

Abstract
We recently described a fluorescence polarization platform for competitive activity-based protein profiling (fluopol-ABPP) that enables high-throughput inhibitor screening for enzymes with poorly characterized biochemical activity. Here, we report the discovery of a class of oxime ester inhibitors for the unannotated serine hydrolase RBBP9 from a full-deck (200,000+ compound) fluopol-ABPP screen conducted in collaboration with the Molecular Libraries Screening Center Network (MLSCN). We show that these compounds covalently inhibit RBBP9 by modifying the enzyme’s active site serine nucleophile and, based on competitive ABPP in cell and tissue proteomes, are selective for RBBP9 relative to other mammalian serine hydrolases.
In both academia and industry, high-throughput screening (HTS) of large compound libraries has emerged as a powerful means to identify lead scaffolds for chemical probes and drugs1, 2. A wide array of HTS-compatible assays have been introduced, ranging from classical in vitro substrate assays for enzyme inhibitors to in situ screens that profile cellular phenotypes. Still, the development of an accurate and reproducible assay, the critical first step in an HTS campaign, can be difficult, even for well-characterized biological systems. For example, a target-based HTS program that seeks to identify modulators of a specific protein must optimize the readout of biochemical activity so that it is consistent between wells and plates, has enough sensitivity to detect compounds with weak activity, and is economically feasible. As a consequence, the estimated 30–50% of the human proteome with poorly characterized biochemical activities has largely remained outside the general scope of HTS.
To address this problem, our laboratory has recently adapted the chemical proteomic technology activity-based protein profiling (ABPP)3, 4 for HTS5. ABPP employs reactive chemical probes to covalently label the active sites of mechanistically related enzymes, regardless of their degree of biochemical annotation, enabling the direct evaluation of the functional state of either purified enzymes or enzymes in complex biological systems. ABPP can be performed in a competitive format to discover lead inhibitors, where compounds are assayed for their ability to impede probe labeling of enzymes6, 7. Importantly, this strategy, when employed in complex proteomes, enables the simultaneous optimization of both the potency and selectivity of inhibitors against many enzymes in parallel. Competitive ABPP has traditionally required a gel-based readout, limiting the throughput to hundreds of compounds, but has nevertheless led to the identification of many selective inhibitors6–10, including several for uncharacterized enzymes8, 10.
To create an HTS-amenable version of competitive ABPP, we modified this platform such that probe labeling of purified enzymes could be monitored by fluorescence polarization (fluopol-ABBP)5. We initially applied fluopol-ABPP to the retinoblastoma-binding protein-9 (RBBP9), a putative serine hydrolase that reacts with reporter-tagged flurophosphonate probes [FP-biotin and FP-rhodamine (FP-Rh)]11, 12. RBBP9, originally discovered as a protein that confers resistance to the growth-inhibitory effects of TGF-β1, has been shown to bind the retinoblastoma (Rb) protein and transform rat epithelial cell lines13. Most recently, RBBP9 has been found to promote anchorage-independent growth and pancreatic carcinogenesis through overriding TGF-β-mediated antiproliferative signaling14. Although these data suggest that RBBP9 plays an important role in cancer, the biochemical function of this enzyme and identity of its endogenous substrate remain unknown.
From an initial ~20,000 compound fluopol-ABPP screen, we identified the natural product emetine as a reversible inhibitor of RBBP9 that selectively blocked FP-Rh labeling of this enzyme compared to other members of the serine hydrolase family5 (Fig. 1). Although emetine does not interact with other serine hydrolases, it is a cytotoxic natural product and has been shown to inhibit translation in vitro, possibly through direct interactions with the ribosome15. Emetine has also been shown to antagonize of α2-adrenergic receptors16. As these additional targets could complicate the use of emetine in biological studies of RBBP9, we sought to identify additional classes of inhibitors for this enzyme. Toward this end, we report herein the discovery of an oxime ester class of inhibitors of RBBP9 from a 200,000+ compound fluopol-ABPP screen. We show that these oxime ester compounds covalently modify the active site serine of RBBP9 and are selective for RBBP9 relative to other mammalian serine hydrolases in proteomes.
