145.8 mmol) was added and the reaction mixture was allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the reaction mixture was poured into 10% aqueous NaHCO3 solution and was extracted with DCM (2×). The organic layer washed again with the NaHCO3 solution, brine washed, and was dried over Na2SO4, filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with 1:10 to 1:1 ethyl acetate : DCM gradient solvent system to give 13.7 g (65%) of 3 as a white solid. Rf = 0.44 (DCM); mp 69–70°C; IR (neat) 1740 (C=O), 1638 (C=N), 1498, 1450, 1257, 1177 cm−1; 1H NMR (300 MHz, CDCl3) 7.82 ( 1 H, dd, J = 8.0, 1.8 Hz), 7.50-7.47 (2H, m), 7.42-7.26 (9H, m), 6.99 ( 2 H, dd, J = 7.7, 6.2 Hz), 5.25 ( 4 H, dd, J = 25.1, 11.8 Hz), 5.01 ( 1 H, dd, J = 10.6, 8.0 Hz), 4.61 ( 2 H, m); 13C NMR (126 MHz, CDCl3) δ171.03, 165.65, 157.56, 136.79, 135.40, 132.64, 131.62, 128.50, 128.36, 128.29, 128.25, 127.53, 126.69, 120.69, 120.67, 117.12, 113.75, 70.55, 69.16, 68.77, 67.12; FABMS: 388 (M+1); HRMS calcd. for C24H21NO4 387.1471, found 387.1458. Elemental calcd. for C24H21NO4 C, 74.40; H, 5.46; N, 3.62; found C, 74.35; H, 5.32; N, 3.60. [α]D25 +1.02 (c = 1.545 g/100 mL CHCl3).
(S)-Benzyl 4,5-dihydro-2-phenyloxazole-4-carboxylate (15)
Benzoyl chloride (4.15 g, 3.4 mL, 28.6 mmol) was dissolved in 120 mL of dichloromethane (DCM, anhydrous) and then L-serine benzyl ester hydrochloride (7.2 g, 30.1 mmol) and diisopropylamine (DIPEA, 9.3 g, 12.5 mL, 71.6 mmol) was added very slowly to the chilled reaction mixture (0°C, ice bath temperature). The reaction mixture was warmed to room temperature and then heated to reflux, 50°C (bath temperature) for 14 h. The reaction mixture was cooled, washed with 10% aqueous NaHCO3 solution (2×), water, then washed with 0.5 N citric acid (2×) and then brine. The organic phase was collected and dried over sodium sulfate (Na2SO4), filtered and then concentrated in vacuo to give 7.15 g (83%) of (S)-benzyl 2-(benzamido)-3-hydroxypropanoate as an oil which became an off white solid upon standing. 1H NMR (300 MHz, CDCl3) δ 7.85-7.79 ( 2 H, m), 7.56-7.30 ( 7 H, m), 7.14 ( 1 H, d, J = 7.1 Hz), 5.25 ( 2 H, s), 4.91 ( 1 H, td, J = 7.1, 3.5, 3.5 Hz), 4.15-4.01 ( 2 H, m).
(S)-Benzyl 2-(benzamido)-3-hydroxypropanoate (7.15 g, 28.5 mmol) was dissolved in DCM (240 mL) and cooled to −78°C (bath temp, dry ice-acetone). Bis(2-methoxyethyl)amino-sulfur trifluoride (DAST, 4.4 g, 3.6 mL, 27.5 mmol) was added slowly to the cold mixture under argon where it stirred for 30 min. Then K2CO3 (8.9 g, 64.4 mmol) was added and the reaction mixture was allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the reaction mixture was poured into 10% aqueous NaHCO3 solution and extracted with DCM (2×). The organic layer was washed again with the NaHCO3 solution, brine, and then dried over Na2SO4, filtered and concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl acetate : DCM gradient solvent system to give 5.7 g (85%) of 15 as a white solid. Rf = 0.25 (DCM); mp 51–52°C; IR (neat) 1741 (C=O), 1643 (C=N), 1188 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.04-7.92 ( 2 H, m), 7.57-7.29 ( 8 H, m), 5.26 ( 2 H, dd, J = 20.9, 12.3 Hz), 4.99 ( 1 H, dd, J = 10.6, 7.9 Hz), 4.80-4.42 ( 2 H, m). 13C NMR (126 MHz, CDCl3) δ 170.42, 163.45, 158.40, 156.25, 135.56, 135.48, 135.14, 128.67, 128.61, 128.49, 126.70, 124.19, 118.88, 118.71, 70.31, 68.83, 67.60. Elemental calcd. for C17H15NO3: C, 72.58; H, 5.37; N, 4.98; found C, 72.44; H, 5.40; N, 4.95; [α]D25 +1.14 (c = 1.54 g/100 mL CHCl3).
