

Abstract
The sarco/endoplasmic reticulum calcium ATPase (SERCA) is essential for the control of intracellular free Ca2+ levels. Recently, SERCA has been identified as an important effector of nitric oxide (NO) action in vascular cells. NO stimulates the uptake of cytosolic Ca2+ via SERCA by adducting glutathione to the reactive cysteine-674 thiol. Mutation of this single amino acid prevents the stimulation of Ca2+ uptake by NO, as well as its ability to decrease Ca2+ and cell migration. NO function is impaired in a variety of cardiovascular diseases, including diabetes and atherosclerosis, which are associated with irreversible oxidation of SERCA cysteine-674. Targeting the sources of oxidants in vascular diseases to prevent SERCA oxidation and/or increasing the expression of SERCA may impede vascular disease.
Introduction
The sarco/endoplasmic reticulum calcium ATPase (SERCA) is a 97–115 kDa membrane protein expressed in the ER/SR of all cells as the product of three genes [1]. SERCA1 is present in fast twitch skeletal muscle, SERCA2a in heart and slow twitch skeletal muscle, SERCA2b in all cells including smooth muscle cells (SMC), endothelial cells (EC), and platelets, and SERCA3 in non-muscle cells including platelets and EC. SERCA2a is also expressed in proliferating SMC [2;3]. The major SMC isoform, SERCA2b, is an alternatively spliced form that differs from SERCA2a by having an elongated C-terminus within the SR lumen. In SERCA3 an even longer C-terminus is thought to decrease the Ca2+ affinity of the protein, enabling it to increase the concentration of Ca2+ in the stores to a greater degree than the other isoforms [4].
The sarcoplasmic reticulum stores contain the bulk of intracellular Ca2+. Although it contains only a small fraction of the total, it is cytosolic free Ca2+ which is accountable for regulating the function of Ca2+-dependent proteins that regulate cell function. As the major mechanism for uptake of Ca2+ into the stores, SERCA is essential. SERCA is also important because the level of Ca2+ in the stores inversely regulates influx of Ca2+ from the extracellular space into the cytosol by a mechanism called store-operated Ca2+ entry. Thus, SERCA-mediated Ca2+ uptake into stores plays a key role in maintenance of intracellular free Ca2+ levels within a physiological range. In addition, during contractions caused by elevated levels of intracellular Ca2+, accelerated sequestration of Ca2+ by SERCA mediates smooth, cardiac, and skeletal muscle relaxation. In non-contractile cells, such as proliferating smooth muscle cells or endothelium, SERCA regulates many cellular processes, including cell growth, apoptosis, and migration.
Nitric oxide (NO), the free radical which is the biologically active component of endothelium-derived relaxing factor, has critical roles in the maintenance of vascular homeostasis. Under normal conditions, a major and immediate action of NO in all cells is to decrease intracellular calcium, which can inhibit cellular functions, including growth, proliferation, adhesion, and contractility. In intact arteries, the immediate consequence of the reduction of intracellular Ca2+ in smooth muscle cells (SMC) by NO is relaxation. NO relaxes vascular SMC via both cyclic GMP-dependent and -independent mechanisms (Figure 1A). By a cyclic GMP-dependent pathway, NO activates guanylyl cyclase to increase cyclic GMP which in turn activates protein kinase G (PKG) and regulates its downstream targets (eg. contractile proteins, ion channels) to relax SMC. NO can also relax SMC by cyclic GMP-independent mechanisms, including by direct modifications of SERCA protein (Figure 1B).
Figure 1.