Figure 1.

Structures of HTS lead RBBP9 inhibitors. Normalized percent inhibition of RBBP9 (compounds at 7.94 µM) from fluopol-ABPP PubChem Bioassay AID 1537 is shown in parenthesis.
In collaboration with the Molecular Libraries Screening Center Network at The Scripps Research Institute, we screened 217,969 individual compounds (7.94 µM) for RBBP9 inhibition by fluopol-ABPP (PubChem AID: 1515). A robust average Z′ factor (0.75) enabled the identification of 445 potential inhibitors, defined as those that reduced the RBBP9-induced fluorescence polarization signal by greater than 14.2%. A confirmation screen, performed identically to the primary screen and assaying hit compounds in triplicate, verified 137 as active (PubChem AID: 1537). Notably, this group included the natural product emetine, confirming the reproducibility of our small- and large-scale screens. The confirmed hit compounds were next analyzed by gel-based competitive ABPP in the membrane fraction of mouse brain doped with exogenous RBBP9 and in the soluble fraction of RBBP9-transfected HEK 293T cells (Fig. 2 A,B). Briefly, the proteomes were incubated with compounds (20 µM) for 30 minutes, followed by addition of FP-Rh (1 µM). The reactions were quenched after 10 minutes, separated by SDS-PAGE, and quantified by in-gel fluorescence scanning. These assays enabled us to discard dozens of weak and/or promiscuous inhibitors and aggregation-based inhibitors (which showed a characteristic profile of blocking probe labeling of most serine hydrolases in the soluble proteome, while exhibiting negligible activity in the membrane proteome5) in favor of compounds that selectively inhibited RBBP9, including three with oxime ester moieties (inhibitors 2–4, Fig. 1).
Figure 2.

Selectivity profiling of oxime ester RBBP9 inhibitors. (A) Evaluation of RBBP9 inhibitors by competitive ABPP with the FP-Rh probe in the RBBP9-transfected HEK 293T soluble proteome at 20 µM (left panel) and at 200 µM (right panel). Fluorescent gels are shown in gray scale. (B) Evaluation of the selectivity of oxime ester 3 at indicated concentrations and (C) oxime ester 18 at 200 µM by competitive ABPP in the mouse brain membrane proteome doped with recombinant RBBP9.
Compounds 2 and 3, differing only in the para substituent of the phenyl ring were highly selective for RBBP9, inhibiting only a single additional hydrolase target (75 kDa) at high concentrations (Fig. 2B). Compound 4, a differently substituted oxime ester possessing two potentially cysteine-reactive chemical moieties (see below), was less selective for RBBP9, inhibiting the same 75 kDa hydrolase described above, as well as 30 and 40 kDa hydrolases in the HEK 293T soluble proteome (Fig. 2A).
The IC50 values of compounds 1–4 against purified RBBP9 as determined by gel-based ABPP correlated closely with the percent inhibition at 7.94 µM observed in the fluopol-ABPP confirmation screen (Fig. 1 and Table 1, Table 2). Emetine (1) inhibited RBBP9 in the initial fluopol-ABPP assay by 51% and, remarkably, inhibited this enzyme in the gel-based assay with an IC50 of 7.8 µM. Compounds 2 and 4 both inhibited RBBP9 in the HTS confirmation screen by ~80% and gave more potent IC50 values (1.9 µM and 1.2 µM, respectively) in the gel-based assay. Compound 3 inhibited RBBP9 by 39% in the confirmation screen and had the lowest gel-based IC50 value (9.2 µM) of these compounds. This demonstrates that the relative potency of inhibitors can be extrapolated from the percent inhibition value in a fluopol-ABPP screen.
Table 1.