(S)-Benzyl 2-(4-fluoro-3-nitrophenyl)-4,5-dihydrooxazole-4-carboxylate (29)
4-Fluoro-3-nitrobenzoic acid (6.65 g, 35.2 mmol) was dissolved in 75 mL of anhydrous DCM and then oxalyl chloride (6.2 mL, 70.4 mmol) was added drop wise followed by the addition of 50 µL of dimethylformamide (DMF) to catalyze the reaction. The reaction mixture bubbled and was stirred at room temperature for four h. During this time it became a homogeneous solution. The reaction mixture was concentrated to give a yellow paste which was subsequently concentrated in vacuo from toluene to give 4-fluoro-3-nitrobenzoyl chloride as a crude yellow solid (7.2 g, 94%) which is used immediately in the subsequent reaction. Rf = 0.5 (EtOAc, red streak) 1H NMR (300 MHz, CDCl3) δ 8.85 (1H, dd, J = 7.0, 2.3 Hz), 8.40 (1H, ddd, J = 8.8, 4.0, 2.4 Hz), 7.57-7.43 (1H, m).
4-Fluoro-3-nitrobenzoyl chloride (7.2 g, 34.1 mmol) was dissolved in 150 mL of DCM and then L-serine benzyl ester hydrochloride (8.56 g, 35.8 mmol) and diisopropylethylamine (14.9 mL, 85.3 mmol) were added while the reaction was chilled in an ice bath (0°C bath temperature). The reaction mixture was warmed to room temperature and then heated to reflux, 50°C (bath temperature) for 14 h. The reaction mixture was cooled, washed with 10% aqueous NaHCO3 solution (2×), water, then washed with 0.5 N citric acid (2×) and then brine. The organic phase was collected and dried over Na2SO4, filtered and then concentrated in vacuo The crude material obtained was purified using silica gel column chromatography with 5% to 50% ethyl acetate : DCM as the solvent system to give (S)-benzyl 2-(4-fluoro-3-nitrobenzamido)-3-hydroxypropanoate (11.1 g, 90%) as a yellow oil which became a yellow solid upon standing. Rf product = 0.1 (DCM). 1H NMR (300 MHz, CDCl3) δ 8.54 ( 1 H, dd, J = 7.0, 2.2 Hz), 8.13 ( 1 H, ddd, J = 8.6, 4.1, 2.3 Hz), 7.44-7.30 ( 5 H, m), 7.19 ( 1 H, d, J = 6.86 Hz), 5.33-5.20 ( 2 H, m), 4.96-4.87 ( 1 H, m), 4.10 ( 2 H, ddd, J = 35.3, 11.2, 3.1 Hz).
(S)-Benzyl 2-(4-fluoro-3-nitrobenzamido)-3-hydroxypropanoate (5.9 g, 16.3 mmol) was dissolved in DCM (160 mL) and cooled to −78°C (bath temp, dry ice-acetone). DAST (2.45 mL, 18.7 mmol) was added slowly to the cold mixture under argon and the reaction was stirred for 30 min. Then K2CO3 (6.1 g, 43.9 mmol) was added and the reaction was allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the mixture was poured into 10% aqueous NaHCO3 and extracted with DCM (2×). The organic layer washed again with the NaHCO3 solution, brine, and then dried over Na2SO4 , filtered and concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl acetate : DCM gradient solvent system to give 4.9 g (87%) of 29 as a yellow solid upon standing. Rf = 0.18 (DCM); mp 77–78°C; IR (neat) 1741 (C=O), 1651, 1541 (C=O), 1350, 1190 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.68 ( 1 H, dd, J = 7.2, 2.2 Hz), 8.28 ( 1 H, ddd, J = 8.7, 4.2, 2.2 Hz), 7.45-7.31 ( 6 H, m), 5.26 ( 2 H, dd, J = 20.8, 12.2 Hz), 5.02 ( 1 H, dd, J = 10.6, 8.0 Hz), 4.78-4.61 ( 2 H, m). 13C NMR (126 MHz, CDCl3) δ 170.42, 163.45, 158.40, 156.25, 135.56, 135.48, 135.14, 128.67, 128.61, 128.49, 126.69, 124.19, 118.88, 118.71, 70.32, 68.83, 67.60. HRMS calcd. For C17H13ClFNO3 345.0887, found 345.0901. Elemental calcd. for C17H13ClFNO3: C, 59.30; H, 3.81; N, 8.14; found C, 59.20; H, 3.77; N, 8.13; [α]D25 +0.94 (c = 1.52 g/100 mL CHCl3).