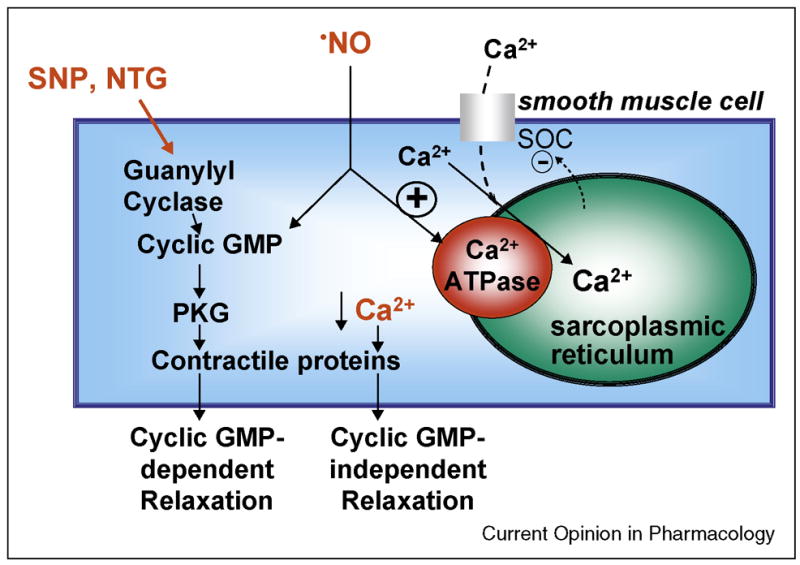
A. NO relaxes vascular smooth muscle (SMC) in part by redox regulation of SERCA. Nitrovasodilators activate guanylyl cyclase and increase cyclic GMP which activates protein kinase G (PKG), which in turn regulates its downstream targets to relax smooth muscle. Unlike guanylyl cyclase-dependent nitrovasodilators, authentic NO activates SERCA and refills intracellular Ca2+ stores. Consequently, store-operated cation channels (SOC) are inhibited and the decrease in intracellular free Ca2+ relaxes smooth muscle. The latter mechanism is predominantly cyclic-GMP-independent. B. Under physiological conditions (left) · NO and O2−• form the reactive nitrogen species, peroxynitrite (OONO−), which increases glutathione (GSH) adducts (GSS-) primarily on cysteine(C)-674 of SERCA. This adduct increases Ca2+ uptake activity of SERCA. Under pathophysiological conditions (right) higher levels of O2−• and H2O2 increase the destruction and consumption of · NO, and produce OONO− which can irreversibly oxidize the reactive SERCA C674 thiol (-SO3H). This prevents its reversible glutathiolation and blocks the stimulation of SERCA by · NO. Thus, the redox status of C674 can determine physiological and pathophysiological changes in vascular tone as well as SMC and endothelial cell (EC) migration. From reference [5].
Redox regulation of SERCA by NO
NO can stimulate the uptake of cytosolic Ca2+ by SERCA [5]. By reacting with superoxide anion, NO forms the more thiol reactive oxidant, peroxynitrite, which in turn can adduct glutathione (GSH) to SERCA cysteine thiols. The reaction of NO with superoxide anion is required as indicated by the fact that mouse arteries in which superoxide dismutase is overexpressed relax less to NO [5]. The majority of the GSH on SERCA is bound to the most reactive thiol on cysteine-674, and mutation of this single cysteine prevents not only formation of most of the GSH adducts, but also stimulation of Ca2+ uptake by NO. In atherosclerotic arteries, NO could not stimulate SERCA activity because of the irreversible oxidation of the cysteine-674 thiol caused by the high levels of oxidants accompanying the disease [5] (Figure 1B).
Injury, such as that induced by a balloon angioplasty catheter, results in SMC dedifferentiation and migration. This process accounts in large part for arterial restenosis after angioplasty. NO donors normally inhibit SMC migration. To test whether NO inhibits cell migration through redox regulation of SERCA activity, expression of either wild type (WT) or a serine mutant of SERCA cysteine-674 (C674S) was accomplished by stable transfection of HEK 293 cells or adenoviral expression of the proteins in rat aortic SMCs. In the cells expressing the mutant SERCA, labeling of free reactive thiols with biotinylated-iodoacetamide (BIAM) was nearly eliminated, validating the mutant. In addition, biotinylated-glutathione adducts of SERCA induced by NO were eliminated, and NO failed to increase SERCA activity or decrease Ca2+ influx, consistent with the single cysteine SERCA mutant mediating NO-dependent activation. In the absence of NO, fetal bovine serum stimulated migration of HEK cells or SMC expressing WT or C674S SERCA at similar rates. The NO donors, S-nitrosopenicillamine (SNAP) or DETA NONOate, inhibited migration of cells with WT SERCA, but had no effect on the migration of either cell type expressing C674S mutant SERCA. When iNOS was induced by interleukin-1β, the endogenous NO inhibited migration of SMC expressing WT, but not the mutant SERCA. This result may be relevant to proliferating SMC in injured arteries in which iNOS expression is induced by cytokines. Blocking cyclic GMP does not prevent the inhibition of migration by NO. These results indicate that the cyclic GMP-independent, redox regulation of SERCA cysteine-674 is required for the inhibition of cell migration by both exogenous and endogenously generated NO [6].