Inhibition of RBBP9 by thiazole-containing ester-oxime compounds.
 | |||
|---|---|---|---|
| Compdsa | R | ABPP IC50 (µM)b |
Inhibition (20 µM)c |
|
1 (emetine) |
- | 7.8 | 62 |
| 2 | 4-Cl-Ph | 1.9 | 78 |
| 3 | 4-OMe-Ph | 9.2 | 60 |
| 5 | 4-F-Ph | 5.7 | 76 |
| 6 | 4-I-Ph | NDd | 66 |
| 7 | 2,5-(F2)-Ph | ND | 66 |
| 8 | 2-F-Ph | ND | 64 |
| 9 | 3-Cl-Ph | ND | 63 |
| 10 | Ph | ND | 62 |
| 11 | 3-CF3-Ph | ND | 46 |
| 12 | 4-NO2-Ph | ND | 15 |
| 13 | 4-Ph-Ph | ND | 4 |
| 14 |  |
ND | 17 |
| 15 |  |
ND | 8 |
| 16 |  |
ND | 14 |
| 17 | ND | 42 | |
| 18 | Cyclohexyl | 0.64 | 91 |
| 19 | Me | ND | 0 |
| 20 | ND | 0 | |
Compounds 6, 10, 12, 13, 17, and 20 were synthesized following previously reported methods17. Emetine was purchased from Sigma, compounds 2–5, 7–9, 11, 14–16, and 18 were purchased from Key Organics, and compound 19 was purchased from ChemBridge.
IC50 values were determined by three independent gel-based competitive ABBP experiments with purified RBBP9.
Percent inhibition of purified RBBP9 at 20 µM.
Not determined.
Table 2.
RBBP9 inhibitory activities of compound 3 and 4 derivatives.
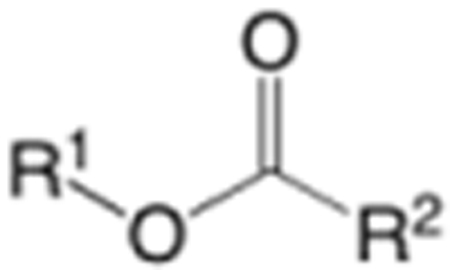 | ||||
|---|---|---|---|---|
| Compdsa | R1 | R2 | ABPP IC50 (µM) |
Inhibition (20 µM) |
| 4 |  |
1.2 | 78 | |
| 21 |  |
4-OMe-Ph | 1.5 | 93 |
| 22 | 4-OMe-Ph | ND | 76 | |
| 23 |  |
4-OMe-Ph | ND | 48 |
| 24 |  |
 |
ND | 42 |
Compounds 4 and 21–23 were purchased from ChemBridge. Compound 24 was synthesized following previously reported methods.17
We hypothesized that these oxime ester inhibitors were acting as “slow-turnover substrate” inhibitors of RBBP9 by covalently acylating the active serine nucleophile (Ser75) of the enzyme. Consistent with a covalent mode of inhibition, and unlike the reversible inhibitor emetine (1), blockade of FP-rhodamine RBBP9 labeling by compound 2 was not reversed by gel filtration of the inhibitor 2-RBPP9 reaction (Fig. 3A). To identify the site of covalent labeling, we incubated RBBP9 with either DMSO or compound 2, digested the enzyme with trypsin, and analyzed the resulting peptides by 1D reverse-phase LC-MS/MS. The MS/MS spectra were searched via the SEQUEST algorithm against a composite database containing the human IPI protein database, allowing for static modification of 57.02 Da on cysteine and differential modification of 138.00 Da on serine and 80.98 Da on cysteine. This analysis revealed an adduct only on the active site serine nucleophile (Ser75) of RBBP9 in the compound 2-treated sample (Fig. 3B). This modification is consistent with nucleophilic attack by Ser75 at the carbonyl carbon of 2 and displacement of the oxime leaving group. These data demonstrate that the oxime ester compounds inhibit RBBP9 by covalently labeling the enzyme’s serine nucleophile.