General Procedure for Preparation of Oxazole Products (compounds 58 – 100)
Oxazoline 8 (165 mg, 0.5 mmol, 1 equiv) was dissolved in 2 mL of dry DCM and chilled in an ice bath (0°C bath temperature). DBU (0.22 mL, 1.5 mmol, 3 equiv) was added followed by slow addition of bromotrichloromethane (0.15 mL, 1.5 mmol, 3 equiv). The reaction was stirred for 1 h cold and then allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the reaction mixture was poured into 10% aqueous NaHCO3 solution and extracted with DCM (2×). The organic layer washed again with the NaHCO3 solution, brine, dried over Na2SO4, filtered and then concentrated in vacuo. Crude oily material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl acetate : DCM gradient solvent system to give solid upon standing.
Benzyl 2-(4-methoxyphenyl)oxazole-4-carboxylate (60)
4-Methoxybenzoyl chloride (4.11 g, 23.4 mmol) was dissolved in 120 mL of DCM and then L-serine benzyl ester hydrochloride (5.86 g, 24.5 mmol) and diisopropylethylamine (10.2 mL, 58.4 mmol) were added while the reaction was chilled in an ice bath (0°C bath temperature). The reaction mixture was warmed to room temperature and then heated to reflux, 50°C (bath temperature) for 14 h. The reaction mixture was cooled, washed with 10% aqueous NaHCO3 solution (2×), water, with 0.5 N citric acid (2×) and then brine. The organic phase was collected dried over sodium sulfate (Na2SO4), filtered and then concentrated in vacuo The crude material obtained was purified using silica gel column chromatography with 5% to 50% ethyl acetate : DCM as the solvent system to give (S)-benzyl 2-(4-methoxybenzamido)-3-hydroxypropanoate (7.65 g, 99%) as a yellow oil which solidified upon standing. Rf = 0.31 (DCM). 1H NMR (300 MHz, CDCl3) δ 1H NMR (300 MHz, CDCl3) δ ppm 7.83-7.74 ( 2 H, m), 7.44-7.30 ( 4 H, m), 7.07 ( 1 H, d, J = 7.0 Hz), 6.95-6.86 ( 2 H, m), 5.24 ( 2 H, s), 4.89 ( 1 H, td, J = 7.1, 3.6, 3.6 Hz), 4.05 ( 2 H, dd, J = 3.6, 1.9 Hz), 3.84 ( 3 H, s).
(S)-Benzyl 2-(4-methoxybenzamido)-3-hydroxypropanoate (7.65 g, 23.3 mmol) was dissolved in DCM (240 mL) and cooled to −78°C (bath temp, dry ice-acetone). DAST (3.6 mL, 4.3 mmol) was added slowly to cold mixture under argon where it stirred for 30 min. Then K2CO3 (8.73 g, 63.1 mmol) was added and the reaction was allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis the mixture was poured into 10% aqueous NaHCO3 solution and was extracted with DCM (2×). The organic layer washed again with the NaHCO3 solution, brine, dried over Na2SO4 , filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl acetate : DCM gradient solvent system to give 6.74 g (93%) of (S)-benzyl 2-(4-methoxyphenyl)-4,5-dihydrooxazole-4-carboxylate, 21, as a white solid upon standing. Rf = 0.40 (DCM). 1H NMR (300 MHz, CDCl3) δ 7.96-7.89 ( 2 H, m), 7.44-7.31 ( 5 H, m), 6.94-6.87 ( 2 H, m), 5.25 ( 2 H, q, J = 12.3, 12.3, 12.3 Hz), 4.95 ( 1 H, dd, J = 10.5, 7.8 Hz), 4.69-4.50 ( 2 H, m), 3.84 ( 3 H, s).
(S)-benzyl 2-(4-methoxyphenyl)-4,5-dihydrooxazole-4-carboxylate (21, 6.74 g, 21.6 mmol) was dissolved in 75 mL of dry DCM and chilled in an ice bath (0°C bath temperature). DBU (9.7 mL, 64.9 mmol) was added followed by slow addition of bromotrichloromethane (6.5 mL, 64.9 mmol). The reaction was stirred for 1 h cold and then allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the reaction mixture was poured into 10% aqueous NaHCO3 solution and extracted with DCM (2×). The combined organic layers were washed again with the NaHCO3 solution and brine then dried over Na2SO4 , filtered and concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl acetate : DCM gradient solvent system to give 5.0 g (75%) of 60 as a white solid. Rf = 0.55 (DCM); mp 111–112°C; IR (neat) 1716 (C=O), 1313, 1263, 1138, 1117 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.23 ( 1 H, s), 8.10-8.01 ( 2 H, m), 7.51-7.30 ( 5 H, m), 7.02-6.94 ( 2 H, m), 5.40 ( 2 H, s), 3.86 ( 2 H, s). 13C NMR (126 MHz, CDCl3) δ 162.86, 162.17, 161.56, 143.65, 135.80, 134.39, 128.88, 128.83, 128.76, 128.66, 119.32, 114.45, 67.00, 55.65. HRMS calcd. For C18H15NO4, 310.1079 found 310.1071. Elemental calcd. for C18H15NO4: C, 69.89; H, 4.89; N, 4.53; found C, 69.67; H, 4.92; N, 4.46.