NO and SERCA dysfunction in cardiovascular diseases
NO-induced vasodilatation is impaired in patients with a variety of cardiovascular diseases, including diabetes, hypercholesterolemia and atherosclerosis [5;7;8]. In diseased blood vessels high levels of reactive oxygen species (ROS) are produced by vascular cells and leukocytes and these contribute importantly to the vascular dysfunction [9–11]. Potential sources of vascular ROS production include NADPH oxidases, xanthine oxidase, lipoxygenase, mitochondrial electron transport, and NO synthases (NOS). NADPH oxidases appear to be the principal source of excess vascular superoxide anion in several animal models of vascular disease. Vascular dysfunction may occur because NO is destroyed or consumed by reactions with ROS. Attempts to restore endothelium-dependent relaxation in arteries from hypercholesterolemic animals by incubating them for a few hours with antioxidants [12;13] or by adenovirus-mediated gene transfer of superoxide dismutase (SOD) [11] have largely failed. This may be due to the fact that the excess oxidants may irreversibly impair the function of proteins that are directly regulated by NO in aortic SMC, such as SERCA [5;13].
Early studies showed that hypercholesterolemia (HC) impairs acetylcholine-induced relaxation of the rabbit aorta but had little effect on that caused by the NO donor, sodium nitroprusside (SNP), suggesting that acetylcholine released less NO from the endothelium in HC[9]. However, the relaxation to authentic NO gas was found to be also impaired in HC aorta, indicating the importance of an abnormal SMC response[13]. A selective guanylyl cyclase inhibitor, ODQ, eliminated SNP-induced relaxation, but only partially blocked NO-induced relaxation of both normal and HC aorta. In arteries treated with ODQ, the residual relaxation to NO was still less in HC and, in both normal and HC aorta, was abolished by concomitant administration of the SERCA inhibitor, cyclopiazonic acid. SERCA activity was markedly decreased in HC, although SERCA 2 protein expression did not change [13]. These studies indicate that while the cyclic GMP pathway is preserved in HC animal arteries, the redox stimulation of SERCA by NO to cause relaxation is impaired, and can account for the abnormal response to acetylcholine-induced release of NO. By using mass spectrometry and sequence-specific antibodies to detect oxidized amino acid residues, the abnormal SERCA function in the hypercholesterolemic rabbit aorta can be attributed to extensive oxidant-induced tyrosine nitration and thiol oxidation of SERCA [8;14]. Both tyrosine nitration and thiol oxidation may be attributed to excess production of peroxynitrite, the chemical product of NO and superoxide [8;14].
Studies in diabetic and insulin-treated diabetic Wistar rat aorta also correlated an increase in tyrosine nitration of aortic SERCA2b with impaired aortic relaxation to acetylcholine which is also a characteristic of the diabetic state [15]. Prolonged treatment of the diabetic rat with insulin also impaired aortic relaxations that were attributed to dysfunction of the smooth muscle, and were caused, at least in part, by SERCA dysfunction [15]. In the streptozotocin-induced diabetic rat model, increased plasma angiotensin II and peroxynitrite levels impaired cyclic GMP-independent aortic relaxation, apparently by inhibiting SERCA [16]. Additionally, diabetic hypercholesterolemic pig aorta contained SERCA2b in which the cysteine-674 thiol was oxidized, and this was prevented in pigs treated with insulin. Interestingly, the irreversible oxidation of SERCA in the diabetic pig aorta corresponded to less intact 110 kDa SERCA protein and a lower molecular mass SERCA protein of approximately 70 kDa [14]. The lower molecular mass SERCA was confirmed by mass spectrometry. These studies suggest that SERCA degradation is associated with its oxidation and may play a role in progression of diabetic vascular disease [14].
A number of studies support the concept that increased aggregation of platelets contribute to the pathogenesis and progression of the vascular complications of diabetes. SERCA2 in platelets from patients with type 2 diabetes mellitus showed increased tyrosine nitration and was accompanied by inactivation of SERCA, elevated platelet free Ca2+ levels, and activated μ-calpain. The tyrosine nitration of SERCA2 and the activation of μ-calpain in platelets from healthy volunteers could be evoked by peroxynitrite in vitro, implicating the importance of the oxidation of SERCA in the diabetic patients [17].
Recent studies indicate that NO fails to inhibit migration of SMC cultured in high glucose (HG) to mimic the diabetic state. This is due to irreversible oxidation of SERCA cysteine-674 caused by ROS produced by upregulated expression of NADPH oxidase [18;19]. After exposure of SMC to HG for 3 days, NO failed to inhibit migration as it does in normal glucose (Figure 3), nor did it decrease Ca2+-dependent association of calmodulin with calcineurin, indicating that NO failed to decrease intracellular free Ca2+ levels. In SMC exposed to HG, NO produced far fewer glutathione adducts on the SERCA C674 thiol. Thiol labeling studies showed that the C674 thiol was oxidized by HG, and the sequence-specific antibody that recognized sulfonic acid oxidation of the C674 SERCA thiol in diseased arteries mentioned above, also detected that the thiol was oxidized in SMC. These results indicate that failure of NO to inhibit migration in SMC exposed to HG is due to irreversible oxidation of the SERCA cysteine-674 reactive thiol [18].