Figure 3.

Oxime ester compounds covalently inhibit RBBP9. (A) Recombinant RBBP9 was incubated with DMSO or inhibitor (100 µM) and each reaction was split into two fractions. One fraction was reacted directly with FP-Rh (left panels), and the other was filtered and then reacted with FP-Rh (right panels) to assess the reversibility of inhibition. (B) RBBP9 was incubated with DMSO or compound 2, digested with trypsin, and analyzed by LC-MS/MS. The spectral counts of peptides with a +138.00 Da modification on Ser75 are shown.
Since the highly similar compounds 2 and 3 were more selective than compound 4 in our initial competitive ABPP experiments performed in cell and tissue proteomes, we next evaluated a panel of thiazole-containing oxime ester analogs for RBBP9 inhibitory activity (Table 1). Neither introduction of an unsubstituted aryl ring (compound 10) nor varying halogen substitution on the aryl ring (compounds 5–9) had much effect on activity. These compounds were all moderately less potent than compound 2, with inhibition at 20 µM ranging from 60–80%. The addition of bulky groups to the aryl ring decreased activity, with a meta CF3 (compound 11) reducing inhibition of RBBP9 to only 46% at 20 µM and a para phenyl (compound 13) completely ablating all inhibitory activity. This suggests that RBBP9 may have a small active site without much room to accommodate large groups in the acyl-enzyme adduct. Consistent with additional steric bulk reducing activity, compounds 14–16 exhibited inhibition of RBBP9 below 20% at 20 µM. In addition, the introduction of a strongly electron-withdrawing para NO2 group (compound 12) also resulted in a loss of RBBP9 inhibitory activity, indicating that a more electrophilic ester may be unable to react with or form a stable adduct with RBBP9.
Interestingly, replacement of the aryl ring with a cylcohexyl ring (compound 18) generated the most potent inhibitor of RBBP9 yet identified (Table 1). Compound 18 exhibited a sub-µM IC50 (0.64 µM) against recombinant RBBP9, 3-fold lower than the hit compound 2, and inhibited 91% of RBBP9 activity at 20 µM. Moreover, this compound did not inhibit any other hydrolases, even at 200 µM, in the mouse brain membrane proteome (Fig. 2C), and was also selective for RBBP9 in HEK 293T cell proteomes (data not shown). The alkyl substituted analog 1-butyne (17), however, was significantly less potent (42% inhibition at 20 µM) and methyl-substituted compound (19) exhibited no detectable inhibitory activity. We speculated the loss of inhibition by compound 19 may be due to the less-hindered methyl group forming an unstable adduct that is quickly hydrolyzed by water. Because even the best oxime ester inhibitors likely also act as slowly turned over substrates for RBBP9, we synthesized the analogous carbamate (compound 20), a chemical class well-known to form stable covalent adducts with serine hydrolases8, 9, of our potent inhibitor 18 to potentially further increase the stability of this compound’s covalent adduct. Unfortunately, however, carbamate 20 exhibited no inhibition of RBBP9.
To more fully explore the potential of oxime ester compounds to inhibit RBBP9, we next investigated the activity of the hit compound 4 (Table 2). As shown above, compound 4 inhibited several serine hydrolases in addition to RBBP9 (Fig. 2A). Since compound 4 also has two potentially thiol-reactive chemical groups, we reasoned that it may also inhibit proteins sensitive to alkylating agents, particularly enzymes with catalytic cysteine nucleophiles. To assay for thiol-reactivity, we profiled our hit compounds by gel-based competitive ABPP in the MDA-MB-231 proteome using the phenylsulfonate-rhodamine (PS-Rh) probe, which reacts with several cysteine-dependent enzymes18 (Fig. 4A). As expected, compounds 1–3 exhibited no significant thiol cross-reactivity as judged by a lack of effect on PS-Rh proteome labeling. However, 4 potently inhibited PS-Rh labeling of glutathione transferase omega 1 (GSTO1), an enzyme with a catalytic cysteine nucleophile19. We believe that this competitive ABPP assay can be used as a general screen to identify cysteine-reactive groups in small molecules.