Benzyl 2-(2-(benzyloxy)phenyl)oxazole-4-carboxylate (63)
(S)-Benzyl 2-(2-(benzyloxy)phenyl)-4,5-dihydrooxazole-4-carboxylate (3, 5.88 g, 15.2 mmol) was dissolved in 100 mL of dry DCM and chilled in an ice bath (0°C bath temperature). Next, DBU (6.8 mL, 45.5 mmol) was added followed by slow addition of bromotrichloromethane (4.5 mL, 45.5 mmol). The reaction was stirred for 1 h cold and then allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the reaction mixture was poured into 10% aqueous NaHCO3 solution and extracted with DCM (2×). The combined organic layers were washed again with the NaHCO3 solution, brine, dried over Na2SO4 , filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl acetate : DCM gradient solvent system to give 4.7 g (81%) of 63 as a white solid. Rf = 0.6 (DCM); mp 74–75°C; IR (neat) 1740 (C=O), 1453, 1319, 1145, 1121 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.31 ( 1 H, d, J = 2.4 Hz), 8.06 ( 1 H, dd, J = 8.1, 1.7 Hz), 7.57-7.27 ( 11 H, m), 7.11-7.03 ( 2 H, m), 5.41 ( 2 H, s), 5.22 ( 2 H, s). 13C NMR (126 MHz, CDCl3) δ 161.32, 156.80, 143.92, 136.55, 135.59, 133.89, 132.44, 131.10, 128.57, 128.50, 128.49, 128.36, 127.72, 126.77, 120.98, 116.17, 113.47, 70.48, 66.68. HRMS calcd. For C24H19NO4, 386.1392 found 386.1385. Elemental calcd. for C24H19NO4: C, 74.79; H, 4.97; N, 3.63; found C, 74.59; H, 4.97; N, 3.66.
Benzyl 2-phenyloxazole-4-carboxylate (65)
(S)-Benzyl 4,5-dihydro-2-phenyloxazole-4-carboxylate (15, 6.27 g, 22.3 mmol) was dissolved in 75 mL of dry DCM and chilled in an ice bath (0°C bath temperature). DBU (10 mL, 66.9 mmol) was added followed by slow addition of bromo-trichloromethane (6.6 mL, 66.9 mmol). The reaction was stirred for 1 h cold and then allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the reaction mixture was poured into 10% aqueous NaHCO3 solution and extracted with DCM (2×). The organic layer washed again with the NaHCO3 solution, brine, dried over Na2SO4, filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl acetate : DCM gradient solvent system to give 5.64 g (91%) of 65 as a white solid. Rf = 0.37 (DCM); mp 94–95°C; IR (neat) 1741 (C=O), 1323, 1144, 1114 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.28 ( 1 H, s), 8.12 ( 2 H, dt, J = 4.9, 4.7, 3.1 Hz), 7.52-7.31 ( 8 H, m), 5.41 ( 2 H, s). 13C NMR (126 MHz, CDCl3) δ 162.49, 161.13, 143.84, 135.47, 134.32, 131.15, 128.96, 128.78, 128.57, 128.50, 128.46, 128.41, 126.86, 126.35, 66.79. HRMS calcd. For C17H13NO3, 280.0974 found 280.0973. Elemental calcd. for C17H13NO3: C, 73.11; H, 4.69; N, 5.02; found C, 72.82; H, 4.67; N, 4.96.
Benzyl 2-(4-fluoro-3-nitrophenyl)oxazole-4-carboxylate (66)
(S)-4-Fluoro-3-nitrobenzyl 2-(benzamido)-3-hydroxypropanoate (29, 5.10 g, 14.8 mmol) was dissolved in 200 mL of dry DCM and chilled in an ice bath (0°C bath temperature). DBU (6.6 mL, 44.4 mmol) was added followed by slow addition of bromo-trichloromethane (4.4 mL, 44.4 mmol). The reaction was stirred for 1 h cold and then allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the reaction mixture was poured into 10% aqueous NaHCO3 solution and extracted with DCM (2×). The combined organic layers were washed again with NaHCO3, brine dried over Na2SO4, filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a1:10 to 1:1 ethyl acetate : DCM gradient solvent system to give 3.1 g (60%) of 66 as a light yellow solid upon standing. Rf = 0.29 (DCM); mp 120–121°C; IR (neat) 1732 (C=O), 1534 (C=O), 1349, 1322, 1151 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.79 ( 1 H, dd, J = 7.0, 2.2 Hz), 8.40 ( 1 H, ddd, J = 8.8, 4.2, 2.3 Hz), 8.33 ( 1 H, d, J = 2.6 Hz), 7.50-7.31 ( 6 H, m), 5.41 ( 2 H, s). 13C NMR (126 MHz, CDCl3) δ 160.62, 159.33, 157.89, 155.74, 144.60, 135.17, 134.85, 133.59, 128.67, 128.62, 124.70, 124.68, 123.53, 123.50, 119.57, 119.40, 67.15. For C17H11FN2O5, 343.0730 found 343.0751. Elemental calcd. for C17H11FN2O5: C, 59.65; H, 3.24; F, 5.55; N, 8.18; found C, 59.83; H, 3.29; N, 8.00.