Figure 3.
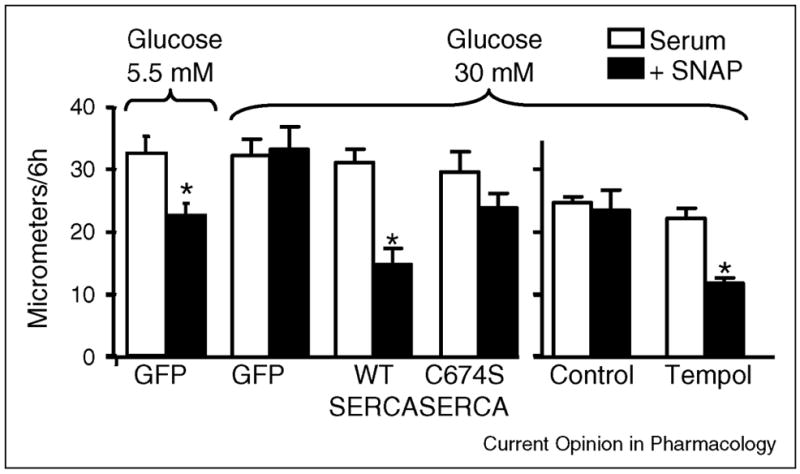
Overexpression of SERCA WT, but not SERCA C674S mutant, or application of the antioxidant, Tempol, preserves the ability of the NO donor, S-N-acetyl penicillamine (SNAP) to inhibit migration of SMC exposed to high glucose. SNAP inhibits serum-induced migration of SMC exposed to normal glucose (5.5 mmol/L), but of those exposed to high glucose (25 mmol/L). Overexpression of SERCA WT, but not SERCA C674S preserves the ability of SNAP to inhibit migration in SMC exposed to HG. N = 6 (mean ± S.E.M.). *P < 0.05, paired t-test between cells treated with or without SNAP. In uninfected SMC exposed to high glucose, treatment with Tempol has no effect on migration of cells in the absence of SNAP, and protects the ability of SNAP to inhibit migration of cells exposed to HG. N = 3, *P < 0.05, two-way ANOVA. Data from reference [18].
In SMC cultured in HG it was also noted that the mRNA levels of Nox1 and Nox4 as well as the protein level of Nox4 were significantly increased. These two proteins are isoforms of gp91phox, termed Nox2, the cytochrome containing component of the leukocyte NADPH oxidase. Studies in which either Nox1 or Nox4 siRNA was used to selectively knockdown protein expression showed that both Nox isoforms played a role in producing oxidants that were responsible for the failure of NO to inhibit SMC migration caused by HG [19].
SERCA as a therapeutic target to prevent vascular disease development
Targeting the oxidant sources in vascular diseases to prevent SERCA oxidation and/or providing new SERCA protein may improve vascular disease development. As an example, in a study of impaired endothelium-dependent relaxations in HC rabbit aorta, the antioxidant t-butylhydroxytoluene (BHT) dramatically improved the smooth muscle response to NO, and this was mirrored by improved endothelium-dependent relaxation to acetylcholine, suggesting that the beneficial effects of the antioxidant depends on improvement in the SMC response to NO (Figure 2) [8]. BHT also restored the decreased aortic SERCA activity in HC and decreased tyrosine-nitrated SERCA without changing SERCA protein expression. These beneficial effects may depend on decreasing the direct effects on SERCA of ROS that are augmented in HC [8]. In addition, improvement of endothelium-dependent relaxation precedes the regression of atherosclerosis caused by cholesterol lowering or antioxidant treatments, suggesting that improvement of the response to NO can limit the progression of atherosclerosis. The improvement in SMC function is associated with decreased oxidative modification of SERCA protein and improved Ca2+ uptake activity. This suggests that preservation of SERCA function may be regarded as a new target for the treatment of impaired vascular function.
Figure 2.