Figure 4.

(A) Selectivity profiling of RBBP9 hit compounds (20 µM) for thiol-crossreactivity using a cysteine-reactive phenylsulfonate-rhodamine (PS-Rh) probe (5 µM, 1 hour). (B) Evaluation of oxime ester 21 by competitive ABPP in the mouse brain membrane proteome doped with recombinant RBBP9.
When the R2 group of inhibitor 4 was replaced with the 4-OMe-Ph group from compound 3, the resulting chimeric compound 21 no longer inhibited GSTO1, indicating that the unsaturated ketone was the site of thiol-reactivity (Table 2, Fig. 4A). Furthermore, compound 21 exhibited potent activity against the recombinant RBBP9 (IC50 = 1.5 µM) and retained good selectivity for RBBP9 in the mouse brain membrane proteome (Fig. 4B). In addition, we observed that repositioning of one methyl group (compound 22) on the dienone closer to the ester resulted in a loss of RBBP9 inhibitory activity, an effect exacerbated by the repositioning of both of the methyl groups (compound 23). This indicates that increased steric bulk on the leaving group side of the molecule near the carbonyl attacked by RBBP9’s serine nucleophile also hinders potency. Finally, we observed that the opposite chimeric compound 24 containing the unsaturated ketone with a thiazole oxime leaving group had significantly reduced activity against RBBP9 (Table 2), suggesting that an unsaturated ketone at the R2 position is sub-optimal for RBBP9 inhibition.
In conclusion, we have discovered a class of oxime ester compounds as selective, covalent inhibitors for the uncharacterized hydrolase RBBP9. We believe that these compounds, as well as the natural product emetine, can serve as useful scaffolds for future optimization of RBPP9 inhibitors. A couple potential challenges, however, should also be mentioned. As activated esters, the oxime esters may only form covalent adducts with RBBP9 for a limited period of time before being hydrolyzed by water. Moreover, these compounds could be degraded by promiscuous esterases in living cells and animals, reducing their bioavailability. Further improvements in oxime ester activity could derive from modifications that maintain binding interactions with RBBP9 while limiting the rate of hydrolysis of the parent compound and acyl-enzyme adduct. In this regard, we should note that ester inhibitors that stably acylate other serine hydrolases with excellent potency have been developed as probes and even drugs (e.g., lactones, such as tetrahydrolipstatin20). We also speculate that the structure-activity relationship analysis garnered in this study could provide lead fragments for incorporation into other inhibitory scaffolds, analogous to the substrate activity screening (SAS) approach for enzyme inhibitor identification pioneered by Ellman and coworkers 21. In this method, active site-binding substrates are converted into more potent inhibitors by combining them with mechanism-based pharmacophores (e.g., electrophilic warheads). Although our initial attempt to convert oxime ester 18 into the analogous carbamate 20 failed to produce an inhibitor of RBBP9, we suspect that future medicinal chemistry efforts could identify a more suitable warhead, for example a boronate, trifluoromethylketone, or α-ketoheterocycle. More generally, we anticipate that continued efforts following the general path outlined in this study—from a primary fluopol-ABPP screen, to selectivity screens by gel-based competitive ABPP in cell and tissues proteomes, to follow up medicinal chemistry efforts to refine potency and selectivity—will find general utility as a platform to discover and optimize novel inhibitors for the large number of uncharacterized enzymes that populate eukaryotic and prokaryotic proteomes.