Benzyl 2-(2-chlorophenyl)oxazole-4-carboxylate (68)
2-Chlorobenzoyl chloride (5.39 g, 29.9 mmol) was dissolved in 150 mL of DCM and then L-serine benzyl ester hydrochloride (7.49 g, 31.4 mmol) and diisopropylethylamine (13 mL, 74.7 mmol) were added while the reaction was chilled in an ice bath (0°C bath temperature). The reaction mixture was warmed to room temperature and then heated to reflux, 50°C (bath temperature) for 14 h. The reaction mixture was cooled, washed with 10% aqueous NaHCO3 solution (2×), water, 0.5 N citric acid (2×) and then brine. The organic phase was collected and dried over sodium sulfate (Na2SO4), filtered and then concentrated in vacuo The crude material obtained was purified using silica gel column chromatography with 5% to 50% ethyl acetate : DCM as the solvent system to give (S)-benzyl 2-(2-chlorobenzamido)-3-hydroxypropanoate (9.9 g, 99%) as a yellow oil which solidified upon standing. Rf product = 0.16 (DCM). 1H NMR (300 MHz, CDCl3) δ 7.72-7.64 ( 1 H, m), 7.46-7.18 ( 8 H, m), 5.28 ( 2 H, d, J = 12.2 Hz), 4.97-4.86 ( 1 H, m), 4.10 ( 2 H, s).
(S)-Benzyl 2-(2-chlorobenzamido)-3-hydroxypropanoate (5.9 g, 16.3 mmol) was dissolved in DCM (160 mL) and cooled to −78 °C (bath temp, dry ice-acetone). DAST (2.4 mL, 18.7 mmol) was added slowly to the cold mixture under argon where it stirred for 30 min. K2CO3 (6.0 g, 43.9 mmol) was added and the reaction was allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the reaction mixture was poured into 10% aqueous NaHCO3 and extracted with DCM (2×). The organic layer washed again with the NaHCO3 solution, brine, dried over Na2SO4, filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl acetate : DCM gradient solvent system to give 4.9 g (87%) of (S)-benzyl 2-(2-chlorophenyl)-4,5-dihydrooxazole-4-carboxylate, 16, as a white solid. Rf product = 0.36 (DCM). 1H NMR (300 MHz, CDCl3) δ 7.82 ( 1 H, dd, J = 7.6, 1.7 Hz), 7.49-7.27 ( 8 H, m), 5.33-5.19 ( 2 H, m), 5.04 ( 1 H, dd, J = 10.6, 7.7 Hz), 4.77-4.58 ( 2 H, m).
(S)-Benzyl 2-(2-chlorophenyl)-4,5-dihydrooxazole-4-carboxylate (16, 5.88 g, 15.2 mmol) was dissolved in 100 mL of dry DCM and chilled in an ice bath (0°C bath temperature). DBU (6.8 mL, 45.5 mmol) was added followed by slow addition of bromo-trichloromethane (4.5 mL, 45.5 mmol). The reaction was stirred for 1 h cold and then allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the reaction mixture was poured into 10% aqueous NaHCO3 solution and extracted with DCM (2×). The organic layer washed again with the NaHCO3 solution, brine, and dried over Na2SO4 , filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl acetate : DCM gradient solvent system to give 4.7 g (81%) of 68 as a white solid. Rf = 0.59 (DCM); mp 62–63°C IR (neat) 1741 (C=O), 1314, 1145, 1118 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.36 ( 1 H, d, J = 1.2 Hz), 8.09-7.99 ( 1 H, m), 7.56-7.21 ( 8 H, m), 5.41 ( 2 H, s). 13C NMR (126 MHz, CDCl3) δ 160.94, 160.67, 144.32, 135.44, 134.14, 132.88, 131.86, 131.64, 131.07, 128.59, 128.53, 128.44, 126.85, 125.51, 66.85. For C17H12ClNO3, 314.0584 found 314.0571. Elemental calcd. for C17H12ClNO3: C, 59.65; H, 3.24; F, 5.55; N, 8.18; found C, 65.08; H, 3.86; N, 4.46; found C, 65.07; H, 3.93; N, 4.42.