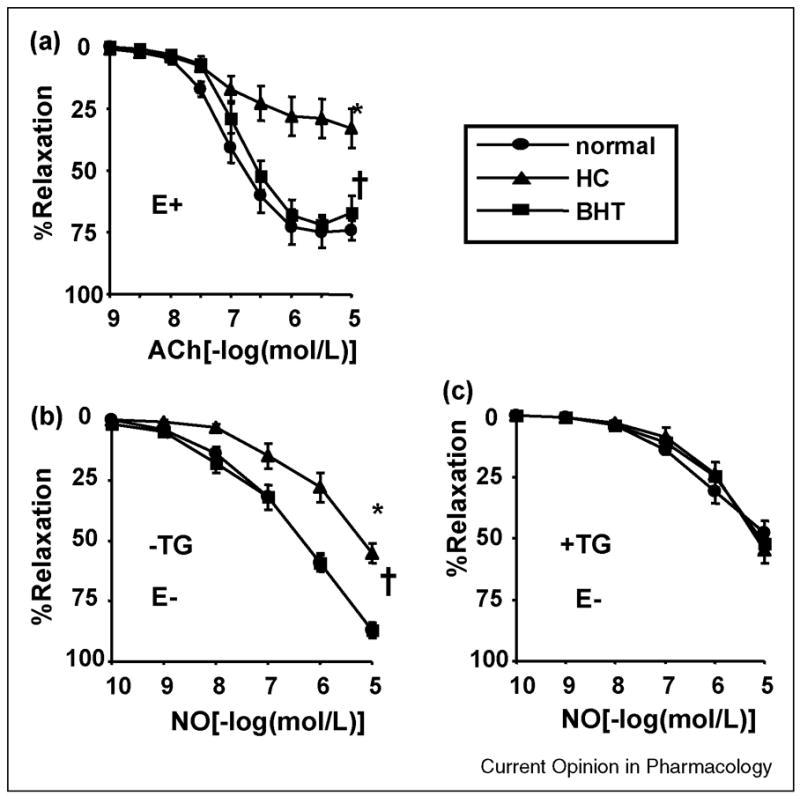
Impaired acetylcholine (ACh)- (A) and exogenous NO-induced (B) relaxation of hypercholesterolemic (HC) rabbit aorta is prevented by treatment with the antioxidant, butylated hydroxytoluene (BHT). Panel A shows ACh (normal [n=12], HC [n=11], and HC+BHT [n=9]), and panel B shows NO (normal [n=12], HC [n=10], and HC+BHT [n=9]). *P<0.05 by ANOVA. Both ACh-induced relaxations in rings with endothelium (E+) and NO-induced relaxations in rings without endothelium (E−) were markedly reduced in HC, and treatment with BHT Prevented the impairment. C. Preincubation with the SERCA inhibitor thapsigargin (10 μmol/L, 1 hour) decreased NO-induced relaxation in aortas from normal and BHT-treated rabbits but not from HC rabbits. Thapsigargin also eliminated the differences among the 3 groups, suggesting that SERCA-dependent relaxation to NO is selectively inhibited in HC and that BHT improves it. Panel C shows NO (normal [n=8], HC [n=8], and HC+BHT [n=8]). *P<0.05 vs normal; †P<0.05 vs HC. Data from reference [8].
In the streptozotocin-induced diabetic rat model, elevated plasma angiotensin II levels are improved by treatment with the angiotensin-converting enzyme inhibitor, enalapril. As angiotensin II is a powerful inducer of NADPH oxidase, it is likely that enalapril helps prevent the diabetes-related impairment of SERCA function in the aorta by suppressing ROS formation [16].
The enhanced tyrosine nitration and inactivation of SERCA2 in platelets from type 2 diabetic patients mentioned above were restored by treating the patients with the peroxisome proliferator-activated receptor-γ agonist, rosiglitazone. This was accompanied by improvements in platelet [Ca2+]i and aggregation, indicating the importance of SERCA oxidation in platelet function in diabetic patients. Rosiglitazone also partially restored inhibition of platelet aggregation by NO, consistent with improvements in SERCA redox function [17].
In a type 2 diabetic rat model, left ventricular diastolic dysfunction characterized by a slow rate of ventricular relaxation is accounted for by decreased SERCA2a protein expression. Transcoronary gene transfer with an adenoviral vector to overexpress SERCA2a increased coronary blood flow and decreased cardiomyocyte hypertrophy [20]. The increased coronary blood flow observed may have been due to the increased SERCA2a gene expression in vascular EC and SMC in coronary arteries.