Acknowledgements
We thank Mr. Pierre Baillargeon and Mrs. Lina DeLuca (Lead Identification Division, TSRI Florida) for their assistance with compound management. This work was supported by the National Institutes of Health (CA132630, MH084512), a National Science Foundation Predoctoral Fellowship (D.A.B.), and the Skaggs Institute for Chemical Biology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Inglese J, Johnson RL, Simeonov A, Xia M, Zheng W, Austin CP, Auld DS. Nat Chem Biol. 2007;3:466. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- 2.Shelat AA, Guy RK. Nat Chem Biol. 2007;3:442. doi: 10.1038/nchembio0807-442. [DOI] [PubMed] [Google Scholar]
- 3.Evans MJ, Cravatt BF. Chem Rev. 2006;106:3279. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]
- 4.Cravatt BF, Wright AT, Kozarich JW. Annu Rev Biochem. 2008;77:383. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 5.Bachovchin DA, Brown SJ, Rosen H, Cravatt BF. Nat. Biotechnol. 2009;27:387. doi: 10.1038/nbt.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leung D, Hardouin C, Boger DL, Cravatt BF. Nat. Biotechnol. 2003;21:687. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- 7.Greenbaum D, Baruch A, Hayrapetian L, Darula Z, Burlingame A, Medzihradszky KF, Bogyo M. Mol. Cell. Proteomics. 2002;1:60. doi: 10.1074/mcp.t100003-mcp200. [DOI] [PubMed] [Google Scholar]
- 8.Li W, Blankman JL, Cravatt BF. J. Am. Chem. Soc. 2007;129:9594. doi: 10.1021/ja073650c. [DOI] [PubMed] [Google Scholar]
- 9.Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Nat Chem Biol. 2009;5:37. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang KP, Niessen S, Saghatelian A, Cravatt BF. Chem Biol. 2006;13:1041. doi: 10.1016/j.chembiol.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y, Patricelli MP, Cravatt BF. Proc. Natl. Acad. Sci. U.S.A. 1999;96:14694. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patricelli MP, Giang DK, Stamp LM, Burbaum JJ. Proteomics. 2001;1:1067. doi: 10.1002/1615-9861(200109)1:9<1067::AID-PROT1067>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 13.Woitach JT, Zhang M, Niu CH, Thorgeirsson SS. Nat Genet. 1998;19:371. doi: 10.1038/1258. [DOI] [PubMed] [Google Scholar]
- 14.Shields DJ, Niessen S, Murphy EA, Mielgo A, Desgrosellier JS, Lau SK, Barnes LA, Lesperance J, Bouvet M, Tarin D, Cravatt BF, Cheresh DA. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0911646107. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grollman AP. Proc Natl Acad Sci U S A. 1966;56:1867. doi: 10.1073/pnas.56.6.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keiser MJ, Roth BL, Armbruster BN, Ernsberger P, Irwin JJ, Shoichet BK. Nat Biotechnol. 2007;25:197. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- 17.Jeong HJ, Park YD, Park HY, Jeong IY, Jeong TS, Lee WS. Bioorg Med Chem Lett. 2006;16:5576. doi: 10.1016/j.bmcl.2006.08.031. [DOI] [PubMed] [Google Scholar]
- 18.Adam GC, Sorensen EJ, Cravatt BF. Nat Biotechnol. 2002;20:805. doi: 10.1038/nbt714. [DOI] [PubMed] [Google Scholar]
- 19.Adam GC, Burbaum JJ, Kozarich JW, Patricelli MP, Cravatt BF. J. Am. Chem. Soc. 2004;126:1363. doi: 10.1021/ja038441g. [DOI] [PubMed] [Google Scholar]
- 20.Hadvary P, Sidler W, Meister W, Vetter W, Wolfer H. J Biol Chem. 1991;266:2021. [PubMed] [Google Scholar]
- 21.Wood WJ, Patterson AW, Tsuruoka H, Jain RK, Ellman JA. J Am Chem Soc. 2005;127:15521. doi: 10.1021/ja0547230. [DOI] [PubMed] [Google Scholar]