General Procedure for Preparation of Threonine Amides (9, open L-Threonine form)
Benzoic acid (414 mg, 3.3 mmol, 1 equiv) and L-threonine benzyl ester hydrochloride (874 mg, 3.5 mmol, 1.05 equiv) were dissolved in 7 mL of acetonitrile (ACN) and treated with N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (EDC-HCl, 1 g, 6.6 mmol, 2 equiv) and N,N-dimethylaminopyridine (DMAP, 1.2 g, 9.9 mmol, 3 equiv). The resulting solution was stirred for 12 h at room temperature. The reaction mixture was diluted with EtOAc, washed with 0.5 N citric acid (2×), water, 10% aqueous NaHCO3 solution (2×). The organic phase was collected, dried over sodium sulfate (Na2SO4), filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl acetate : dichloromethane (DCM) gradient solvent system.
Method 2
Benzoyl chloride (449 mg, 0.4 mL, 3.1 mmol, 0.95 equiv) was dissolved in 6 mL of DCM and then L-threonine benzyl ester hydrochloride (711 mg, 3.3 mmol, 1 equiv) and triethylamine (1 g, 1.4 mL, 9.9 mmol, 3 equiv) were added. The reaction mixture was warmed to room temperature and then heated to reflux, 50°C (bath temperature) for 14 h. The reaction mixture was cooled, washed with 10% aqueous NaHCO3 solution (2×), water, 0.5 N citric acid (2×) and then brine. The organic phase was collected and dried over Na2SO4, filtered and then concentrated in vacuo The crude material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl acetate : DCM gradient solvent system.
General Procedure for Preparation of Threonine Derived Oxazoline Products (compounds 10, 101 – 105)
L-Threonine amide 9 (330 mg, 1.1 mmol, 1 equiv) was dissolved in DCM (5 mL) and cooled to −78°C (bath temp, dry ice-acetone). DAST (0.16 mL, 1.3 mmol, 1.2 equiv) was added slowly to the cold mixture under argon and the reaction was stirred for 30 min. Then K2CO3 (393 mg, 2.8 mmol, 2.7 equiv) was added and the reaction was allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the reaction mixture was poured into 10% aqueous NaHCO3 solution and extracted with DCM (2×). The combined organic layers were washed with the NaHCO3 solution, brine, dried over Na2SO4 , filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl acetate : DCM as the gradient solvent system.
General Procedure for Preparation of Threonine Derived Oxazoles (compounds 11, 106 – 111)
Threonine derived oxazoline 10 (195 mg, 0.66 mmol, 1 equiv) was dissolved in 2 mL of dry DCM and chilled in an ice bath (0°C bath temperature). DBU (0.3 mL, 2 mmol, 3 equiv) was added followed by slow addition of bromotrichloromethane (0.2 mL, 2 mmol, 3 equiv). The reaction was stirred for 1 h cold and then allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the reaction mixture was poured into 10% aqueous NaHCO3 solution and extracted with DCM (2×). The combined organic layers were washed again with the NaHCO3 solution, brine, dried over Na2SO4, filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a 1:10 to 1:1 ethyl acetate : DCM gradient solvent system.
Anti-tuberculosis and Cytotoxicity Assays
Activity against replicating Mycobacterium tuberculosis H37Rv (ATCC 27294, American Type Culture Collection, Rockville, MD) was determined using a fluorescence readout in the Microplate Alamar Blue Assay (MABA) [32] following incubation for one week with test compounds in glycerol-alanine-salts medium (GAS) as well as in medium without added iron but with Tween 80 (GAST) [33]. The MIC was defined as the minimum concentration inhibiting fluorescence by 90% relative to bacteria-only controls. Activity against non-replicating M. tuberculosis was determined using the Low Oxygen Recovery Assay (LORA) [34] by exposing low oxygen-adapted M. tuberculosis containing a luciferase gene to test compounds under anaerobic conditions for 10 days. After a 28 hour recovery in air, the luciferase signal was measured. MIC was defined as the lowest concentration inhibiting recovery of luciferase signal by ≥ 90% relative to bacteria-only controls. Toxicity to African green monkey kidney cells (VERO) was determined using a colorimetric assay as previously described [32].
Simulated Gastric Fluid Stability
Compounds were incubated in simulated gastric fluid [35] (consisting of 3.2 mg/mL pepsin in 0.03 M sodium chloride at pH 1.2) at 37°C at a concentration of 50 µM. Aliquots (500 µL) were taken at 0 hr and 2 hr, 500 µL of 10% sodium bicarbonate containing internal standard was added, followed by centrifugation for 5 min at 12,000 rpm. Aliquots of the supernatants were analyzed by reversed-phase HPLC with UV detection at 254 nm. The mobile phase consisted of elution on a YMC 5µ-basic 4.6 mm i.d. × 15 cm column at 1 mL/min with 5-min isocratic elution with 10% acetonitrile/90% water, 20-min linear gradient from 10% acetonitrile to 90% acetonitrile, and 10-min isocratic elution with 90% acetonitrile/10% water. The peak area ratio relative to internal starndard was measured and compared to that at time 0 and reported as percent remaining.