SERCA 2a and SERCA 2b isoforms are expressed in a ratio of 30:70 in the media of carotid artery of adult male Sprague-Dawley rats[21]. However, two weeks after balloon-catheter injury, SERCA2a protein expression fell dramatically in both the media and the neointima, while SERCA2b was unchanged [22]. Preventing the loss of SERCA2a in the injured carotid arteries by infection with adenoviral-SERCA2a blocked the neointima formation observed in uninfected injured carotids or those infected with a β-galactosidase control vector [22]. Furthermore, overexpression of SERCA2a prevented the proliferation-induced changes in phenotype and apoptosis compared with the controls [22]. By lowering the cytosolic Ca2+ level, increased SERCA2a expression and activity can inactivate calcium/calmodulin-dependent serine/threonine-specific protein phosphatase 2b and its downstream signaling, resulting in decreased nuclear factor of activated T lymphocytes (NFAT) transcriptional activity [22]. Although SERCA 2a and 2b do not differ in structure around cysteine-674 and would therefore both be expected to be similarly redox-regulated, studies in the authors’ laboratory have been performed with the SERCA 2b isoform. The stability of its expression in injured arteries suggest that redox regulation of the 2b isoform may be key to SERCA function in diseased arteries in which it may be the sole isoform. Currently, there are a number of clinical trials of SERCA2a gene therapy for the treatment of patients with heart failure using adeno-associated vectors [23]. Even though these trials are targeting patients with heart failure, their safety profile may pave the way for SERCA2a gene therapy for the treatment of vascular diseases.
In studies of SMC exposed to HG, the antioxidant, Tempol, or adenoviral mediated overexpression of SOD prevented the effects of HG (Figure 3) [18]. Overexpression of SERCA2b WT, but not the SERCA2b C674S mutant, also preserved the ability for NO to inhibit migration despite exposing the cells to HG (Figure 3). This indicates that despite the fact that the C674S mutant has normal basal activity, that the redox regulation of cysteine-674 is key to the restoration of NO function. Interestingly, HG still oxidized 70% of the cysteine-674 thiol in the overexpressed SERCA WT protein. However, the remaining 30% amounted to a 2.5-fold excess in the total reduced thiol on SERCA that was available for S-glutathiolation and stimulation of activity compared to uninfected cells. These experiments indicate that overexpression of SERCA may achieve part of its therapeutic effect by mechanisms involving the redox regulation of cysteine-674, which can be preserved by antioxidants such as Tempol and BHT, increasing the expression of antioxidant enzymes, or inhibiting the expression of NADPH oxidase. Notably, Tempol has been shown to prevent the excess restenosis observed in the fructose-induced Type 2 diabetic rat [24] perhaps in part via its effects on SERCA.
Recent experiments performed on insulin-resistant Zucker obese rats indicate that upregulation of NADPH oxidase expression in vivo leads to oxidation of SERCA cysteine-674, making it insensitive to NO. Interestingly, the increased oxidants and impaired responsiveness to NO persisted when the SMC from the Zucker obese rats were cultured, whereas SMC from lean Zucker control rats functioned normally. This phenomenon appears to be explained by persistent excess production of ROS that oxidize SERCA.
Conclusions
In summary, NO normally stimulates Ca2+ uptake activity of SERCA by increasing S-glutathione adducts on the reactive cysteine-674 thiol. This reactive SERCA thiol also undergoes irreversible oxidation caused by increased ROS in vascular disease. Antioxidant scavengers or inhibiting excess ROS production can prevent the irreversible oxidation of SERCA and maintain vascular function. Expressing new functional SERCA protein in diseased cardiovascular tissues may improve function, and may in part rely upon restoration of sufficient reduced thiol on cysteine-674 to allow for its redox regulation. Recognition that both physiological and pathophysiological levels of ROS regulate cell functions by direct modifications of SERCA not only brings new understanding of the way in which ROS regulate cell function, but also may suggest new strategies to prevent vascular disease development.
Acknowledgments
This work was supported by NIH grants: R01 HL031607, PO1 HL081587, R01 AG27080 and P01 HL068758. A. Evangelista is supported by an NIH training grant T32 HL 007501-26. The authors thank Roger Hajjar, Director of the Cardiovascular Research Center, Mount Sinai Medical Center, New York for his helpful suggestions for this review.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.MacLennan DH, Rice WJ, Green NM. The mechanism of Ca2+ transport by sarco(endo)plasmic reticulum Ca2+-ATPases. J Biol Chem. 1997;272:28815–28818. doi: 10.1074/jbc.272.46.28815. [DOI] [PubMed] [Google Scholar]
- 2.Magnier-Gaubil C, Herbert JM, Quarck R, Papp B, Corvazier E, Wuytack F, Levy-Toledano S, Enouf J. Smooth muscle cell cycle and proliferation. Relationship between calcium influx and sarco-endoplasmic reticulum Ca2+ATPase regulation. J Biol Chem. 1996;271:27788–27794. doi: 10.1074/jbc.271.44.27788. [DOI] [PubMed] [Google Scholar]
- 3.Magnier C, Papp B, Corvazier E, Bredoux R, Wuytack F, Eggermont J, Maclouf J, Enouf J. Regulation of sarco-endoplasmic reticulum Ca(2+)-ATPases during platelet-derived growth factor-induced smooth muscle cell proliferation. J Biol Chem. 1992;267:15808–15815. [PubMed] [Google Scholar]
- 4.Dode L, Vilsen B, Van BK, Wuytack F, Clausen JD, Andersen JP. Dissection of the functional differences between sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) 1 and 3 isoforms by steady-state and transient kinetic analyses. J Biol Chem. 2002;277:45579–45591. doi: 10.1074/jbc.M207778200. [DOI] [PubMed] [Google Scholar]
- 5•.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. This paper demonstrates that S-glutathiolation of SERCA cysteine-674 by peroxynitrite increases SERCA Ca2+ uptake activity. The paper also shows that glutathiolation of SERCA occurs during arterial relaxation to NO, but that in atherosclerotic rabbit aorta, cysteine-674 is oxidized, preventing the glutathiolation and stimulation of SERCA activity. [DOI] [PubMed] [Google Scholar]
- 6•.Ying J, Tong X, Pimentel DR, Weisbrod RM, Trucillo MP, Adachi T, Cohen RA. Cysteine-674 of the sarco/endoplasmic reticulum calcium ATPase is required for the inhibition of cell migration by nitric oxide. Arterioscler Thromb Vasc Biol. 2007;27:783–790. doi: 10.1161/01.ATV.0000258413.72747.23. The authors demonstrate that the cyclic GMP-independent, redox regulation of SERCA cysteine-674 is required for the inhibition of smooth muscle cell migration by both exogenous and endogenously generated NO. This is the first study that shows that redox regulation of SERCA cysteine-674 by NO is involved in smooth muscle cell migration. [DOI] [PubMed] [Google Scholar]
- 7.Williams SB, Cusco JA, Roddy MA, Johnstone MT, Creager MA. Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J Am Coll Cardiol. 1996;27:567–574. doi: 10.1016/0735-1097(95)00522-6. [DOI] [PubMed] [Google Scholar]
- 8.Adachi T, Matsui R, Xu S, Kirber M, Lazar HL, Sharov VS, Schoneich C, Cohen RA. Antioxidant improves smooth muscle sarco/endoplasmic reticulum Ca(2+)-ATPase function and lowers tyrosine nitration in hypercholesterolemia and improves nitric oxide-induced relaxation. Circ Res. 2002;90:1114–1121. doi: 10.1161/01.res.0000019757.57344.d5. [DOI] [PubMed] [Google Scholar]
- 9.Ohara Y, Peterson TE, Harrison DG. Hypercholesterolemia increases endothelial superoxide anion production. J Clin Invest. 1993;91:2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guzik TJ, West NE, Black E, McDonald D, Ratnatunga C, Pillai R, Channon KM. Vascular superoxide production by NAD(P)H oxidase: association with endothelial dysfunction and clinical risk factors. Circ Res. 2000;86:E85–E90. doi: 10.1161/01.res.86.9.e85. [DOI] [PubMed] [Google Scholar]
- 11.Miller FJ, Jr, Gutterman DD, Rios CD, Heistad DD, Davidson BL. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res. 1998;82:1298–1305. doi: 10.1161/01.res.82.12.1298. [DOI] [PubMed] [Google Scholar]
- 12.Adachi T, Cohen RA. Decreased aortic glutathione levels may contribute to impaired nitric oxide-induced relaxation in hypercholesterolaemia. Br J Pharmacol. 2000;129:1014–1020. doi: 10.1038/sj.bjp.0703127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adachi T, Matsui R, Weisbrod RM, Najibi S, Cohen RA. Reduced sarco/endoplasmic reticulum Ca2+ uptake activity can account for the reduced response to NO, but not sodium nitroprusside, in hypercholesterolemic rabbit aorta. Circ. 2001;104:1040–1045. doi: 10.1161/hc3501.093798. [DOI] [PubMed] [Google Scholar]