Metabolic Stability
Compounds (20 µM) were mixed with NADPH (0.5 mM) in potassium phosphate buffer (50 mM, pH 7.4) in a total volume of 0.5 mL and preincubated at 37 ° C. The reaction was started by adding pooled male Sprague-Dawley liver microsomes (0.2 mg protein). After 15-min incubation, the reaction was quenched with 0.5 mL acetonitrile containing internal standard and centrifuged to remove protein. A second incubation was terminated at time 0. An aliquot of the supernatant was analyzed by reversed-phase HPLC with UV detection at 254 nm. The chromatographic conditions were the same as for the gastric fluid stability. The peak area ratio relative to internal standard was measured and compared relative to that at time 0 and reported as percent remaining.
HPLC chromatography
The HPLC system consisted of a Perkin-Elmer series 200 quaternary pump (Perkin-Elmer Corp., Norwalk, CT), a Perkin-Elmer UV detector series 200, and a Perkin-Elmer auto sampler series 200. Samples were analyzed on a YMC 5 µm basic 4.6 mm i.d. × 15 cm column (YMC Inc., Wilmington, NC) connected to a YMC 5 µm basic 4.0 mm i.d. × 2 cm guard cartridge.
Supplementary Material
Acknowledgment
This work was supported by Grant PHS 398/2590 from the National Institutes of Health. We would like to thank the University of Notre Dame especially the MS facility (Bill Boggess, Nonka Sevova) and Prof. Jennifer DuBois for profound scientific discussion. The excellent technical assistance of Baojie Wan and Yuehong Wang with anti-TB assays at UIC and Uta Wohfield with microbial assays at the HKI is greatly appreciated. Adriel Villegas-Estrada was a Fellow of the Chemistry-Biochemistry-Biology Interface (CBBI) Program at the University of Notre Dame, supported by training grant T32GM075762 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information Available. Analytical data for the following compounds (3, 4, 12 – 111) can be found. This material is available free of charge via the Internet at http://www.sciencedirect.com
References
- 1.WHO. Anti-tuberculosis Drug Resistance in the World: The WHO/IUATLD Global Project on Anti-tuberculosis Drug Resistance Surveillance 1994–1997. 1997 WHO/TB/97.229.
- 2.Daniel TM, Bates JH, Downes KA. Tuberculosis: Pathogenesis, Protection, and Control, Barry Bloom editor. Washington, D.C.: ASM Press; 1994. History of Tuberculosis; pp. 13–24. [Google Scholar]
- 3.WHO. Global tuberculosis control : surveillance, planning, financing : WHO report 2008. 2008 WHO/HTM/TB/2008.393.
- 4.Basso LA, Blanchard JS. Adv. Exp. Med. Biol. 1998;456:115–144. doi: 10.1007/978-1-4615-4897-3_7. [DOI] [PubMed] [Google Scholar]
- 5.Bradford WZ, Daley CL. Infectious Dis. Clin. North Am. 1998;12:157–172. doi: 10.1016/s0891-5520(05)70415-3. [DOI] [PubMed] [Google Scholar]
- 6.Bastian I, Colebunders R. R. Drugs. 1999;58:633–666. doi: 10.2165/00003495-199958040-00005. [DOI] [PubMed] [Google Scholar]
- 7.Cohn DL, Bustreo F, Raviglione MC. Clin. Infect. Dis. 1997;24:S121–S130. doi: 10.1093/clinids/24.supplement_1.s121. [DOI] [PubMed] [Google Scholar]
- 8.Snider DE, Castro KG. New. Engl. J. Med. 1998;338:1689–1690. doi: 10.1056/NEJM199806043382309. [DOI] [PubMed] [Google Scholar]
- 9.Tuberculosis: A Global Emergency. 1993 World Health Forum 14:438. [Google Scholar]
- 10.Dorman SE, Chaisson RE. Nature Med. 2007;13:295–298. doi: 10.1038/nm0307-295. [DOI] [PubMed] [Google Scholar]
- 11.Girijavallabhan V, Miller MJ. Therapeutic Uses of Iron(III) Chelators and their Antimicrobial Conjugates. In: Crosa JH, Mey AR, Payne SM, editors. Iron Transport in Bacteria. Chapter 27. 2004. pp. 413–433. [Google Scholar]
- 12.Roosenberg JM, II, Lin Y-M, Lu Y, Miller MJ. Curr. Med. Chem. 2000;7:159–197. doi: 10.2174/0929867003375353. [DOI] [PubMed] [Google Scholar]