- 14.Ying J, Sharov V, Xu S, Jiang B, Gerrity R, Schoneich C, Cohen RA. Cysteine-674 oxidation and degradation of sarcoplasmic reticulum Ca(2+) ATPase in diabetic pig aorta. Free Radic Biol Med. 2008;45:756–762. doi: 10.1016/j.freeradbiomed.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kobayashi T, Taguchi K, Takenouchi Y, Matsumoto T, Kamata K. Insulin-induced impairment via peroxynitrite production of endothelium-dependent relaxation and sarco/endoplasmic reticulum Ca(2+)-ATPase function in aortas from diabetic rats. Free Radic Biol Med. 2007;43:431–443. doi: 10.1016/j.freeradbiomed.2007.04.019. [DOI] [PubMed] [Google Scholar]
- 16.Taguchi K, Kobayashi T, Hayashi Y, Matsumoto T, Kamata K. Enalapril improves impairment of SERCA-derived relaxation and enhancement of tyrosine nitration in diabetic rat aorta. Eur J Pharmacol. 2007;556:121–128. doi: 10.1016/j.ejphar.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 17•.Randriamboavonjy V, Pistrosch F, Bolck B, Schwinger RH, Dixit M, Badenhoop K, Cohen RA, Busse R, Fleming I. Platelet sarcoplasmic endoplasmic reticulum Ca2+-ATPase and mu-calpain activity are altered in type 2 diabetes mellitus and restored by rosiglitazone. Circulation. 2008;117:52–60. doi: 10.1161/CIRCULATIONAHA.107.719807. The authors demonstrate that in type 2 diabetic patients, the peroxisome proliferator-activated receptor-γ agonist rosiglitazone increases platelet SERCA2 expression and Ca2+-ATPase activity, decreases SERCA2 tyrosine nitration, and normalizes platelet intracellular calcium. This is the first study to show that targeting SERCA oxidation to restore platelet function in diabetes. [DOI] [PubMed] [Google Scholar]
- 18•.Tong X, Ying J, Pimentel DR, Trucillo M, Adachi T, Cohen RA. High glucose oxidizes SERCA cysteine-674 and prevents inhibition by nitric oxide of smooth muscle cell migration. J Mol Cell Cardiol. 2008;44:361–369. doi: 10.1016/j.yjmcc.2007.10.022. The authors demonstrate that there is abnormal migration response to NO in smooth muscle cell cultured in high glucose, which is involved in the oxidation of SERCA2b cysteine 674. This is the first study to show that the oxidation of SERCA by high glucose causes NO dysfunction, and overexpression SERCA wide type can restore NO function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19•.Tong X, Schroder K. NADPH oxidases are Responsible for the Failure of Nitric Oxide to Inhibit Migration of Smooth Muscle Cells exposed to High Glucose. Free Radic Biol Med. 2009 doi: 10.1016/j.freeradbiomed.2009.08.026. The authors demonstrate that the upregulated NADPH oxidases are responsible for the failure of NO to inhibit SMC migration caused by high glucose. This is the first study to show that targeting NADPH oxidase in high glucose can decrease SERCA oxidation and restore NO function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakata S, Lebeche D, Sakata Y, Sakata N, Chemaly ER, Liang L, Nakajima-Takenaka C, Tsuji T, Konishi N, del MF, Hajjar RJ, Takaki M. Transcoronary gene transfer of SERCA2a increases coronary blood flow and decreases cardiomyocyte size in a type 2 diabetic rat model. Am J Physiol Heart Circ Physiol. 2007;292:H1204–H1207. doi: 10.1152/ajpheart.00892.2006. [DOI] [PubMed] [Google Scholar]
- 21.Levitsky DO, Clergue M, Lambert F, Souponitskaya MV, Le Jemtel TH, Lecarpentier Y, Lompre AM. Sarcoplasmic reticulum calcium transport and Ca(2+)-ATPase gene expression in thoracic and abdominal aortas of normotensive and spontaneously hypertensive rats. J Biol Chem. 1993;268:8325–8331. [PubMed] [Google Scholar]
- 22•.Lipskaia L, del Monte F, Capiod T, Yacoubi S, Hadri L, Hours M, Hajjar RJ, Lompre AM. Sarco/endoplasmic reticulum Ca2+-ATPase gene transfer reduces vascular smooth muscle cell proliferation and neointima formation in the rat. Circ Res. 2005;97:488–495. doi: 10.1161/01.RES.0000180663.42594.aa. The authors demonstrate that infection of SERCA2a in injured rat common carotid artery prevents neointima formation. This is the first study to show that overexpression of SERCA in vivo prevents neointima formation. [DOI] [PubMed] [Google Scholar]
- 23.Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail. 2009;15:171–181. doi: 10.1016/j.cardfail.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jagadeesha DK, Lindley TE, Deleon J, Sharma RV, Miller F, Bhalla RC. Tempol therapy attenuates medial smooth muscle cell apoptosis and neointima formation after balloon catheter injury in carotid artery of diabetic rats. Am J Physiol Heart Circ Physiol. 2005;289:H1047–H1053. doi: 10.1152/ajpheart.01071.2004. [DOI] [PubMed] [Google Scholar]