- 13.Lu Y, Miller MJ. Bioorg. Med. Chem. 1999;7:3025–3038. doi: 10.1016/s0968-0896(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 14.Vergne AF, Walz AJ, Miller MJ. Nat. Prod. Rep. 2000;17:99–116. doi: 10.1039/a809397k. [DOI] [PubMed] [Google Scholar]
- 15.Miller M, Zhu H, Xu Y, Wu C, Walz A, Vergne A, Roosenberg J, Moraski G, Minnick A, McKee-Dolence J, Hu J, Fennell K, Dolence K, Dong L, Franzblau S, Malouin F, Mollmann U. Biometals. 2009;22:61–75. doi: 10.1007/s10534-008-9185-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu Y, Miller MJ. J. Org. Chem. 1998;63:4314–4322. [Google Scholar]
- 17.Hu J, Miller MJ. Am. Chem. Soc. 1997;119:3462–3468. [Google Scholar]
- 18.Moraski G, Miller MJ, Franzblau SG. Hetrocycles. 2009;80 doi: 10.3987/COM-09-S(S)69. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.7.7 µM is the average value from multiple tests (15 in total) of ND-005859 in the GAST (low iron Tween 80 containing media) run in Scott Franzblau’s lab at the University of Illinois at Chicago. The MIC has ranged from 2.2 µM to 13.7 µM in this assay.
- 20.Snow GA. Bacteriol. Rev. 1970;34:99–125. doi: 10.1128/br.34.2.99-125.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Assay data from level two testing at the Tuberculosis Antimicrobial Acquisition and Coordinating Facility (TAACF) Bethesda, MD: [Google Scholar]
- 22.Ferreras JA, Ryu J-S, Lello FD, Tan DS, Quadri LE. Nat. Chem. Biol. 2005;1:29–32. doi: 10.1038/nchembio706. [DOI] [PubMed] [Google Scholar]
- 23.Somu RV, Boshoff H, Qiao C, Bennett EM, Barry CE, III, Aldrich CC. J. Med. Chem. 2006;49:31–34. doi: 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]
- 24.Miethke M, Bisseret P, Beckering CL, Vignard D, Eustache J, Marahiel MA. FEBS J. 2006;273:409. doi: 10.1111/j.1742-4658.2005.05077.x. [DOI] [PubMed] [Google Scholar]
- 25.Vannada J, Bennett EM, Wilson DJ, Boshoff H, Barry CE, III, Aldrich CC. Org. Lett. 2006;21:4707–4710. doi: 10.1021/ol0617289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barry CE, III, Slayden RA, Sampson AE, Lee RE. Biochem. Pharm. 2000;59:221–231. doi: 10.1016/s0006-2952(99)00253-1. [DOI] [PubMed] [Google Scholar]
- 27.Phillips AJ, Uto Y, Wipf P, Reno MJ, Williams DR. Org. Lett. 2000;2:1165–1168. doi: 10.1021/ol005777b. [DOI] [PubMed] [Google Scholar]
- 28.Williams DR, Lowder PD, Gu YG, Brooks DA. Tet. Lett. 1997;38:331–334. [Google Scholar]
- 29.Rakesh, Sun D, Lee RB, Tangallapally RP, Lee RE. Euro. J. Med. Chem. 2009;44:460–472. doi: 10.1016/j.ejmech.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lilienkampf A, Mao J, Wan B, Wang Y, Franzblau SG, Kozikowski AP. J. Med. Chem. 2009;52:2109–2118. doi: 10.1021/jm900003c. [DOI] [PubMed] [Google Scholar]
- 31.Boshoff HI, Myers TG, Copp BR, NcNeil MR, Wilson MA, Barry CE., III J. Bio. Chem. 2004;279:40174–40184. doi: 10.1074/jbc.M406796200. [DOI] [PubMed] [Google Scholar]
- 32.(a) Collins L, Franzblau SG. Antimicrob. Agents Chemother. 1997;41:1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, et al. Proc. Nat. Acad. Sci. USA. 2000;97:1252–1257. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho SH, Wan B, Di W, Hwang CH, Franzblau SG. Keystone Symposium: Tuberculosis: Integrating Host and Pathogen Biology: Whistler, British Columbia, Canada. 2005 [Google Scholar]
- 34.Falzari K, Zhou Z, Pan D, Liu H, Hongmanee P, Franzblau SG. Antimicrob Agents Chemother. 2005;49:1447–1454. doi: 10.1128/AAC.49.4.1447-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.The United States Pharmacopeia 23, The National Formulary 18. Rockville, MD: The United States Pharmacopeial Convention, Inc; 1995. Simulated gastric fluid and simulated intestinal fluid; p. 2053. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.